

Abstract
Liver cirrhosis is the main risk factor for the development of hepatocellular carcinoma (HCC). Activated hepatic stellate cells (HSC) are the effector cells of hepatic fibrosis and also infiltrate the HCC stroma where they might play a critical role in HCC progression. Here we aimed to analyze the effects of activated HSC on the proliferation and growth of HCC cell lines in vitro and in vivo. Conditioned media (CM) collected from HSC significantly induced proliferation and migration of HCC cells cultured in monolayers. In a 3‐dimensional spheroid coculture system, HSC promoted HCC growth and diminished the extent of central necrosis. In accordance, in vivo simultaneous implantation of HSC and HCC cells into nude mice promoted tumor growth and invasiveness, and inhibited necrosis formation. As potential mechanism of the tumorigenic effects of HSC we identified activation of NFkappaB and extracellular‐regulated kinase (ERK) in HCC cells, two signaling cascades that play a crucial role in HCC progression. In summary, our data indicate that stromal HSC promotes HCC progression and suggest the HSC–HCC interaction as an interesting tumor differentiation‐independent target for therapy of this highly aggressive cancer. (Cancer Sci 2009; 100: 646–653)
Primary hepatocellular carcinoma (HCC) is one of the most fatal cancers in humans with rising incidence in many regions around the world.( 1 ) The majority of HCC patients have a background of chronic liver disease, and the presence of liver cirrhosis is the main risk factor for the development of HCC.( 2 , 3 )
The activation of hepatic stellate cells (HSC) is recognized as a central event in the development of hepatic fibrosis and lastly, cirrhosis. Upon hepatic injury, HSC transform to an activated, myofibroblast‐like phenotype that is responsible for the excessive hepatic matrix deposition in chronically damaged livers.( 4 , 5 ) Moreover, in HCC the stroma is infiltrated by activated HSC/myofibroblasts. They are located around tumor sinusoids, fibrous septa and capsule, if the latter is present.( 6 , 7 , 8 )
The interaction between tumor cells and their microenvironment has been recognized to fundamentally affect cancer development by triggering cell proliferation and survival as well as the capability to invade the surrounding tissue for subsequent spreading and colonization. In the majority of tumors, the aberrant networks of growth factors, cytokines and chemokines including their cognate receptors, are critically involved in cancer progression. Paracrine and autocrine mechanisms are responsible for the communication between cancer and surrounding neighboring cells, collectively known as tumor‐stroma.( 9 , 10 )
Yet, the molecular framework of this crosstalk in the specific tissue context and its consequences on carcinogenesis are largely unknown and rather, are based on observations from individual cell types. Especially, the role of activated HSC/myofibroblasts in HCC development and progression is largely unknown.
In this study we tackled this issue by applying three different models. First, we studied the influence of conditioned medium (CM) collected from in vitro activated human HSC on different human HCC cell lines cultured in monolayers. Next, we used a three‐dimensional coculture system to study the effect of activated HSC on HCC cells. Finally, we applied simultaneous xenografting of HCC cells and activated HSC. In summary, we provide evidence that paracrine feedback mechanisms regulated by activated HSC strongly affect the malignant progression of HCC cells in vitro and in vivo, and that induction of extracellular‐regulated kinase (ERK) and NFkappaB activation may be responsible for these effects.
Materials and Methods
Cells and cell culture. The HCC cell lines HepG2 (ATCC HB‐8065), PLC (ATCC CRL‐8024), and Hep3B (ATCC HB‐8064) were used. Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with penicillin (400 U/mL), streptomycin (50 µg/mL), L‐glutamine (300 µg/mL) and 10% fetal calf serum (FCS; Sigma, Deisenhofen, Germany) and passaged at a 1:5 ratio every 3 days. Spheroid cell culture was performed as described previously.( 11 )
Human liver tissue for cell isolation was obtained according to the guidelines of the charitable state‐controlled foundation Human Tissue & Cell Research (HTCR) with the patient's informed consent. Isolation and culture of hepatic stellate cells (HSC) were performed as described previously.( 12 , 13 ) In vitro activation of HSC was achieved by cell culture on uncoated tissue culture dishes for 20 days as described previously.( 13 )
To clearly characterize these cells as activated HSC we analyzed neurotrophin‐3 (NT‐3) mRNA expression since it has been shown previously that HSC express NT‐3 while NT‐3 expression cannot be detected in other subpopulations of myofibroblasts (MF), as interface MF, septal MF or portal MF.( 14 , 15 ) Reverse transcription – polymerase chain reaction (RT‐PCR) analysis applying primers specific for NT‐3 (NT3‐forw: 5′‐GTC ATC GGC CAT CGA CAT TCG; NT3‐rev: 5′‐GTT CTC TGA AGT CAG TGC TCG) revealed an equal and strong band in cDNA isolated from activated HSC isolated from three different human donors (Supporting Information Fig. S1A). Furthermore, immunohistochemical analysis applying anti‐alpha‐smooth muscle actin (alpha‐sma) antibodies showed the typical morphology of activated HSC and confirmed the purity of the cell culture (Supporting Information Fig. S1B).
Collection of conditioned medium (CM) from activated HSC. One day after seeding into T75 flasks (2 × 106 cells), activated HSC were washed twice with serum‐free DMEM, and then incubated for 24 h with serum‐free DMEM (15 mL/T75).
For collection of CM from nonactivated HSC, freshly isolated cells were seeded into T25 flasks (0.7 × 106 cells). At the next day, cells were washed with serum‐free DMEM, and incubated for another 24 h with complete medium (supplemented with 10% FCS). At day 2 after isolation, cells were washed again twice with serum‐free DMEM, and then incubated for 24 h with serum‐free DMEM (5 mL/T25).
Serum‐free DMEM incubated for 24 h in cell culture flasks without cells served as the control.
For individual experiments, CMs were preincubated with antihuman hepatocyte growth factor (HGF), antihuman tumor growth factor (TGF)‐beta, or isotope‐control antibodies (100 µg/mL; all from R & D Systems, Wiesbaden, Germany).
Tumor cell inoculation and measurement of tumor growth in Naval Medical Research Institute (NMRI) (nu/nu) mice. A model of inoculation of tumor cells into NMRI (nu/nu) mice to monitor tumor growth in vivo was performed as described previously.( 16 ) Briefly, HCC cells and activated HSC were harvested after incubation with phosphate buffered saline (PBS) containing 0.05% trypsin and 0.04% ethylenediaminetetraacetic acid (EDTA) (Sigma). Cells were washed twice with serum‐free DMEM (Gibco, Karlsruhe, Germany) at room temperature and resuspended in DMEM at a concentration of 1 × 107 cells/mL. For each experimental setting a group of 10 NMRI (nu/nu) mice with a mean body weight of 32 g was formed. Mice were injected subcutaneously with a cell suspension of 0.1 mL containing either: (i) 5 × 105 HCC cells; (ii) 5 × 105 activated HSC; or (iii) a mixture of 5 × 105 HCC cells and 5 × 105 activated HSC. Tumor growth kinetics were recorded on a regular basis by measurement of tumor diameters at the inoculation site (region of the thoracic mammary fat pad) with an electronic caliper. Tumor areas were calculated as the product of two perpendicular diameters, one measured across the greatest width of the tumor. For ethical reasons the mice were killed at day 16 after the first tumors underwent ulceration, and the tumors were taken out and stored for subsequent analysis.
Expression analysis. Isolation of total cellular RNA from cultured cells was performed as described previously.( 13 , 16 ) Quantitative real time‐PCR was performed with specific primers for interleukin (IL)‐8 applying LightCycler technology (Roche, Mannheim, Germany) as described.( 17 )
Protein analysis. Protein extraction and Western blotting were performed as described previously( 16 ) applying the following primary antibodies: monoclonal anti‐ERK antibody, or antiphospho‐ERK‐antibody (both from Calbiochem, Darmstadt, Germany).
Immunohistochemical staining of paraformaldehyde (PFA) fixed cells or 5 µm sections of tumor‐tissue, respectively, was performed using a mouse monoclonal anti‐alpha‐sma antibody (M0634, 1:50 dilution; DAKO, Glostrup, Denmark), and an indirect immunoperoxidase protocol according to the LSAB2‐kit (Dako, Hamburg, Germany) as described.( 18 ) Further, PFA‐fixed tissue sections were stained against CD31 (PECAM‐1) (Dako Cytomation, Hamburg, Germany) on a Dako Autostainer (Dako Cytomation).
The HGF level in conditioned medium of activated HSC was analyzed using an enzyme‐linked immunosorbent assay (ELISA) from R & D Systems (Minneapolis, MN, USA) according to the manufacturer's instructions.
Sirius red staining. PFA‐fixed tissue was embedded in paraffin and 10 µm sections were mounted on glass slides. Sections were deparaffinized and the slides were incubated for 30 min in a solution of saturated picric acid containing 0.1% sirius red and 0.1% fast green.
Quantification of activated nuclear NFkappaB concentration. NFkappB was quantified in nuclear extracts with the ELISA‐based kit TransAm from Active Motif (Rixensart, Belgium) according to the manufacturer's instructions as described.( 17 )
Proliferation assay. HCC cells were seeded into 96‐well plates (4000 cells/well) in DMEM supplemented with 10% FCS. One day later seeding cells were washed twice with serum‐free DMEM, and subsequently, cells were cultured in CM from activated HSC or control medium. Cell proliferation was measured using the XTT assay (Roche, Mannheim, Germany) according to the manufacturer's instructions.( 16 ) Experiments were carried out in triplicate and were repeated three times with consistent results.
In defined functional assays HCC cells were preincubated with inhibitors of iNOS (aminoguanidin; 500 µM), NFkappaB (MG132; 2 µM) or ERK‐signaling (SUO 5402; 15 µM; all from Calbiochem) for 30 min prior to stimulation with CM or control medium.
Migration assays. Migration of HCC cells was assessed applying two different assays.
Boyden chambers containing polycarbonate filters with 8 µm pore size (Costar, Bodenheim, Germany) were used, as described previously.( 16 ) Briefly, filters were coated with gelatine (5 mg/L) to improve cell attachment. The lower compartment was filled with DMEM supplemented with CM from activated HSC or control medium. HCC cells were harvested, resuspended in DMEM without FCS at a density of 2 × 105 cells/mL and placed in the upper compartment. After incubation at 37°C for 4 h, the filters were collected and cells adhering to the lower surface fixed, stained and counted. Experiments were carried out in triplicate and were repeated three times with consistent results.
Further, migration was assessed applying time‐lapse scratch assays (‘wound‐healing‐assay’). Here, HCC cells were seeded in high density into 6‐well plates. After adherence, cells were washed with serum‐free DMEM and either in CM from activated HSC or control medium. Subsequently, the cell layer was scratched by a pipette tip in a definite array, and the migration into this array was documented and measured after 24 and 48 h. Each analysis was performed in triplicate and repeated twice with consistent results.
Statistical analysis. Results are expressed as mean ± standard error (range) or percent. Comparison between groups was made using the Student's unpaired t‐test. A P‐value < 0.05 was considered statistically significant. All calculations were performed by using the GraphPad Prism Software (GraphPad Software, Inc., San Diego, CA, US).
Results
Effect of conditioned medium collected from activated HSC on HCC cells in vitro. To study the interaction between activated HSC and HCC cells, we first incubated three different HCC cell lines with conditioned medium (CM) collected from activated HSC. Analysis of cell proliferation revealed that CM from activated HSC significantly enhanced the mitogenic activity of all three HCC cell lines (PLC, HepG2 and Hep3B) analyzed (Fig. 1A).
Figure 1.
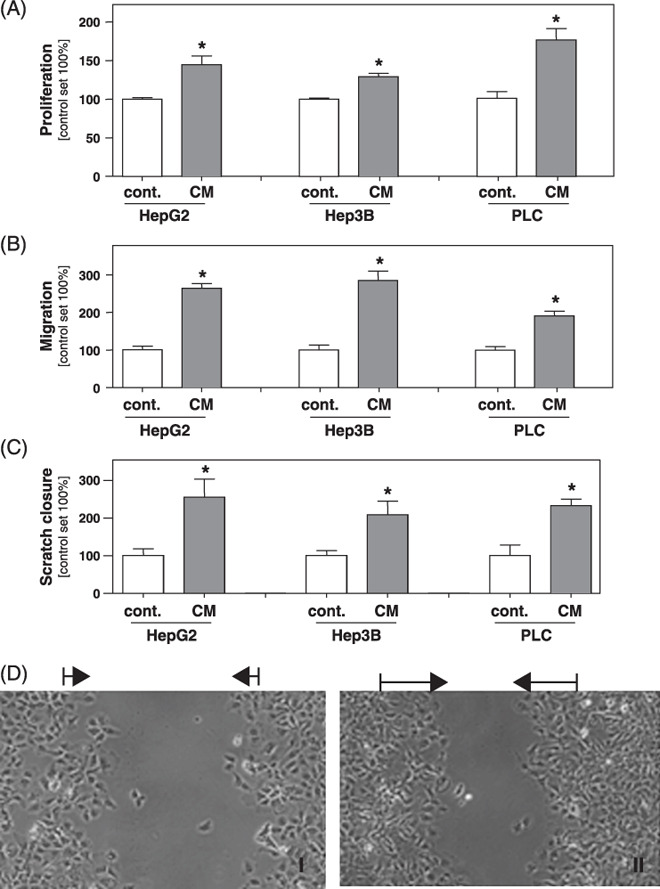
Effect of conditioned medium collected from activated hepatic stellate cells (HSC) on hepatocellular carcinoma (HCC) cells in vitro. HCC cell lines (HepG2, Hep3B and PLC) were stimulated with conditioned medium (CM) collected from in vitro activated HSC. (A) Proliferation was analyzed applying XTT‐assays. Migratory potential analyzed in (B) Boyden chamber and (C) monolayer scratch assays. (D) Representative pictures of the areas between scratch fronts after 2 days (I: Hep3B; II: Hep3B plus CM). (*P < 0.05 compared to control).
Previous studies have shown that activated HSC/myofibroblasts constitute the HCC stroma.( 6 ) Still, it is interesting to note that CM from early cultures of nonactivated HSC exhibited no effects on HCC proliferation (Supporting Information Fig. S2). This finding prompted us further to focus our analysis on the effect of activated HSC and CM from activated HSC, respectively, on HCC cells.
Here, we next analyzed the migratory activity of CM‐stimulated HCC cells in comparison to control cells. Boyden chamber assays demonstrated that CM from activated HSC significantly stimulated the migratory potential of HCC cells (Fig. 1B). These data were confirmed by time‐lapse scratch assays that revealed significantly faster wound closure in HCC cells treated with CM from HSC (Fig. 1C,D).
Effect of activated HSC and HCC cells in a three dimensional in vitro coculture system. Next, we applied the spheroid system to analyze the effect of activated HSC on HCC cells. Seeding of HCC cells or activated HSC alone resulted in spheroid formation after 2–3 days. Seeding of the same number of both cell types together lead to formation of mixed spheroids after the same time period. However, subsequently these mixed spheroids revealed significantly faster growth. Figure 2(A) depicts the spheroid volumes after 10 days. The dashed bar indicates the sum (5.1 ± 0.2 mm3 × 10−3) of the mean volumes of the pure HSC (0.3 ± 0.1 mm3 × 10−3) and Hep3B (4.7 ± 0.2 mm3 × 10−3) spheroids, being significantly smaller than the mean volume of the mixed spheroids formed by both activated HSC and HCC cells (7.2 ± 0.3 mm3 × 10−3; P = 0.0002).
Figure 2.
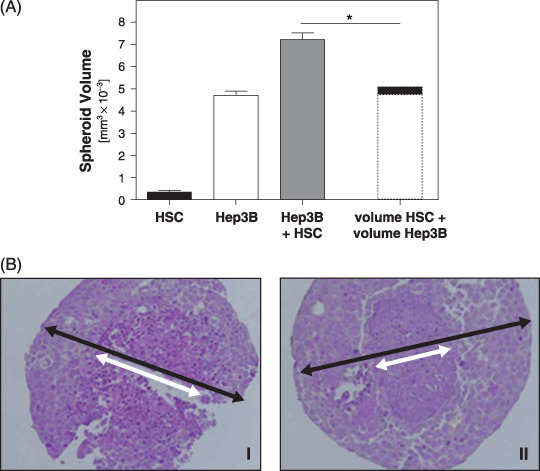
Effect of activated hepatic stellate cells (HSC) on hepatocellular carcinoma (HCC) cells in a three dimensional in vitro coculture system. (A) Volume of spheroids formed by in vitro activated HSC, Hep3B cells, and a 1:1 mixture of activated HSC and Hep3B cells after 10 days. The dashed bar indicates the virtual sum of the mean volumes of pure HSC and Hep3B spheroids. (*P < 0.05). (B) Histochemical staining (hematoxylin and eosin) of spheroids formed by pure Hep3B cells or a 1:1 mixture of activated HSC and Hep3B cells after 10 days. The diameter of central necrosis (white arrow) and the outer diameter of the spheroid (black arrow), respectively, are indicated. (I: HCC; II: HCC plus HSC).
Interestingly, despite their larger size, mixed spheroids revealed smaller areas of central necrosis in histological analysis (Fig. 2B).
Effect of activated HSC on HCC cells in vivo. Next, we sought to investigate the effect of HSC on HCC cells in vivo in a xenograft model. HCC cells were implanted either alone or together with activated HSC into nude mice (500 000 cells/mouse). Implantation of pure activated HSC (500 000) did not result in tumor formation within an observation period of 3 months. Also, after implantation of PLC cells and Hep3B cells alone no tumor formation was observed in our experimental setting. However, if activated HSC were injected simultaneously with PLC or Hep3B cells, both HCC cell lines formed tumors in all mice (n = 10/group).
HepG2 cells were capable of forming tumors also in the absence of activated HSC. However, if injected together with HSC these cells exhibited a more rapid tumor growth (Fig. 3A and 83.1 ± 19.8 mm3 vs. 28.7 ± 10.6 mm3 16 days after injection).
Figure 3.
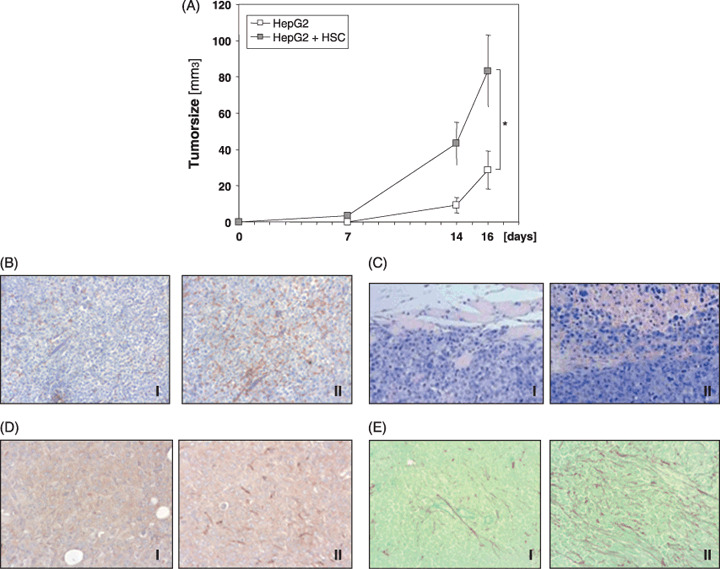
Effect of activated hepatic stellate cells (HSC) on hepatocellular carcinoma (HCC) cells in vivo. (A) Growth kinetic of tumors formed in nude mice after implantation of pure HepG2 cells or HepG2 cells together with activated HSC. (*P < 0.05). (B–E) (Immuno‐)histochemical staining pictures of tumors formed in nude mice after implantation of pure HepG2 cells (I) or HepG2 cells together with activated HSC (II) (day 16): (B) CD31 (C) hematoxylin and eosin (D) alpha‐smooth muscle actin, and (E) sirius‐red fast green staining. (100×; representative staining results are depicted).
Similarly as observed in the in vitro spheroid system, mixed tumors of HepG2 and activated HSC exhibited smaller central necrosis than tumors formed by HepG2 cells alone (data not shown). To analyze whether in vivo this phenomenon is also caused by increased angiogenesis we performed immunohistochemical analysis applying anti‐CD31 antibodies. It is noteworthy that mixed tumors of HepG2 and activated HSC revealed more intense CD31 staining, indicating advanced angiogenesis as compared to tumors formed by HepG2 cells alone (Fig. 3B).
Furthermore, histopathological analysis revealed that tumors that developed after simultaneous injection of HepG2 cells and activated HSC show diffuse growth, while tumors formed by pure HepG2 cells showed a more nodular growth pattern. Representative pictures are shown in Fig. 3(C).
Immunohistochemical analysis of alpha‐sma, an established marker of activated HSC, revealed no staining signal in the stroma of tumors formed by HepG2 cells alone (Fig. 3D, panel I). However and interestingly, also in the stroma of mixed tumors of HepG2 and activated HSC, only sparsely alpha‐sma positive cells could be detected (Fig. 3D, panel II). In vitro HepG2 cells grow significantly faster than HSC (doubling time approximately 2–3 days vs. 8–10 days; data not shown), and it appears that 16 days after implantation of equal numbers of HSC and HepG2 cells into nude mice the faster growing HCC cells compete with HSC in the tumor stroma of nude mice. Still, sirius‐red fast green staining which specifically detects extracellular matrix (ECM) proteins revealed significantly more ECM deposition in mixed tumors of HepG2 and activated HSC than in tumors formed by HepG2 cells alone (Fig. 3C).
In summary and in line with the in vitro results, data obtained in nude mice indicate that activated HSC promote tumorigenicity of HCC cells. In human HCC where HSC significantly represent the tumor stroma, these effects may be even more pronounced than in the nude mouse model where tumor‐promoting effects of HSC may diminish with time due to numerical vanishing from the stroma.
Effect of activated HSC on activation of ERK and NFkappaB in HCC cells. To get insight into the molecular mechanisms causing the tumorigenic effect of activated HSC on HCC cells, we analyzed ERK and NFkappaB activation, two major signaling pathways known to play an important role in hepatocancerogenesis.
Western blot analysis revealed that stimulation with CM from activated HSC induced a strong phosphorylation of p42/p44 Mitogen‐activated protein kinase (MAPK) in all three HCC cell lines (Fig. 4A).
Figure 4.
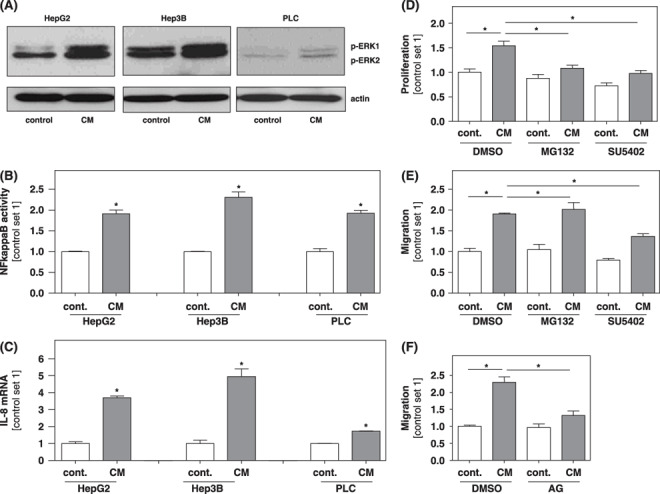
Effect of activated hepatic stellate cells (HSC) on activation of extracellular‐regulated kinase (ERK) and NFkappaB in hepatocellular carcinoma (HCC) cells. HCC cells were stimulated with conditioned medium (CM) collected from in vitro activated HSC. Non‐stimulated cells served as control. (A) Western blot analysis using an antibody against phosphorylated ERK, p44 and p42, designed ERK1 and ERK2. Analysis of activated nuclear NFkappaB (B) and (C) interleukin (IL)‐8 mRNA expression. (*P < 0.05 compared to control). (D–F) Prior to stimulation with CM Hep3B cells were incubated with MG132 (2 µM), SU 5402 (15 µM), or aminoguanidin (AG; 500 µM) for 30 min. Subsequently, proliferation was analyzed applying XTT‐assays (D), and the migratory potential was analyzed in Boyden chamber assays (E, F). (*P < 0.05).
Next, we analyzed the concentration of activated NFkappaB in nuclear extracts in HCC cells stimulated with CM from activated HSC and controls using an ELISA‐based technique. Here, a significantly higher NFkappaB activity was observed in CM‐stimulated HCC cells (Fig. 4B).
The pro‐inflammatory chemokine IL‐8 is known to be regulated by NFkappaB and ERK, and to be expressed by HCC cells.( 19 , 20 ) Stimulation of HCC cells with CM from activated HSC enhanced IL‐8 expression as demonstrated by quantitative RT‐PCR analysis (Fig. 4C).
This finding further indicated that CM from activated HSC exerts its pro‐tumorigenic effect via activation of NFkappaB and ERK. To further confirm this hypothesis HCC cells were incubated with specific inhibitors of NFkappaB (MG132) and ERK (SU5402) signaling prior to incubation with CM from HSC, and subsequently, functional assays were performed. Both inhibitors significantly impaired the growth‐promoting effect of CM from activated HSC (Fig. 4D). Further, SU5402 significantly inhibited the migration‐promoting effect of CM from activated HSC while the migratory potential of HCC cells was not affected by MG132 (Fig. 4E).
A recent study indicated that inducible nitric oxide synthase (iNOS) crosstalk with NFkappaB and Ha‐RAS/ERK cascades influences HCC progression.( 21 ) This prompted us to study whether suppression of iNOS signaling in HCC cells affects the pro‐tumorigenic effect of CM from activated HSC. Interestingly, preincubation of HCC cells with aminoguanidine, an inhibitor of iNOS signaling, inhibited the migration‐inducing (Fig. 4E) but not the growth‐promoting (data not shown) effect of CM from activated HSC.
Effect of activated HSC on tumorigenicity of HCC cells in vitro is in part mediated by HGF. Next, we wanted to identify the humoral factors in the CM of activated HSC responsible for the tumorigenic effect observed upon stimulation of HCC cells. Here, we focused on HGF, a factor known to act protumorigenically during hepatocancerogenesis.( 22 ) Moreover, it has been shown that all three cell lines used in the present study (HepG2, PLC and Hep3B) express c‐met, the receptor for HGF( 23 ) and that NFkappaB and ERK activation are known downstream regulators of HGF/c‐met signaling in HCC.( 22 , 24 ) Applying ELISA technology we found high HGF levels in the CM of activated HSC (≈ 20 ng/mL). It is noteworthy that preincubation of CM from activated HSC with anti‐HGF antibodies markedly reduced the growth‐promoting effect on Hep3B cells ( Fig. 5A ). Similarly, the migration‐promoting effect of CM from activated HSC was significantly impaired by preincubation with anti‐HGF antibodies ( Fig. 5B ).
Figure 5.
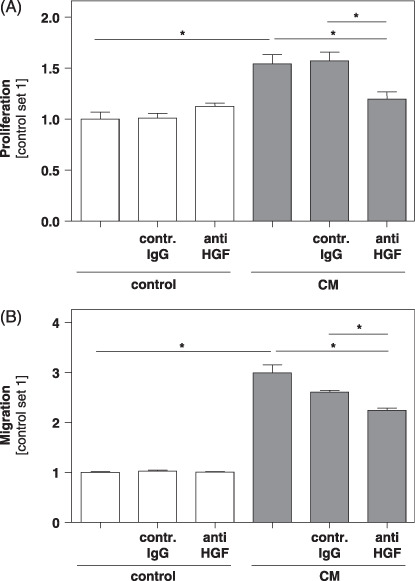
Effect of anti‐hepatocyte growth factor (HGF) antibodies on the proliferation and migration of hepatocellular carcinoma (HCC) cells in the presence of conditioned medium from activated hepatic stellate cells (HSC). Conditioned medium (CM) from activated HSC was preincubated with anti‐HGF antibodies or isotype matched control antibodies (contr. IgG) before addition to HCC cells. Subsequently (A) proliferation was analyzed applying XTT‐assays, and (B) migratory potential was analyzed in Boyden chamber assays. (*P < 0.05).
In contrast, preincubation of CM from activated HSC with anti‐TGF‐beta antibodies neither impaired the growth nor the migration‐promoting effect on any of the three HCC cell lines analyzed (data not shown).
Discussion
In this study we aimed to analyze the effects of activated HSC on the tumorigenicity of HCC cells in vitro and in vivo. Our experiments show that activated human HSC induce proliferation and migration in three different human HCC cell lines in vitro. Furthermore, activated HSC promote growth and invasiveness of human HCC cells in nude mice. Together these data indicate that activated HSC enhance tumorigenicity of HCC.
As potential molecular mechanisms mediating the tumorigenic effect of HSC on HCC cells we identified increased activity of NFkappaB and ERK in HCC cells induced by conditioned medium (CM) collected from activated HSC.
Aberrant activation of the MAP kinase/ERK pathway is involved in the progression of human HCC. Increased ERK activation is known to induce proliferation of HCC cells and to protect HCC cells from apoptosis.( 25 ) Furthermore, increased ERK activity has been shown to affect the migratory activity and invasiveness of HCC cells, suggesting that this molecular pathway may be critical in intrahepatic metastasis of HCC.( 26 )
Activation of the transcription factor NFkappaB is one of the early key events involved in neoplastic progression of the liver, and plays a crucial role in bridging the action of growth factors and inflammation to hepatic oncogenesis.( 27 , 28 ) Constitutive as well as induced activation of NFkappaB has been shown to induce survival of HCC cells in vitro and in vivo, and cell culture studies have established NFkappaB's critical role in cancer progression.( 28 , 29 )
Interestingly, a recent study indicates that the crosstalk between iNOS and NF‐kappaB and ERK cascades promotes HCC progression( 21 ) and here, we found that suppression of iNOS signaling inhibited the migration‐inducing, but not the growth‐promoting effect of CM from activated HSC. These findings further underscore that pleiotropic signaling pathways are dysregulated in HCC, and that the resultant functional effects are modulated by multiple crosstalks between these different signaling pathways.
Thus, nitric oxide as well as NFkappaB and ERK activity promote transcription of IL‐8, a chemokine that is constitutively expressed in HCC cells.( 30 , 31 ) Here, we found that activated HSC stimulate IL‐8 expression in HCC cells. Previous studies have shown that IL‐8 may be directly or indirectly involved in the progression of HCC.( 30 , 32 ) Thus, IL‐8 may contribute to cancer progression through potential mitogenic, angiogenic or motogenic functions. In the present study, mixed tumors of activated HSC and HCC cells revealed smaller areas of central necrosis than tumors formed by pure HCC cells in a xenograft model. As potential explanation we found enhanced CD31 staining in mixed tumors, indicating improved angiogenesis and blood supply, respectively. However, central necrosis of spheroids formed by activated HSC and HCC cells was smaller compared to pure HCC cell spheroids. In this 3‐dimensional cell culture system, supply of oxygen and nutrients is independent of angiogenesis and purely limited by diffusion. Further, a recent study by Kubo et al. showed that IL‐8 in human HCC correlates with cancer cell invasion but not with tumor angiogenesis.( 30 ) In summary, these findings indicate that in addition to induction of angiogenesis, activated HSC promote HCC growth by enhancement of the misbalance between proliferation and cell death that represents a protumorigenic principle in hepatocarcinogenesis.
We applied different models to study the interaction between activated HSC and HCC cells. In the first experimental setting enhanced NFkappaB and ERK activity as well as induced proliferation, migration, and expression of IL‐8 were observed in HCC cells stimulated with conditioned medium collected from activated HSC. These findings indicated that soluble factors released by activated HSC are responsible for the tumorigenic effects observed. Several different growth factors, matrix metalloproteinases (MMPs), chemokines and cytokines are potential candidates.
In search for the relevant factor or factors, respectively, we initially focused on HGF since it is known to act protumorigenically during hepatocancerogenesis( 20 ) and previous studies have shown that HGF secreted by activated HSC promotes invasiveness of HCC cell lines in vitro. ( 33 ) We found that HSC secrete relevant HGF levels, and applying inhibitory anti‐HGF antibodies we demonstrated that the growth and migration‐promoting effect of CM from activated HSC was in part mediated by HGF.
In contrast, anti‐TGF‐beta antibodies did not affect the protumorigenic effect of CM from activated HSC. This finding is in line with a previous report by Neaud et al. who used HepG2 cells for their study( 34 ) but is in contrast to a study by Mikula et al. who demonstrated that HSC augment HCC progression via TGF‐beta.( 34 ) However, the authors of the latter study used immortalized murine hepatic cell lines while we and Neaud et al. applied human HCC cell lines and primary human HSC, respectively. Thus, species differences or divergent experimental settings may account for the discrepant results regarding the effect of TGF‐beta.
Still it has to be noted that several further growth factors are known to be expressed by activated HSC and to affect hepatocancerogenesis as fibroblast growth factor (FGF) 1 and 2, platelet‐derived‐growth‐factor (PDGF) or insulin‐like growth factor (IGF).( 4 ) It appears more than likely that (a combination of) several of these as well as so far not‐identified factors secreted by activated HSC caused the tumorigenic effects on HCC cells observed in the present as well as other previous studies. Further, it can be speculated that these factors affect different pathophysiological mechanisms, and that the same factor may exhibit diverse effects in different HCC cell lines or HCC patients, respectively.
Subsequent analysis of the interaction between activated HSC and HCC cells in a 3‐dimensional cell coculture system and in an in vivo model, further confirmed the tumorigenic effect of activated HSC on HCC. In these models, besides soluble factors released by activated HSC, further mechanisms may have caused the cancerogenic effects observed. For example, previous studies have shown that HSC affect progression of hepatic tumors or metastasis by ECM remodeling or expression of chemotactic factors.( 35 , 36 ) These and related studies in other tumors even show that tumor cells further induce the expression of tumorigenic factors by activated HSC, indicating a mutual interaction of both cancer cells and stromal (myo)fibroblasts in promoting carcinogenesis.
In summary, the present study further supports the impact of stromal activated HSC/myofibroblasts on HCC progression in vitro and in vivo. This interaction may be an interesting tumor cell differentiation‐independent target for therapy. Furthermore, quantification of activated HSC/myofibroblasts in HCC may serve as a prognostic marker.
Supporting information
Fig. S1. (A) Neurotrophin‐3 (NT‐3) mRNA expression of activated hepatic stellate cells (HSC) isolated from three different human donors.
(B) alpha‐sma immunohistochemical staining of activated HSC (100×).
Fig. S2. Effect of conditioned medium collected from quiescent and activated hepatic stellate cells (HSC) on proliferation of hepatocellular carcinoma (HCC) cells in vitro.
Hep3B cells were stimulated with conditioned medium (CM) collected from HSC 2 days after isolation (quiescent HSC; Q‐HSC) and in vitro activated HSC (A‐HSC). Proliferation of HCC cells was analyzed applying XTT‐assays. (*P < 0.05).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Acknowledgments
We are indebted to Gertrud Lallinger, Birgitta Ott‐Rötzer and Diane Lochbaum for excellent technical assistance. This work was supported by grants from the German Research Association (DFG) to P.K., A.K.B and C.H., and the Medical Faculty of the University of Regensburg (ReForM) to A.K.B and C.H.
References
- 1. El Serag HB. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology 2004; 127: S27–S34. [DOI] [PubMed] [Google Scholar]
- 2. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006; 6: 674–87. [DOI] [PubMed] [Google Scholar]
- 3. Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell 2004; 5: 215–9. [DOI] [PubMed] [Google Scholar]
- 4. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005; 115: 209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Friedman SL. Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol 2004; 1: 98–105. [DOI] [PubMed] [Google Scholar]
- 6. Faouzi S, Le Bail B, Neaud V et al . Myofibroblasts are responsible for collagen synthesis in the stroma of human hepatocellular carcinoma: an in vivo and in vitro study. J Hepatol 1999; 30: 275–84. [DOI] [PubMed] [Google Scholar]
- 7. Le Bail B, Faouzi S, Boussarie L et al . Osteonectin/SPARC is overexpressed in human hepatocellular carcinoma. J Pathol 1999; 189: 46–52. [DOI] [PubMed] [Google Scholar]
- 8. Dubuisson L, Lepreux S, Bioulac‐Sage P et al . Expression and cellular localization of fibrillin‐1 in normal and pathological human liver. J Hepatol 2001; 34: 514–22. [DOI] [PubMed] [Google Scholar]
- 9. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6: 392–401. [DOI] [PubMed] [Google Scholar]
- 10. Desmouliere A, Guyot C, Gabbiani G. The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol 2004; 48: 509–17. [DOI] [PubMed] [Google Scholar]
- 11. Tatzel J, Poser I, Schroeder J, Bosserhoff AK. Inhibition of melanoma inhibitory activity (MIA) expression in melanoma cells leads to molecular and phenotypic changes. Pigment Cell Res 2005; 18: 92–101. [DOI] [PubMed] [Google Scholar]
- 12. Weiss TS, Jahn B, Cetto M, Jauch KW, Thasler WE. Collagen sandwich culture affects intracellular polyamine levels of human hepatocytes. Cell Prolif 2002; 35: 257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Muhlbauer M, Fleck M, Schutz C et al . PD‐L1 is induced in hepatocytes by viral infection and by interferon‐alpha and ‐gamma and mediates T cell apoptosis. J Hepatol 2006; 45: 520–8. [DOI] [PubMed] [Google Scholar]
- 14. Cassiman D, Denef C, Desmet VJ, Roskams T. Human and rat hepatic stellate cells express neurotrophins and neurotrophin receptors. Hepatology 2001; 33: 148–58. [DOI] [PubMed] [Google Scholar]
- 15. Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol 2002; 36: 200–9. [DOI] [PubMed] [Google Scholar]
- 16. Hellerbrand C, Amann T, Schlegel J et al . The novel gene MIA2 acts as a tumour suppressor in hepatocellular carcinoma. Gut 2008; 57: 243–51. [DOI] [PubMed] [Google Scholar]
- 17. Muhlbauer M, Allard B, Bosserhoff AK et al . Differential effects of deoxycholic acid and taurodeoxycholic acid on NF‐kappa B signal transduction and IL‐8 gene expression in colonic epithelial cells. Am J Physiol Gastrointest Liver Physiol 2004; 286: G1000–G1008. [DOI] [PubMed] [Google Scholar]
- 18. Steiling H, Muhlbauer M, Bataille F, Scholmerich J, Werner S, Hellerbrand C. Activated hepatic stellate cells express keratinocyte growth factor in chronic liver disease. Am J Pathol 2004; 165: 1233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joshi‐Barve S, Barve SS, Amancherla K et al . Palmitic acid induces production of proinflammatory cytokine interleukin‐8 from hepatocytes. Hepatology 2007; 46: 823–30. [DOI] [PubMed] [Google Scholar]
- 20. Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte growth factor/scatter factor‐induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin‐8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res 2001; 61: 5911–8. [PubMed] [Google Scholar]
- 21. Calvisi DF, Pinna F, Ladu S et al . Aberrant iNOS signaling is under genetic control in rodent liver cancer and potentially prognostic for the human disease. Carcinogenesis 2008; 29: 1639–47. [DOI] [PubMed] [Google Scholar]
- 22. Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006; 25: 3787–800. [DOI] [PubMed] [Google Scholar]
- 23. Lee HS, Huang AM, Huang GT et al . Hepatocyte growth factor stimulates the growth and activates mitogen‐activated protein kinase in human hepatoma cells. J Biomed Sci 1998; 5: 180–4. [DOI] [PubMed] [Google Scholar]
- 24. Wang SW, Pan SL, Peng CY et al . CHM‐1 inhibits hepatocyte growth factor‐induced invasion of SK‐Hep‐1 human hepatocellular carcinoma cells by suppressing matrix metalloproteinase‐9 expression. Cancer Lett 2007; 257: 87–96. [DOI] [PubMed] [Google Scholar]
- 25. Fabregat I, Roncero C, Fernandez M. Survival and apoptosis: a dysregulated balance in liver cancer. Liver Int 2007; 27: 155–62. [DOI] [PubMed] [Google Scholar]
- 26. Honma N, Genda T, Matsuda Y et al . MEK/ERK signaling is a critical mediator for integrin‐induced cell scattering in highly metastatic hepatocellular carcinoma cells. Lab Invest 2006; 86: 687–96. [DOI] [PubMed] [Google Scholar]
- 27. Arsura M, Cavin LG. Nuclear factor‐kappaB and liver carcinogenesis. Cancer Lett 2005; 229: 157–69. [DOI] [PubMed] [Google Scholar]
- 28. Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int 2006; 26: 1163–74. [DOI] [PubMed] [Google Scholar]
- 29. Qiao L, Zhang H, Yu J et al . Constitutive activation of NF‐kappaB in human hepatocellular carcinoma: evidence of a cytoprotective role. Hum Gene Ther 2006; 17: 280–90. [DOI] [PubMed] [Google Scholar]
- 30. Kubo F, Ueno S, Hiwatashi K et al . Interleukin 8 in human hepatocellular carcinoma correlates with cancer cell invasion of vessels but not with tumor angiogenesis. Ann Surg Oncol 2005; 12: 800–7. [DOI] [PubMed] [Google Scholar]
- 31. Heo SK, Yun HJ, Noh EK, Park WH, Park SD. LPS induces inflammatory responses in human aortic vascular smooth muscle cells via Toll‐like receptor 4 expression and nitric oxide production. Immunol Lett 2008; 120: 57–64. [DOI] [PubMed] [Google Scholar]
- 32. Ren Y, Poon RT, Tsui HT et al . Interleukin‐8 serum levels in patients with hepatocellular carcinoma. correlations with clinicopathological features and prognosis. Clin Cancer Res 2003; 9: 5996–6001. [PubMed] [Google Scholar]
- 33. Neaud V, Faouzi S, Guirouilh J et al . Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: evidence for a role of hepatocyte growth factor. Hepatology 1997; 26: 1458–66. [DOI] [PubMed] [Google Scholar]
- 34. Mikula M, Proell V, Fischer AN, Mikulits W. Activated hepatic stellate cells induce tumor progression of neoplastic hepatocytes in a TGF‐beta dependent fashion. J Cell Physiol 2006; 209: 560–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olaso E, Santisteban A, Bidaurrazaga J, Gressner AM, Rosenbaum J, Vidal‐Vanaclocha F. Tumor‐dependent activation of rodent hepatic stellate cells during experimental melanoma metastasis. Hepatology 1997; 26: 634–42. [DOI] [PubMed] [Google Scholar]
- 36. Theret N, Musso O, Turlin B et al . Increased extracellular matrix remodeling is associated with tumor progression in human hepatocellular carcinomas. Hepatology 2001; 34: 82–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A) Neurotrophin‐3 (NT‐3) mRNA expression of activated hepatic stellate cells (HSC) isolated from three different human donors.
(B) alpha‐sma immunohistochemical staining of activated HSC (100×).
Fig. S2. Effect of conditioned medium collected from quiescent and activated hepatic stellate cells (HSC) on proliferation of hepatocellular carcinoma (HCC) cells in vitro.
Hep3B cells were stimulated with conditioned medium (CM) collected from HSC 2 days after isolation (quiescent HSC; Q‐HSC) and in vitro activated HSC (A‐HSC). Proliferation of HCC cells was analyzed applying XTT‐assays. (*P < 0.05).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item