

Abstract
Vascular endothelial growth factor (VEGF) is an important regulator of angiogenesis, vasculogenesis and vascular permeability. Edema in glioma tumors is considered one of the most pathological characteristics, but the mechanism of regulating vascular permeability is still unclear. In the present study, tumorigenic mice were generated by subcutaneous injection of glioma cell lines, C6‐null cells and stable transfected‐C6 cells overexpressing mock vector (C6‐mock) and antisense VEGF (C6‐VEGF−/–). Overexpression of antisense VEGF (C6‐VEGF−/– mice) significantly suppressed tumor growth, decreased angiogenesis and reduced tumoral edema. Further studies by electron microscope revealed that tumor‐induced hyperpermeability was mediated by formation of vesiculo‐vacuolar organelles (VVO), specifically reducing the number of vesicle and caveolae in VVO, and this effect was blocked, at least partially, by antisense VEGF. These data show a possible mechanism of tumor‐induced hyperpermeability and indicate that blockage of VEGF might contribute to therapeutical strategies for tumor edema. (Cancer Sci 2008; 99: 2540–2546)
Glioma is the most common primary central nervous system (CNS) tumor in adults and is a very aggressive, invasive and destructive malignancy. Its proliferation rates are two to five times higher than other brain tumors.( 1 ) Histopathologically, the features of glioma are characterized by high proliferation, cellular polymorphism, necrosis, hypermeability and massive neovascularization.( 2 ) During the progression of glioma, a crucial step is the so‐called angiogenic switch, marking the predominance of pro‐angiogenic factors that subsequently induce the proliferation and activation of vascular endothelial cells (VEC). This process is mediated by various growth factors including vascular endothelial growth factor (VEGF). VEGF is expressed in a wide spectrum of brain tumors and is related to tumor degree; it is expressed at relatively low levels in the normal brain, up‐regulated in low‐grade glioma and highly expressed in high‐grade glioma.( 3 , 4 ) In addition, overexpression of VEGF has been associated with the formation of new tumor blood vessels,( 5 ) suggesting a direct correlation between the VEGF expression level and glioma prognosis.( 6 , 7 )
VEGF is known as a growth regulator of VEC, as well as a vascular permeability factor( 8 , 9 , 10 ) that is responsible for plasma extravasation, leading to edema in tumoral tissues and increased vessel permeability.( 11 ) It has been shown that overexpression of VEGF directly and rapidly induces plasma extravasation in the endothelium of skeletal muscle and skin( 12 ) and in the neovasculature of VEGF‐secreting tumors.( 13 ) The molecular mechanisms underlying this function are still uncertain. Several endothelial subcellular structures have been involved in these processes: caveolae, transendothelial channels, fenestrae, vesiculo‐vacuolar organelles (VVO), sinusoidal gaps and intercellular junctions.( 14 , 15 ) Recentely, Dvorak et al.( 14 ) demonstrated that VEGF induces leakage of macromolecules from venules through extravasation via VVO,( 16 , 17 ) the fused clustered vesicles of caveolae.( 18 )
The bunches‐of‐grape‐like clusters of VVO are present in the cytoplasm of vascular endothelial cells.( 18 ) They locate near lateral borders of endothelial cells and extend to both the lumen and albumen at multiple sites.( 16 , 17 , 18 ) Individual vesicles and vacuoles comprise VVO and interconnect with each other such that they may open stomata and therefore allow macromolecular components to cross the vascular endothelial membrane. VVO provide the major route of extravasation at sites of vessels, that was induced by vascular permeability factor VEGF, serotonin and histamine in animal models.( 16 , 19 ) Tumor‐induced VEGF secretion is found to localize on the surface of tumor vascular endothelium cells as well as in association with VVO in their cytoplasm.( 17 )
The aim of the present study is to detect the effects of down‐regulation of VEGF in glioma cell proliferation. Moreover, we investigate the molecular mechanism and ultrastructural basis of tumor‐induced hyperpermeability, aiming to elucidate the molecular target of tumor treatment.
Materials and Methods
Plasmids. Total RNA was isolated from 1‐day‐old Sprague Dawley rat brain using Trizol reagent (Invitrogen, Calsbad, CA, USA). Antisense VEGF164 complementary DNA (cDNA) was synthesized using the Titan One‐Step reverse transcriptase–polymerase chain reaction (RT‐PCR) Kit (Roche, Indianapolis, IN, USA) following the manufacturer's instructions. Primers were designed using Primer Express 1.5a software and were as follows: forward, CCCAAGCTTTCACCGCCTTGGCTTGTC; reverse, CGCGGATCCATGAACTTTCTGCTCTCTTG. Plasmid pBudCE4.1/VEGF−/– was generated containing a 640‐bp BamHI/HindIII amplicon cloned into a pBudCE4.1 expression vector (Invitrogen) under the control of human cytomegalovirus (CMV) promoter.
Cell culture. Rat glioma cell C6 (Cell Biology Research Institute of Shanghai, Shanghai, China) was cultured in RPMI‐1640 medium (1640 M) (Invitrogen) supplemented with fetal calf serum (10%). C6 cells (2 × 105) were placed in 35‐mm dishes and transfected with plasmids, pBudCE4.1/VEGF−/– (1 µg) or vector control pBudCE4.1 (1 µg), respectively, using Lipofectamine 2000 (Invitrogen). Medium was replaced with 1640 M containing Zeocin (1 mg/mL, Invitrogen) after 18 h post‐transfection. After 4 weeks, surviving clones were isolated, analyzed using PCR and selected for a transfected cell line that demonstrated the highest expression of genes to generate heterogeneously overexpressing antisense VEGF (C6‐VEGF−/–) and vector control (C6‐mock).
For cell proliferation assay, 2 × 104 cells were placed in a 6‐well plate and were counted after 24 h, 48 h, 72 h, 96 h, 96 h, 120 h and 144 h culture by hemocytometer.
Semi‐quantitative polymerase chain reaction. Total RNA was isolated and cDNA was synthesized as previously described. The sequences of primer sets were: VEGF, forward: CCCAAGCTTATGAACTTTCTGCTCTCTTG, reverse: CGCGGATCCTCACCGCCTTGGCTTGTC; and β‐actin, forward: GAGGCATCCTGACCCTGAAG, reverse: CATCACAATGCCAGTGGTACG. The calculation of expression levels of VEGF was normalized by β‐actin.
Immunostaining. To detect VEGF expression in vitro, cells were fixed in 4% paraformaldehyde and blocked with 3% normal goat serum for 2 h at room temperature for immunocytochemistry.
To determine VEGF and its receptors expression in vivo, anesthetized mice were killed by decapitation. Tumor tissues were removed and frozen in liquid nitrogen quickly. Sections (18‐µm thick) were cut using a cryostat and were rehydrated, treated with 0.3% hydrogen peroxide (H2O2) in methanol for 30 min to inactivate endogenous peroxidase, rinsed with 0.1 M phosphate buffer (PB) for 10 min and exposed to blocking serum (3% normal goat serum) for 2 h at room temperature for immunohistochemistry.
Immunoreaction was performed using antibodies against VEGF (1:150 dilution, United States Biological, Swampscott, MA, USA), VEGF receptor 1 (VEGFR‐1, 1:150, Santa Cruz Biotechnology, Santa Cruz, CA, USA), VEGF receptor 2 (VEGFR‐2, 1:150, Santa Cruz Biotechnology), phospho‐VEGF receptor 1 (pVEGF‐1, 1:100, Abcam, Cambridge, MA, USA) and phospho‐VEGF receptor 2 (pVEGF‐2, 1:100, Abcam) as described.( 20 ) The slices were rinsed with 0.1 M PB and exposed to anti‐rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP; 1:500 dilution, Maixin, Fuzhou, China) for 1 h. After an additional 10‐min rinse, the slices were treated with VectaStain Elite ABC reagent (Maixin) for 30 min and developed with diaminobenzidine (DAB) detection kit (Maixin). The sections were counterstained by hematoxylin and mounted using Permount (Maixin).
Vessel density detection. The tumor sections were treated with primary antibody against CD31 antigen (1:150 dilution, Santa Cruz Biotechnology) overnight at 4°C, then stained using a streptavidin–peroxidase (SP) conjugation kit (Maixin) according to the manufacturer's instructions. Immunoreaction was visible by DAB detection kit (Maixin) under light microscopy and counterstained by hematoxylin (Maixin).
Microvessel density (MVD) was determined under a confocal microscope (Nikon, Japan). The microvessels were carefully counted in five fields (200×) of areas around the basal part of tumor tissues. Single cells, a cluster of cells and branches of vascular trunks were all regarded as a blood vessel if they were CD31‐positive.
Enzyme‐linked immunosorbent assay (ELISA). To measure VEGF secretion in vitro, 5 × 105 cells were placed in 6‐well plate and treated with serum‐free 1640 M medium. Medium was collected after 24 h and 48 h culture. Debris was removed by centrifugation at 2000 × g for 5 min and supernatant was collected for ELISA.
To measure VEGF levels in vivo, tumor tissues (0.1 g) were homogenized in Tris‐HCl buffer (25 mM, pH 7.6) containing 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) and 1 mM phenylmethanesulfonylfluoride (PMSF). Debris was removed by centrifugation at 2000 × g for 5 min, followed with centrifugation at 20 000 × g for 20 min, and supernatants were collected for ELISA. Protein concentration was measured using a protein assay kit (Bio‐rad, Hercules, CA, USA).
Serial dilutions of samples with the highest and the lowest expected values were performed to determine VEGF expression level using a commercial VEGF ELISA kit (R & D Systems, Minneapolis, MN, USA) following the manufacturer's instructions. VEGF expression levels were calculated by standard curve available from the manufacturer. All experiments were performed in triplicate reactions.
Protein analysis. Tumor tissues were homogenized in Tris‐HCl buffer (50 mM, pH 8.0) containing protease inhibitor cocktail V (Calbiochem, San Diego, CA, USA). Homogenate (20 µg) was electrophoresed on 10% sodium dodecylsulfate (SDS) polyacrylamide gel and transferred onto Immobilon membranes (Millipore, Billerica, MA, USA). Western blot analyses were conducted using antibodies against VEGFR‐1:(1:200), VEGFR‐2 (1:200), pVEGFR‐1 (1:100), pVEGFR‐2 (1:100) and β‐actin (1:2000, Neomarker, Fremont, CA, USA). Bands were visualized using an electrochemiluminescence (ECL) kit (Amersham Biosciences, Piscataway, NJ, USA).
VEGF activity assay. Confluence (80–90%) of C6 cells, C6‐mock cells and C6‐VEGF−/– cells were treated with serum‐free 1640 M for 48 h. Media were collected and VEGF concentration were measured by ELISA. HUVEC cells (2 × 104, Cell Biology Research Institute of Shanghai, Shanghai, China) were placed in a 6‐well plate and treated with 100 ng of VEGF secreted from three C6 cell lines. The HUVEC cell growth curve was monitored by cell count.
In vivo tumorigenesis animals. Male 4‐to‐6‐weeks‐old BALB/c (nu/nµ) mice (SLAC, Shanghai, China) were randomized into three groups (n = 8): C6, C6‐mock and C6‐VEGF−/–. Tumorigenic mice were established by subcutaneous (s.c.) flank inoculation of 1.5 × 106 cells in serum‐free 1640 M. After 20 days post‐implantation, tumors were removed for further analysis. All procedures met the national guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the Fujian Medical University, Fujian, China.
Electron microscopy studies. Tumors were rapidly removed, immersed into 3% paraformaldehyde containing 1.5% glutaraldehyde for 4 h and post‐fixed in 1% osmium tetroxide (OsO4). Specimens were dehydrated in gradient alcohols and embedded. Sections (50 nm) were cut and stained with uranyl acetate and lead citrate for morphologic analysis under HU‐12 A transmission electron microscope (TEM; Hitachi, Tokyo, Japan).
Numbers of VVO were observed in 10 microvascular endothelial cells with an intact structure at a magnification of 5000× under TEM. The VVO were counted using the following scale: (0) <5 bunchy vesicles; (1) 6–10 bunchy vesicles; (2) 11–20 bunchy vesicles; and (3) >20 bunchy vesicles.
Tumor vessel permeability. Tumor‐bearing mice received a 0.1‐mL/kg, i.v. injection of Evans blue dye (1% in saline; Sigma‐Aldrich, St. Louis, MO, USA). After 6 h, the animals were killed and Evans blue was extracted from tumors as described.( 21 ) Briefly, tumors were removed and homogenized using 3 mL of N,N‐dimethylformamide (Sigma‐Aldrich), then incubated at 57°C for 12 h. The solutions were vortexed, then 2 mL of l N HCL were added, vortexed again and centrifuged at 2000 × g for 15 min. Supernatant was collected and measured at 620 nm by spectrophotometer (Bechman Coulter Fullerton, CA, USA). Concentrations were calculated using a standard curve for Evans blue dye.
Edema assays. Water content of the tumor represents the degree of edema and was calculated as previously described.( 22 ) Briefly, water content was assessed by subtracting the weight of dry tumor (incubated at 100°C vacuum oven for 24 h) from the weight of the fresh tumor.
Statistical analysis. Data were analyzed by using one‐way ANOVA followed by Tukey's post hoc test for multiple comparisons with the control groups. Differences were considered significant if P < 0.05.
Results
Effect of antisense VEGF in cell proliferation. We first verified VEGF expression in the heterogeneously overexpressing antisense VEGF cells (C6‐VEGF−/–). Immunoreaction with antibody against VEGF showed that endogenous VEGF expression was reduced in C6‐VEGF−/– cells compared with C6‐null cells and C6‐mock cells after 24 h serum deprivation (Fig. 1a–c) as well as that after 48 h serum deprivation (Fig. 1d–f). Simultaneously, we determined VEGF protein secretion in those cells and found that the levels of VEGF in medium from C6‐VEGF−/– cells were significantly lower than that from C6‐null cells and C6‐mock cells (Fig. 1g). These changes were observed similarly after 24 h and 48 h serum deprivation (Fig. 1g), suggesting that antisense VEGF blocked endogenous VEGF expression in glioma tumor cells.
Figure 1.
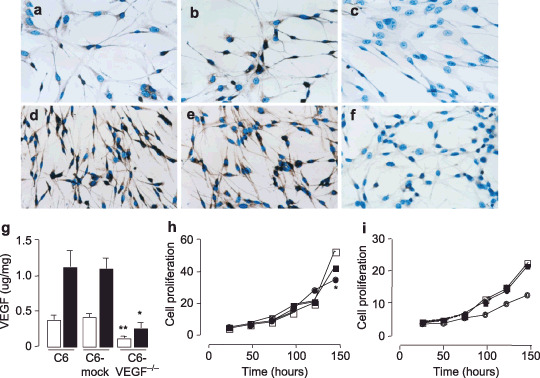
Characterization of antisense vascular endothelial growth factor (VEGF) in vitro. (a–f) Immunocytochemical staining with antibody against VEGF antigen in C6‐null cells (a, d), C6‐mock cells (b, e) and C6‐VEGF−/– cells (c, f) after 24 h (a–c) and 48 h (d–f) serum deprivation. (g) Enzyme‐linked immunosorbent assay (ELISA) of VEGF levels in condition media of C6‐null cells, C6‐mock cells and C6‐VEGF−/– cells after 24 h (open bars) and 48 h (closed bars) serum deprivation. (h) Time course of cell proliferation of C6‐null cells (open squares), C6‐mock cells (closed squares) and C6‐VEGF−/– cells (closed circles). (i) Time course of HUVEC cell growth curve with treatment of VEGF released from C6‐null cells (open squares), C6‐mock cells (closed squares), C6‐VEGF−/– cells (closed circles) and non‐treated (open circles). *, P < 0.05, **, P < 0.01; n = 4–6.
VEGF is considered to be a trigger of cell proliferation. To further investigate the effect of antisense VEGF in cell proliferation, we placed C6‐null cells, C6‐mock cells and C6‐VEGF−/– cells into 6‐well plates at a density of 2 × 104 cells per well, then counted cell numbers after 24 h, 48 h, 72 h, 96 h, 120 h and 144 h culture. Data showed regressive proliferation in C6‐VEGF−/– cells compared with the two controls, C6‐null cells and C6‐mock cells (Fig. 1h); morphology did not change in any cell line (data not shown). Moreover, we examined whether biological activity of VEGF released from different cell lines was altered. We treated HUVEC cells with the same amount VEGF secreted from three C6 cell lines and monitored the HUVEC cell growth chart. We found that HUVEC cells grew similarly in the treatment groups, whereas cells grew apparently slowly in the non‐VEGF‐treatment (Fig. 1i). Together, these data suggested that down‐regulation of VEGF expression inhibited cell proliferation.
Effect of antisense VEGF in tumorigenesis. VEGF plays a key role in tumorigenesis. We sought to determine whether down‐regulation of VEGF blocked its impacts, initiated tumor formation. To evaluate the anti‐tumor action, we injected glioma cells, C6‐null cells (Fig. 2a), C6‐mock cells (Fig. 2b) and C6‐VEGF−/– cells (Fig. 2c), directly into the right inguinal area of BALB/c mice to induce tumor formation (Fig. 2). After 20 days inoculation, tumor size from C6‐VEGF−/– mice (1.687 ± 0.189 cm3) was notably smaller than that from two controls, C6‐null mice (5.299 ± 0.656 cm3) and C6‐mock mice (5.534 ± 0.62 cm3) (Fig. 2d). Also, tumor weight was significantly decreased in C6‐VEGF−/– mice (1.018 ± 0.112 g) compared with C6 mice (2.81 ± 0.187 g) and C6‐mock mice (2.79 ± 0.221 g) (Fig. 2e).
Figure 2.
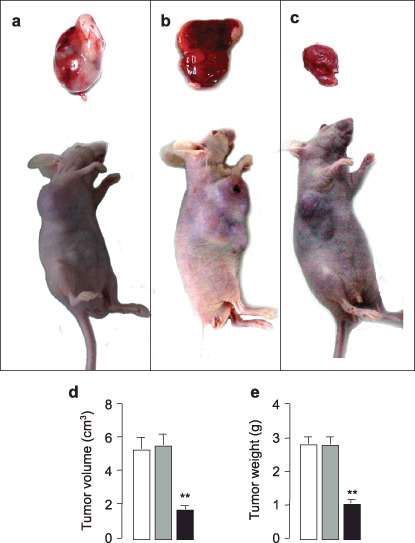
Tumor growth inhibition by antisense vascular endothelial growth factor (VEGF). (a–c) Representative tumorigenic mice implanted with C6‐null cells (a), C6‐mock cells (b) and C6‐VEGF−/– cells (c). Analyses of tumor volume (d) and tumor weight (e) in C6‐null mice (open bars), C6‐mock mice (gray bars) and C6‐VEGF−/– mice (closed bars). **P < 0.01; n = 7.
Characterization of tumorigenic mice. To test the effect of antisense VEGF in vivo as well as in vitro, we determined VEGF levels by ELISA (Fig. 3a) and semi‐quantitative PCR (Fig. 3b). ELISA analysis showed that the levels of VEGF protein were markedly decreased in the tumors from C6‐VEGF−/– mice compared with two control mice, C6 mice and C6‐mock mice (Fig. 3a). Similar results were obtained by semi‐quantitative PCR (Fig. 3b).
Figure 3.
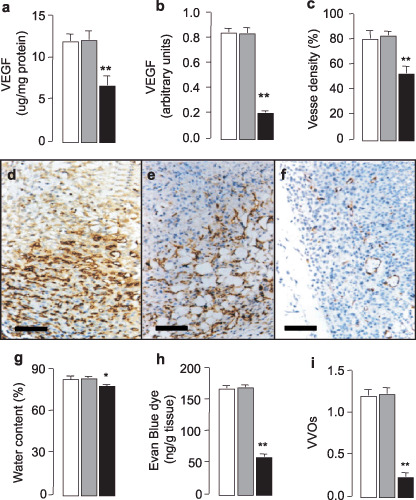
Reduction of neovascularization and edema by antisense vascular endothelial growth factor (VEGF). The expression levels of VEGF protein and mRNA in tumor tissue were assessed by enzyme‐linked immunosorbent assay (ELISA) (a) and semi‐quantitative PCR (b). (c) Quantification of the brown staining revealed the percentage area occupied by vasculature in tumor sections. d–f, Relative area of vascular density of the tumor section from C6‐null mice (d), C6‐mock mice (e) and C6‐VEGF−/– mice (f). Brown, CD31‐positive endothelial cells. (g) Measurement of water content in tumor tissues. (h) Quantitative vascular leakage assessed by Evan Blue dye tracer. (i) Observation of number of vesiculo‐vacuolar organelles (VVO) under electron microscope in tumor slices. Open bars, C6‐null mice; gray bars, C6‐mock mice; closed bars, C6‐VEGF−/– mice. *P < 0.05, **P < 0.01, n = 4–6, scale bar = 100 µm.
Up‐regulation of VEGF shown in brain glioma tumor is associated with angiogenesis.( 2 , 7 , 23 ) Here, we tested whether blockage of VEGF by antisense VEGF contributed anti‐angiogenesis properties. Immunohistochemistry using anti‐CD31 antibody stained microvasculature showed dramatically lower staining in the tumor tissue from C6‐VEGF−/– mice (Fig. 3c,f) compared with C6‐null mice (Fig. 3d) and C6‐mock mice (Fig. 3e), indicating that tumor‐induced angiogenesis was inhibited in C6‐VEGF−/– mice.
A common feature of malignant brain tumors is their ability to increase capillary permeability, subsequently leading to edema. The mechanism of tumor‐induced permeability is complex. Evidence implicating VEGF as an agent in tumor‐generated edema has been reported.( 23 , 24 ) In the present study, we demonstrated that tumoral edema in C6‐VEGF−/– mice was decreased compared with control mice, C6‐null mice and C6‐mock mice (Fig. 3g). Next, to confirm that the edema was due to vascular hyperpermeability, we examined vascular extravasation by using a dye tracer.( 21 ) Results showed that vascular leakage was markedly reduced in C6‐VEGF−/– mice compared with controls (Fig. 3h). These data imply that inhibition of VEGF expression suppresses tumor growth and vascular hyperpermeability, suggesting that a targeted VEGF pathway may be considered as a therapy for brain glioma tumor.
The effect of antisense VEGF on anti‐angiogenesis is mediated by tyrosine kinase receptors. VEGF binds to endothelial cells via interaction with high‐affinity tyrosine kinase receptors, VEGFR‐1 and VEGFR‐2. The selective expression of VEGF receptors ensures that VEGF action is confined to the endothelial cells( 25 , 26 ) and that additional signals are required to trigger angiogenesis in gliomas tumor. Here, we showed that lower production of VEGF was accompanied by lower expression of its receptors, VEGFR‐1 (Fig. 4a) and VEGFR‐2 (Fig. 4b). Furthermore, we examined the expression of phospho‐VEGFR in those tumors and showed that phospho‐VEGFR‐1 (Fig. 4c) and phospho‐VEGFR‐2 (Fig. 4d) were correlated with VEGF levels. Quantitative analysis showed that the expression levels of tyrosine receptors, normalized by β‐actin, markedly decreased in the C6‐VEGF−/– mice (Fig. 4e). Similar observations were obtained by immunohistochemistry studies. 5, 6 demonstrate that C6‐VEGF−/– mice (5, 6) had less staining of VEGF‐1 receptor (Fig. 5c), VEGF‐2 receptor (Fig. 5f), phospho‐VEGF‐1 receptor (Fig. 6c) and phospho‐VEGF‐2 receptor (Fig. 6f) compared with C6‐null mice (5, 6) and C6‐mock mice (5, 6). Interestingly, VEGFR were not only present in vascular endothelial cells, but were also found in the tumor cell body that possible engages with tumor proliferation. This evidence suggests that VEGF acting on vasculogenesis and angiogenesis require its receptor tyrosine kinases.
Figure 4.
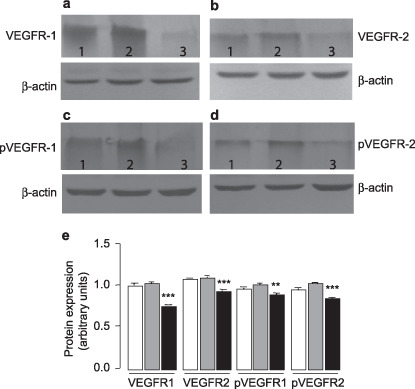
The expression of vascular endothelial growth factor receptors (VEGFR) decreased in C6‐VEGF−/– mice. The expression levels, assessed by Western blot, of VEGF receptors using antibody against (a) VEGFR‐1, (b) VEGFR‐2, (c) phospho‐VEGFR‐1 and (d) phospho‐VEGFR‐2. 1, C6‐null mice; 2, C6‐mock mice; 3, C6‐VEGF−/– mice. (e) Quantitative expression levels of VEGFR‐1, VEGFR‐2, phos‐VEGFR1, and phos‐VEGFR2. Open bars, C6‐null mice; gray bars, C6‐mock mice; closed bars, C6‐VEGF−/– mice. ***P < 0.001; **P < 0.01.
Figure 5.
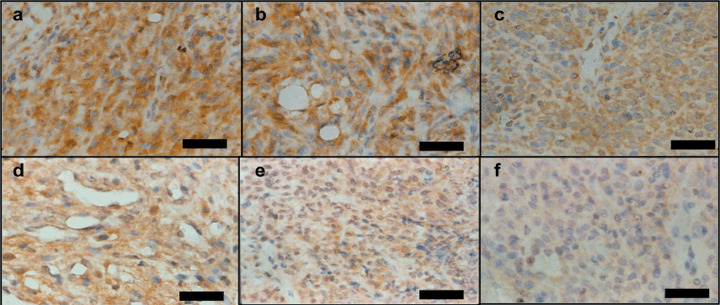
Down‐regulation of vascular endothelial growth factor receptors (VEGFR) expression by antisense vascular endothelial growth factor (VEGF). Immunoreaction with antibody against VEGFR‐1 (a–c) and VEGFR‐2 (d–f) in C6‐null mice (a, d), C6‐mock mice (b, e) and C6‐VEGF−/– mice (c, f). Scale bar = 50 µm.
Figure 6.
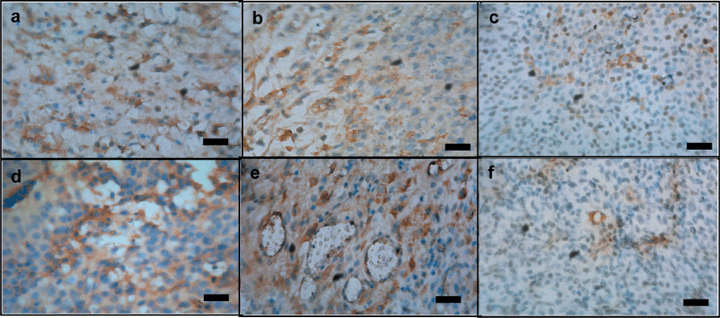
Down‐regulation of phospho‐VEGFR expression by antisense vascular endothelial growth factor (VEGF). Immunoreaction with antibody against phospho‐VEGFR‐1 (a–c) and phospho‐VEGFR‐2 (d–f) in C6‐null mice (a, d), C6‐mock mice (b, e), and C6‐VEGF−/– mice (c, f). Scale bar = 50 µm.
The mechanism of tumor‐induced edema. Previous data showed that the expression level of VEGF in tumors was correlated with the levels of extravasation.( 6 ) To elucidate the structural basis for this effect, we determined the morphological structure of tumors from C6 mice, C6‐mock mice and C6‐VEGF−/– mice by TEM. TEM analyses showed that opened intercellular tight junctions and endothelium fenestration (or cleft) (Fig. 7a–c) were observed in tumor tissues from C6 mice, C6‐mock mice and C6‐VEGF−/– mice; however, these structural changes were not different among those animals. In contrast, there were conspicuous VVO in tumor VEC from C6 mice and C6‐mocks mice (Fig. 7d,e), whereas few VVO were observed in tumor VEC from C6‐VEGF−/– mice (Fig. 7f) that develops attenuated extravasation and edema in tumor tissues. Based on ultrastructural analyses of 10 vascular endothelial cells (VEC), 0.23 ± 0.05 VVO were found in cytoplasm of C6‐VEGF−/– mice (Fig. 3i). However, in the same sections of C6 mice and C6‐mock mice, 1.2 ± 0.07 and 1.23 ± 0.05 VVO were observed, respectively (Fig. 3i). The number of vesicles and vacuoles per individual VVO were significantly reduced in C6‐VEGF−/– mice compared with that in C6 mice and C6‐mock mice (Fig. 7d–f).
Figure 7.
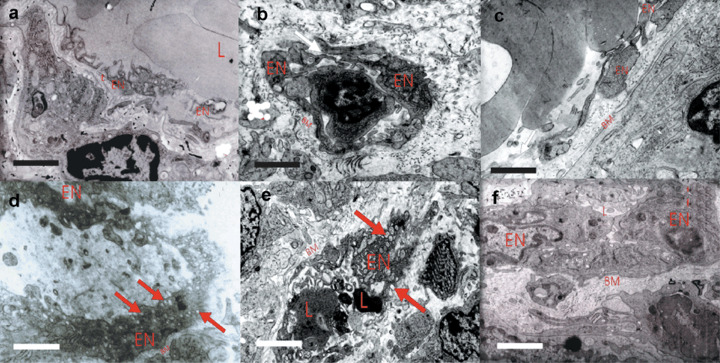
High‐magnification electron micrographs illustrate the reduction of vesiculo‐vacuolar organelles (VVO) structure in C6‐VEGF−/– mice. (a–c) The morphological features of endothelium intercellular junctions (black arrows) and fenestrae (red arrows) between vascular endothelial cells (VEC) in tumor tissue from C6 mice (a), C6‐mock mice (b) and C6‐VEGF−/– mice (c). (d–e) Electron microscope reveals VVO structure (red arrows) in tumor tissue from C6 mice (d), C6‐mock mice (e) and C6‐VEGF−/– mice (f). L, lumens, EN; vascular endothelial cells; BM, base membrane. Scale bar = 1 µm.
Discussion
Determining the contribution of VEGF to tumorigenesis and tumoral angiogenesis is important for developing novel therapeutic approaches for brain gliomas. In the present work, we demonstrated that down‐regulated VEGF expression reduced cell proliferation of glioma C6 cells and we established tumorigenic animals subcutaneously implanted with C6‐null cells, C6‐mock cells and C6‐VEGF−/– cells that were grown in orthotropic sites in mice. In our models, the contribution of VEGF to tumor growth and vascularization was dependent on the expression levels of tumor‐cell‐derived VEGF. Compared with C6‐null mice or C6‐mock mice, tumor size and neovascularization were markedly decreased in C6‐VEGF−/– mice. This result is consistent with previousreports.( 28 ) In addition to the reduction in tumor size, angiogenesis is an extremely important process for sustained tumor growth. Our studies demonstrated lower staining of CD31 in tumor sections from C6‐VEGF−/– mice compared with that in C6‐null cell mice and C6‐mock mice. These data clearly indicate that neomicrovascular formation was decreased in tumors after blockage of VEGF, subsequently reducing the blood flow in the tumor area and limiting tumor growth. Blockage of the VEGF pathway predominantly inhibits tumor endothelial cell proliferation, migration and survival.
Initially, Senger et al.( 24 ) discovered VEGF as a permeability factor and found that tumor‐derived VEGF induced vascular leakage, causing extravasation. The mechanism of VEGF‐induced vascular leakage has not been fully elucidated. Our present in vivo studies demonstrated that the inhibition of tumor growth and edema by down‐regulated VEGF was mediated by tyrosine kinase receptors. These results are in agreement with previously published data.( 28 , 29 ) However, our study is the first demonstration that glioma‐induced edema is caused by fusion of clustered caveolae, referred to as vesiculo‐vacuolar organelles (VVO).( 16 , 18 )
Tumor vasculature has long been characterized as hyperpermeable.( 30 ) However, there are some controversies regarding the mechanisms and structures responsible for the increase permeability of tumor vessels. Earlier reports suggested that open endothelial gaps/fenestration resulted in tumor vessel hyperpermeability induced by VEGF.( 5 , 13 ) However, our studies demonstrated that intercellular fenestrae was observed between VEC of tumors from C6 mice, C6‐mock mice and C6‐VEGF−/– mice and there was no structural difference among them. Moreover, our previous work on microecosystems in human glioma had shown that fenestrae are only observed in a limited number of individual vascular endothelial cells,( 31 ) whereas they are commonly observed in meningioma. It had been reported that fenestrae are formed and maintained by VVO, which increase blood vessel permeability.( 32 ) Feng et al.( 16 ) found that VEGF induced permeability of tumor blood vessels by increasing VVO function such that individual VVO extend across endothelial cells and interconnect with each other to open stomata. These stomata likely provide the structural basis for extravasation acrossed the microvascular endothelium. The function of VVO in tumor vessels is regulated by vasoactive mediators that in some way open the stomata that connected individual VVO vesicles and vascuoles.( 14 ) VEGF is likely one of vasoactive mediators responsible for opening stomata in hyperpermeable tumor vessels.( 24 , 33 , 34 ) In fact, this proves to be the case. Our electron microscopic studies demonstrated that tumor tissues from C6‐VEGF−/– mice prevented extravasation and VVO formation. Intense immunostaining for VEGF was observed on the plasma membrane of tumor‐associated microvascular endothelial cells and VVO are present in these same endothelial cells.( 17 ) Thus, VVO might serve as the major pathway in response to VEGF‐induced vessel hyperpermeability.
In conclusion, our findings demonstrate the effects of VEGF signaling in malignant glioma and it can be interpreted that VVO formation possibly participates in tumor‐induced edema. Blockage of VEGF suppressed tumor growth and prevented tumor hyperpermeability, suggesting an important role of this system in tumor invasion. Assessing VEGF in malignant glioma on a functional level may enable a closer understanding of glioblastoma angiogenesis and lead to more effective therapeutical approaches for brain tumors.
Acknowledgments
We thank Professor Lin Xu for primer design, Professor Tian Jun for assistance in data processing and statistical analysis and Dr Ana Guiano for critical reading. This work was support by a grant (To ZX Lin, No: 01Z034) from the Science and Technology Commission, Fujian, China.
References
- 1. Gurney JG, Kadan‐Lottick N. Brain and other central nervous system tumors: rates, trends, and epidemiology. Curr Opin Oncol 2001; 13: 160–6. [DOI] [PubMed] [Google Scholar]
- 2. Torp SH, Helseth E, Dalen A, Unsgaard G. Epidermal growth factor receptor expression in human gliomas. Cancer Immunol Immunother 1991; 33: 61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Criscuolo GR, Merrill MJ, Oldfield EH. Further characterization of malignant glioma‐derived vascular permeability factor. J Neurosurg 1988; 69: 254–62. [DOI] [PubMed] [Google Scholar]
- 4. Strugar JG, Criscuolo GR, Rothbart D, Harrington WN. Vascular endothelial growth/permeability factor expression in human glioma specimens: correlation with vasogenic brain edema and tumor‐associated cysts. J Neurosurg 1995; 83: 682–9. [DOI] [PubMed] [Google Scholar]
- 5. Roberts WG, Palade GE. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res 1997; 57: 765–72. [PubMed] [Google Scholar]
- 6. Carlson MR, Pope WB, Horvath S et al . Relationship between survival and edema in malignant gliomas: role of vascular endothelial growth factor and neuronal pentraxin 2. Clin Cancer Res 2007; 13: 2592–8. [DOI] [PubMed] [Google Scholar]
- 7. Leon SP, Folkerth RD, Black PM. Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer 1996; 77: 362–72. [DOI] [PubMed] [Google Scholar]
- 8. Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res 1986; 46: 5629–32. [PubMed] [Google Scholar]
- 9. Keck PJ, Hauser SD, Krivi G et al . Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989; 246: 1309–12. [DOI] [PubMed] [Google Scholar]
- 10. Senger DR, Van de Water L, Brown LF et al . Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev 1993; 12: 303–24. [DOI] [PubMed] [Google Scholar]
- 11. Brown LF, Yeo KT, Berse B et al . Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med 1992; 176: 1375–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000; 407: 249–57. [DOI] [PubMed] [Google Scholar]
- 13. Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci 1995; 108: 2369–79. [DOI] [PubMed] [Google Scholar]
- 14. Dvorak AM, Feng D. The vesiculo‐vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem 2001; 49: 419–32. [DOI] [PubMed] [Google Scholar]
- 15. Stan RV. Structure and function of endothelial caveolae. Microsc Res Tech 2002; 57: 350–64. [DOI] [PubMed] [Google Scholar]
- 16. Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo‐vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med 1996; 183: 1981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qu H, Nagy JA, Senger DR, Dvorak HF, Dvorak AM. Ultrastructural localization of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) to the abluminal plasma membrane and vesiculovacuolar organelles of tumor microvascular endothelium. J Histochem Cytochem 1995; 43: 381–9. [DOI] [PubMed] [Google Scholar]
- 18. Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF. The vesiculo‐vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol 1996; 59: 100–15. [PubMed] [Google Scholar]
- 19. Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995; 146: 1029–39. [PMC free article] [PubMed] [Google Scholar]
- 20. Munaut C, Noel A, Hougrand O, Foidart JM, Boniver J, Deprez M. Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1‐MMP, MMP‐2 and MMP‐9 in human glioblastomas. Int J Cancer 2003; 106: 848–55. [DOI] [PubMed] [Google Scholar]
- 21. Roberts WG, Hasan T. Tumor‐secreted vascular permeability factor/vascular endothelial growth factor influences photosensitizer uptake. Cancer Res 1993; 53: 153–7. [PubMed] [Google Scholar]
- 22. Huang F, Xi G, Keep R, Hua Y, Nemoianu A, Hoff J. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg 2002; 96: 287–93. [DOI] [PubMed] [Google Scholar]
- 23. Connolly DT. Vascular permeability factor: a unique regulator of blood vessel function. J Cell Biochem 1991; 47: 219–23. [DOI] [PubMed] [Google Scholar]
- 24. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983; 219: 983–5. [DOI] [PubMed] [Google Scholar]
- 25. Shibuya M, Yamaguchi S, Yamane A et al . Nucleotide sequence and expression of a novel human receptor‐type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990; 5: 519–24. [PubMed] [Google Scholar]
- 26. Millauer B, Wizigmann‐Voos S, Schnurch H et al . High affinity VEGF binding and developmental expression suggest Flk‐1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993; 72: 835–46. [DOI] [PubMed] [Google Scholar]
- 27. Connolly DT, Heuvelman DM, Nelson R et al . Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest 1989; 84: 1470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verheul HM, Hammers H, Van Erp K et al . Vascular endothelial growth factor trap blocks tumor growth, metastasis formation, and vascular leakage in an orthotopic murine renal cell cancer model. Clin Cancer Res 2007; 13: 4201–8. [DOI] [PubMed] [Google Scholar]
- 29. Cantarella G, Risuglia N, Dell’eva R et al . TRAIL inhibits angiogenesis stimulated by VEGF expression in human glioblastoma cells. Br J Cancer 2006; 94: 1428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jain RK. Delivery of novel therapeutic agents in tumors: physiological barriers and strategies. J Natl Cancer Inst 1989; 81: 570–6. [DOI] [PubMed] [Google Scholar]
- 31. Lin Z, Zhang P, Jiang C, Chen Z, He L, Chen J. Morphological observation of microecosystem in human brain glioma invasion. Acta Anat Sinica 2002; 33: 355–9. [Google Scholar]
- 32. Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol 1998; 140: 947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dvorak HF, Dvorak AM, Manseau EJ, Wiberg L, Churchill WH. Fibrin gel investment associated with line 1 and line 10 solid tumor growth, angiogenesis, and fibroplasia in guinea pigs. Role of cellular immunity, myofibroblasts, microvascular damage, and infarction in line 1 tumor regression. J Natl Cancer Inst 1979; 62: 1459–72. [PubMed] [Google Scholar]
- 34. Gospodarowicz D, Abraham JA, Schilling J. Isolation and characterization of a vascular endothelial cell mitogen produced by pituitary‐derived folliculo stellate cells. Proc Natl Acad Sci USA 1989; 86: 7311–15. [DOI] [PMC free article] [PubMed] [Google Scholar]