

Abstract
To elucidate the molecular mechanisms of mammary carcinogenesis and discover novel therapeutic targets for breast cancer, we previously carried out genome‐wide expression profile analysis of 81 breast cancer cases by means of cDNA microarray coupled with laser microbeam microdissection of cancer cells. Among the dozens of transactivated genes, in the present study we focused on the functional significance of kinesin family member 2C (KIF2C)/mitotic centromere‐associated kinesin (MCAK) in the growth of breast cancer cells. Northern blot and immunohistochemical analyses confirmed KIF2C/MCAK overexpression in breast cancer cells, and showed that it is expressed at undetectable levels in normal human tissues except the testis, suggesting KIF2C/MCAK to be a cancer–testis antigen. Western blot analysis using breast cancer cell lines revealed a significant increase in the endogenous KIF2C/MCAK protein level and its phosphorylation in G2/M phase. Treatment of breast cancer cells with small interfering RNA against KIF2C/MCAK effectively suppressed KIF2C/MCAK expression and inhibited the growth of the breast cancer cell lines T47D and HBC5. In addition, we found that KIF2C/MCAK expression was significantly suppressed by ectopic introduction of p53. These findings suggest that overexpression of KIF2C/MCAK might be involved in breast carcinogenesis and is a promising therapeutic target for breast cancers. (Cancer Sci 2008; 99: 62–70)
- Abbreviations: β2MG
β2‐microgloblin
- FACS
fluorescence‐activated cell sorting
- HMEC
human mammalian epithelial cell
- HRP
horseradish peroxidase
- KIF2C
kinesin family member 2C
- MCAK
mitotic centromere‐associated kinesin
- MTT
3‐(4,5‐dimethylthiaxol‐2‐yl)‐2,5‐diphenyltetraozolium bromide
- NP
Nonidet P
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PRC
protein regulator of cytokinesis
- RT
reverse transcription
- SDS
sodium dodecylsulfate
- siRNA
small interfering RNA.
Breast cancer is one of the most common cancers in women worldwide and its incidence has been increasing significantly. According to an estimation in 2002, more than 1 100 000 patients were newly diagnosed with breast cancer, and approximately 410 000 patients died of the disease.( 1 ) Although early detection with mammography and the development of molecular‐targeted therapeutic drugs, such as tamoxifen, aromatase inhibitors, and trastuzumab, have contributed to a reduction in the mortality rate and are improving the quality of life of women diagnosed with this disease, treatment options are still limited to patients at an advanced stage. Hence, novel molecular‐targeted drugs to provide better management to such patients are still eagerly awaited.
Gene‐expression profiles obtained by cDNA microarray analysis have been proven to provide a detailed characterization of individual cancers, and such information should contribute to choosing more appropriate clinical strategies for individual patients through the development of novel drugs and providing the basis for personalized treatment.( 2 ) Through genome‐wide expression analysis we have isolated a number of genes that function as oncogenes in the development and progression of breast cancers,( 3 , 4 , 5 ) bladder cancers,( 6 , 7 ) synovial sarcomas,( 8 , 9 ) testicular seminomas,( 10 ) and renal cell carcinomas.( 11 , 12 ) Such molecules are considered to be candidate targets for the development of new therapeutic modalities.
Because cytotoxic drugs often cause severe adverse reactions, it is obvious that thoughtful selection of novel target molecules on the basis of well‐characterized mechanisms of action should be very helpful for developing effective anticancer drugs with the minimum risk of adverse reactions. Toward this goal, we previously analyzed the genome‐wide expression profile of 81 breast cancers by means of a combination of cDNA microarray and laser microbeam microdissection.( 13 ) Among the genes upregulated in breast cancers, we focused on KIF2C/MCAK as a novel molecular‐target for breast cancer therapy.
KIF2C/MCAK was previously identified as a kinesin in a screen using affinity‐purified antipeptide sera raised against the conserved region of the kinesin motor domain.( 14 ) This protein is known to be a member of the kinesin‐13 subfamily of kinesin‐related proteins that share high homology with other members of the kinesin superfamily.( 14 , 15 , 16 ) It is also thought that its activity is regulated by aurora kinase‐B, and it plays important roles in chromosome segregation and the correction of improper kinetochore–microtubule interactions during mitosis.( 17 , 18 ) Recently, we reported that the interaction between KIF2C/MCAK and PRC1 plays an important role in cytokinesis.( 4 ) However, its pathophysiological roles in mammary carcinogenic cells remain uninvestigated.
Here we report overexpression of KIF2C/MCAK in breast cancer cells and its undetectable level of expression in normal human tissues except the testis. We further demonstrate that downregulation of KIF2C/MCAK expression results in significant growth suppression of breast cancer cells. Moreover, we found that KIF2C/MCAK is negatively regulated by p53. Our findings suggest that KIF2C/MCAK is likely to play critical roles in tumor cell growth and be a promising target for the development of anticancer drugs to breast cancer.
Materials and Methods
Breast cancer cell lines and clinical samples. The human cancer cell lines BT‐20, BT‐474, MCF‐7, SK‐BR‐3, HCC1937, MDA‐MB‐231, MDA‐MB‐435S, T47D (breast cancer), H1299 (human lung carcinoma), and U373MG (glioblastoma) were purchased from American Type Culture Collection (Rockville, MD, USA). The HMEC line was purchased from Cambrex Bio Science (Walkersville, MD, USA). The HBC4 and HBC5 breast cancer cell lines were kindly provided by Dr T. Yamori (Molecular Pharmacology, Cancer Chemotherapy Center of the Japanese Foundation for Cancer Research). The HCT116 (p53 +/+, p53 −/–) cell lines were kindly provided by Dr B. Vogelstein (Johns Hopkins University, Baltimore, MD, USA). All cells were cultured in appropriate media: RPMI‐1640 (Sigma‐Aldrich, St Louis, MO, USA) for HBC4, HBC5, HCC1937, and T47D (with 2 mM l‐glutamine); Dulbecco's modified Eagle's medium (Sigma‐Aldrich) for BT‐474 and HCT116; Eagle's minimum essential medium (EMEM) (Sigma‐Aldrich) for BT‐20, MCF‐7, and U373MG (with 0.01 mg/mL insulin); McCoy (Sigma‐Aldrich) for SK‐BR‐3 (with 1.5 mM l‐glutamine); L‐15 (Roche, Basel, Switzerland) for MDA‐MB‐435S and MDA‐MB‐231; and mammary epithelial growth medium (MEGM; Cambrex Bio Science) for HMEC. Each medium was supplemented with 10% fetal bovine serum (Cansera International, Ontario, Canada) and 1% antibiotic and antimycotic solution (Sigma‐Aldrich). MDA‐MB‐231 cells were maintained at 37°C in an atmosphere of humidified air without CO2. The other cell lines were maintained at 37°C in an atmosphere of humidified air with 5% CO2. Tissue samples from surgically resected breast cancers and their corresponding clinical information were obtained from the Department of Breast Surgery, Cancer Institute Hospital, Tokyo, and the Division of Breast and Endocrine Surgery, Department of Surgery, St Marianna University School of Medicine after obtaining written informed consent.
Semiquantitative RT‐PCR analysis. Microdissection of breast cancer cells was carried out as described previously.( 13 ) Total RNA was extracted from each of the microdissected breast cancer clinical samples, microdissected normal breast ductal cells, and breast cancer cell lines using RNeasy Micro Kits (Qiagen, Valencia, CA, USA) and polyA+ RNA isolated from mammary gland (Takara Clontech, Kyoto, Japan). Subsequently, T7‐based amplification and RT were carried out as described previously.( 13 ) We prepared appropriate dilutions of each single‐stranded cDNA for subsequent PCR by monitoring β2MG as a quantitative control. The sequences of each primer set were as follows; 5′‐ACTCTAGGACTTGCATGATTGCC‐3′ and 5′‐TGGGTGT CAAACCAAACAGA‐3′ for KIF2C/MCAK (GenBank accession number NM_006845), and 5′‐AACTTAGAGGTGGGAGCAG‐3′ and 5′‐CACAACC ATGCCTTACTTTATC‐3′ for β2MG.
Northern blot analysis. Breast cancer northern blot membrane was prepared as described previously.( 3 ) Human multiple‐tissue northern blots (Takara Clontech, Kyoto, Japan) were hybridized with the [α32P]‐dCTP‐labeled PCR products of KIF2C/MCAK (GenBank accession number; NM_006845) prepared by RT‐PCR (see below) or with [α32P]‐dCTP‐labeled β‐actin cDNA as a loading control. Prehybridization, hybridization, and washing were carried out according to the supplier's recommendations. The blots were autoradiographed with intensifying screens at –80°C for 14 days. Specific probe for KIF2C/MCAK (548 bp) was prepared by RT‐PCR using the following primer set: 5′‐ACTCTAGGACTTGCATGATTGCC‐3′ and 5′‐TGGGTGTCAAACCAAAC AGA‐3′. It was radioactively labeled with the megaprime DNA labeling system (GE Healthcare, Buckinghamshire, UK).
Generation of anti‐KIF2C/MCAK‐specific polyclonal antibody. Plasmids designed to express two fragments of KIF2C/MCAK (amino acids 86–239 and 124–239) with a His‐tag at their C‐terminus were prepared using pET21a vectors (Novagen, Madison, WI, USA). The two recombinant peptides were expressed in the Escherichia coli BL21 codon‐plus strain (Stratagene, La Jolla, CA, USA), and purified using Ni‐NTA resin agarose according to the supplier's protocols (Qiagen). The purified recombinant proteins were mixed together and then used for immunization of rabbits (Medical and Biological Laboratories, Nagoya, Japan). Subsequently, the immune sera was purified on antigen affinity columns using Affigel 15 gel according to supplier's instructions (Bio‐Rad, Hercules, CA, USA). We confirmed that this antibody could specifically recognize endogenous KIF2C/MCAK proteins in MCF‐7 breast cancer cells by western blotting analysis. This antibody was used for western blotting, immunocytochemial staining, and immunohistochemical staining analyses as described below.
Western blot analysis. To detect the endogenous KIF2C/MCAK protein in breast cancer cell lines (BT‐20, HBC4, HBC5, HCC1937, MCF‐7, MDA‐MB‐231, MDA‐MB‐435S, SK‐BR‐3, T47D, and BT474) and HMEC, each of these cells was lysed in lysis buffer (50 mM Tris‐HCl [pH 8.0], 150 mM NaCl, 0.5% NP‐40) including 0.1% protease inhibitor cocktail III (Calbiochem, San Diego, CA, USA). The amount of total protein was estimated by protein assay kit (Bio‐Rad), and then proteins were mixed with SDS sample buffer and boiled for 3 min before loading on 10% SDS‐PAGE gels. After electrophoresis, the proteins were blotted onto nitrocellulose membranes (GE Healthcare), which were blocked with 4% BlockAce (Dainippon Pharmaceutical, Osaka, Japan). The membranes were then incubated with anti‐KIF2C/MCAK polyclonal antibody for the detection of endogenous KIF2C/MCAK proteins. Finally, the membrane was incubated with HRP‐conjugated secondary antibody, and bands were visualized using ECL detection reagents (GE Healthcare). β‐Actin served as a loading control.
Lambda phosphatase assay. To examine the phosphorylation status of KIF2C/MCAK protein in breast cancer cells, we treated the cellular extracts from MCF‐7 cells with lambda phosphatase (New England Biolabs, Beverly, MA, USA). Cells were lysed with lysis buffer (50 mM Tris‐HCl [pH 8.0], 150 mM NaCl, 0.5% NP‐40) and the cell lysates were treated for 2 h at 30ºC with 400 U protein phosphatase (New England Biolabs) in phosphatase buffer containing 50 mM Tris‐HCl (pH 7.5), 0.1 mM Na2EDTA, 5 mM dithiothreitol, 2 mM MgCl2, and 0.01% Brij‐35.
Immunocytochemical staining analysis. To examine the subcellular localization of endogenous KIF2C/MCAK protein in the breast cancer cell lines T47D and MCF‐7, we seeded the cells at 1 × 105 cells per well (Laboratory‐Tek II chamber slide; Nalgen Nunc International, Naperville, IL, USA). After 24‐h culture, cells were fixed with PBS (–) containing 4% paraformaldehyde for 15 min, and rendered permeable with PBS containing 0.1% Triton X‐100 at 4°C for 2.5 min. Subsequently, the cells were covered with 3% bovine serum albumin (BSA) in PBS at 4°C for 12 h to block non‐specific antibody binding followed by incubation with a rabbit affinity‐purified anti‐KIF2C/MCAK polyclonal antibody diluted 1:100. After washing with PBS, the cells were stained with an Alexa488‐conjugated antirabbit secondary antibody (Molecular Probes, Eugene, OR, USA) diluted 1:1000. Nuclei were counterstained with 4′,6′‐diamidine‐2′‐phenylindole dihydrochloride. Fluorescence images were obtained under a TCS SP2 AOBS microscope (Leica, Tokyo, Japan).
Immunohistochemical staining analysis. The expression patterns of KIF2C/MCAK in breast cancer and normal human tissues were examined as described previously.( 3 ) Slides of paraffin‐embedded breast cancer specimens (samples 234, 240, and 067), normal mammary tissue (sample 679), and other normal human tissues (heart, liver, kidney, lung, and testis) (Biochain, Hayward, CA, USA) were processed for antigen retrieval by autoclave (108°C, 15 min) in antigen‐retrieval solution, high pH (Dako Cytomation, Carpinteria, CA, USA), and treated with peroxidase blocking regent (Dako Cytomation). Tissue sections were incubated with the affinity‐purified anti‐KIF2C/MCAK polyclonal antibody (1:100) followed by HRP‐conjugated secondary antibody (Dako Cytomation). Antigen was visualized with substrate chromogen (Dako liquid DAB chromogen; Dako Cytomation). Finally, tissue specimens were stained with hematoxylin to discriminate the nucleus from the cytoplasm.
Fluorescence‐activated cell sorting analysis. The cell cycles of cultured HBC5 breast cancer cells were synchronized by treatment with 0.2 µg/mL aphidicolin (Sigma‐Aldrich) for 12 h. They were then washed five times with PBS, and fresh culture medium was added to release them from cell‐cycle arrest. After release from cell‐cycle arrest, the cells were collected and fixed with 70% ethanol, then kept at 4°C before use. The cells were incubated with 10 mg/mL RNaseI in PBS at 37°C for 30 min and stained with 50 µg propidium iodide at room temperature for 30 min. The cell suspensions at each time point were analyzed using FACscan (Becton Dickinson, Franklin Lakes, NJ, USA). Additionally, to detect the expression of endogenous KIF2C/MCAK protein in cells at various cell‐cycle points, we carried out western blotting analysis using anti‐KIF2C/MCAK polyclonal antibody as described in the western blotting analysis section.
Gene‐silencing effect of KIF2C/MCAK by siRNA. We established a vector‐based RNA interference expression system using psiU6BX3.0 siRNA expression vectors.( 19 ) The siRNA expression vectors against KIF2C/MCAK (psiU6BX3.0‐KIF2C/MCAK) and a mock control (psiU6BX3.0‐Mock) were prepared by cloning double‐stranded oligonucleotides into the BbsI site of the psiU6BX3.0 vector. The target sequences of the synthetic oligonucleotides for siRNA were as follows: 5′‐GTGTCTTCAAGCTTGAAGA‐3′ for siMock, 5′‐TAAACCCAGAACTCTTACA‐3′ for Si‐#1, 5′‐GAGAAGAAGGCCCAGAACT‐3′ for Si‐#2, 5′‐CAAAGTATCTGGAGAACCA‐3′ for Si‐#3, 5′‐TAGAAGAAGCCCCAGAACA‐3′ for Si‐m#1, and 5′‐TAGAAGAAGACCCAGAACA‐3′ for Si‐m#2 (bold letters indicate mismatched sequence in Si‐#2). All constructs were confirmed by DNA sequencing (ABI3700; PE Applied Biosystems, Foster, CA, USA).
The human breast cancer cells lines T47D and HBC5 were plated onto 10‐cm dishes (1 × 106 cells/dish) and transfected with 8 µg each of psiU6BX3.0‐Mock (without insertion; siMock) and psiU6BX3.0‐KIF2C/MCAK (Si‐#1, Si‐#2, Si‐#3, and two constructs [Si‐m#1 and Si‐m#2], including three base substitutions in Si‐#2) using FuGENE6 transfection reagent (Roche) as described in the section above. At 24 h after transfection, cells are reseeded for RT‐PCR (1 × 106 cells/10‐cm dish), western blotting (1 × 106 cells/10‐cm dish), colony formation assay (1 × 106 cells/10‐cm dish) and MTT assay (2 × 105 cells/well). We selected the psiU6BX3.0‐introduced T47D or HBC5 cells with medium containing 0.6 or 0.4 mg/mL neomycin (geneticin; Invitrogen, Carlsbad, CA, USA), respectively. We changed culture medium twice a week. To evaluate the knockdown effect of siRNA, we carried out semiquantitative RT‐PCR using total RNA extracted from the cells after a 5‐day incubation with neomycin, and western blot analysis with anti‐KIF2C/MCAK antibody using cell extracts harvested after a 7‐day incubation with neomycin. The specific primer sets for semiquantitative RT‐PCR were as follows: 5′‐ACTCTAGGACTTGCATGATTGCC‐3′ and 5′‐TGGGTGTCAAA CCAAACAGA‐3′ for KIF2C/MCAK; and 5′‐AACTTAGAGGTGGGAGCAG‐3′ and 5′‐CACAACCATGCCTTACTTTATC‐3′ for β2MG as an internal control. Transfectants expressing siRNA were grown for 4 weeks in selective media containing neomycin, and then fixed with 4% paraformaldehyde for 15 min before staining with Giemsa solution (Merck, Whitehouse Station, NJ, USA) to assess colony number. To quantify cell viability, MTT assays were carried out with cell‐counting kit‐8 (Wako, Osaka, Japan) according to the manufacturer's recommendations. Absorbance at 570 nm was measured using Microplate Reader 550 (Bio‐Rad). These experiments were carried out in triplicate. Furthermore, we verified the knockdown effects of KIF2C/MCAK on cell morphology by microscopy and immunocytochemical staining analysis with aphidicollin. At 5 days after the transfection into HBC5 cells, we examined the morphological changes of siKIF2C/MCAK‐si#2 or siMock‐transfected HBC5 cells.
Preparation of recombinant adenovirus and infection of cells. The replication‐deficient recombinant adenoviruses Ad‐p53 and Ad‐LacZ as a control, encoding p53 and LacZ, respectively, were generated and purified as described previously.( 20 ) U373MG or H1299 cells that were seeded in 10‐cm dishes (2 × 106 cells per dish) and incubated at 37°C until the time of harvest before adenoviral infection were infected with 20 multiplicity of infection (MOI) Ad‐p53 or Ad‐LacZ. We carried out semiquantitative RT‐PCR and western blotting analyses to verify the expression of KIF2C/MCAK, p53, and p21WAF1 using mRNA and cell lysates that were extracted from Ad‐p53‐ or Ad‐LacZ‐infected cells as described in the semiquantitative RT‐PCR analysis section. The primer sequences were as follows: 5′‐TTCTGGGACAGCCAAGTCTG‐3′ and 5′‐CACGCACCTCAAAGCTGTTC‐3′ for p53; 5′‐GTTCCTTGTGGAGCCGGAGC‐3′ and 5′‐GGTACAAGACAGTGACAGGTC‐3′ for p21WAF1 . Western blotting analysis using rabbit polyclonal anti‐KIF2C/MCAK and mouse monoclonal antip53 Ab‐6 (clone DO‐1; Calbiochem, San Diego, CA, USA) antibodies were carried out as described in the western blot analysis section.
Luciferase assay. A DNA fragment of KIF2C/MCAK promoter, including potential p53‐binding sites, was amplified by PCR, and inserted into the MluI and XhoI sites of the pGL3‐basic vector (Promega, Madison, WI, USA). The primer sequences for cloning were as follows: PrimerF, 5′‐AAAACGCGTGGCAACATGCTTCTGCACTA‐3′; and PrimerR: 5′‐AAACTCGAGCGGAGAGTCAGCAAGGAAGA‐3′. (The underlining indicates restriction enzyme sites.) U373MG and H1299 cells were seeded on 12‐well plates (1 × 105 cells/well) 24 h prior to transfection. Cells were transfected with 125 ng reporter vector, 25 ng pRL‐CMV (Promega), and either 125 ng wild‐type or mutant p53 (R175H), or Mock vector, using FuGENE6 transfection reagent. Thirty‐six hours after transfection, their luciferase activities were measured using the PicaGene Dual kit according to the manufacturer's recommendation (TOYO B‐Net, Tokyo, Japan).
Response of KIF2C/MCAK to genotixic stress. Cells were seeded at 24 h before treatment and were 60–70% confluent at the time of treatment. To examine the expression of KIF2C/MCAK in response to genotoxic stress, HCT116 (p53+/+ , p53−/– ) and HBC4 were treated continuously with 1 µg/mL adriamycin for 2 h and incubated at 37°C until the time of harvest.
Knockdown of p53 with siRNA oligonucleotides. To further investigate the effect of p53 on KIF2C/MCAK repression, we used siRNA oligonucleotides against p53 (sip53) (Sigma Aldrich Japan KK, Tokyo, Japan). The DNA sequences targeting each gene were as follows: sip53, 5′‐GACUCCAGUGGUAAUCUAC‐3′; siEGFP (control), 5′‐GCAGCACGACUUCUUCAAG‐3′; and siLuc (control), 5′‐GUGCGCUGCUGGUGCCAAC‐3′. HBC4 cells (1 × 106 cells/10‐cm dish) were transfected with each of the siRNA using lipofectamine (Invitrogen) in Opti‐MEM medium (Invitrogen) according to the instructions of the manufacturer. Twenty‐four hours after transfection, we carried out western blot analysis to examine the expression of KIF2C/MCAK and p53 as described in the western blot analysis sections.
Statistical analysis. Statistical significance was calculated using Student's t‐test, with Statview 5.0 software (SAS Institute, Cary, NC, USA). A difference of P < 0.05 was considered to be statistically significant.
Results
Overexpression of KIF2C/MCAK in breast cancers. We previously carried out genome‐wide gene‐expression profile analysis of 81 breast cancer cases using a cDNA microarray representing 27 648 genes or expressed sequence tag (EST).( 13 ) Among the genes upregulated in breast cancers, we identified KIF2C/MCAK, whose expression was increased significantly in the great majority of breast cancer cases. Subsequently, we confirmed overexpression of KIF2C/MCAK in 12 of 14 clinical breast cancer cases by semiquantitative RT‐PCR analysis (Fig. 1a). Northern blot analysis revealed significant upregulation of KIF2C/MCAK in all of the eight breast cancer cell lines examined (Fig. 1b), whereas its expression was hardly detectable in normal human tissues, except in the testis (Fig. 1c).
Figure 1.
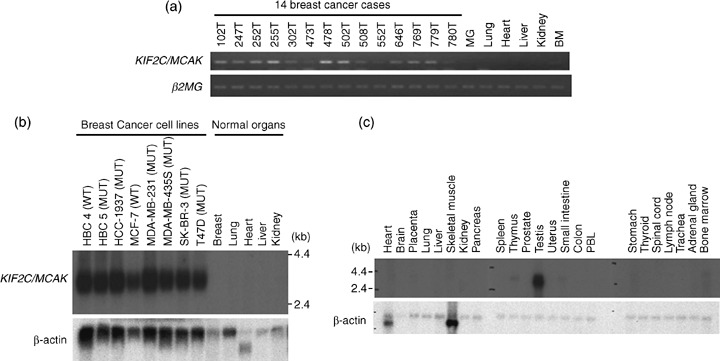
Kinesin family member 2C (KIF2C)/mitotic centromere‐associated kinesin (MCAK) overexpression in breast cancer cells. (a) Expression of KIF2C/MCAK in 14 microdissected breast cancer cases (102T, 247T, 252T, 255T, 302T, 473T, 478T, 502T, 508T, 552T, 646T, 769T, 779T, and 780T) and normal human tissues (mammary gland, lung, heart, liver, kidney, and bone marrow) by semiquantitative reverse transcription–polymerase chain reaction. β2‐Microgloblin (β2MG) served as a loading control. BM, bone marrow; MG, mammary gland. (b) Northern blot analysis of the KIF2C/MCAK transcript in eight breast cancer cell lines (HBC4, HBC5, HCC1937, MCF‐7, MDA‐MB‐231, MDA‐MB‐435S, SK‐BR‐3, and T47D) and normal human tissues, including breast, lung, heart, liver, and kidney. MUT, p53 mutant cell line; WT, p53 wild‐type cell line. A high‐intensity band was observed in breast cancer cell lines, but no signal was seen in normal tissues. β‐Actin was served as a loading control. (c) Northern blot analysis of the KIF2C/MCAK transcript in various normal human tissues. A band was observed specifically in the testis. PBL, peripheral blood leukocyte. β‐Actin served as a loading control.
To further investigate the expression level of endogenous KIF2C/MCAK protein in breast cancer cells, we developed a polyclonal antibody to KIF2C/MCAK. Western blot analysis using cell lysates from 10 breast cancer cell lines (BT‐20, HBC4, HBC5, HCC1937, MCF‐7, MDA‐MB‐231, MDA‐MB‐435S, SKBR3, T47D, and BT‐474) as well as HMEC detected a high level of KIF2C/MCAK expression in 9 of the 10 breast cancer cell lines, but no expression was detected in HMEC in agreement with the results of northern blot analysis (Fig. 2a). In addition, we noticed the presence of two bands that reacted with anti‐KIF2C/MCAK antibody when the proteins were separated better by longer electrophoresis. Therefore, to examine the possible modification of KIF2C/MCAK, we treated cellular extracts from MCF‐7 cells with lambda phosphatase and analyzed the molecular weight of KIF2C/MCAK protein by western blot analysis. Because the upper band disappeared when the cell extracts were incubated with phosphatase, we suspected that the KIF2C/MCAK protein was possibly phosphorylated in breast cancer cells (Fig. 2b).
Figure 2.
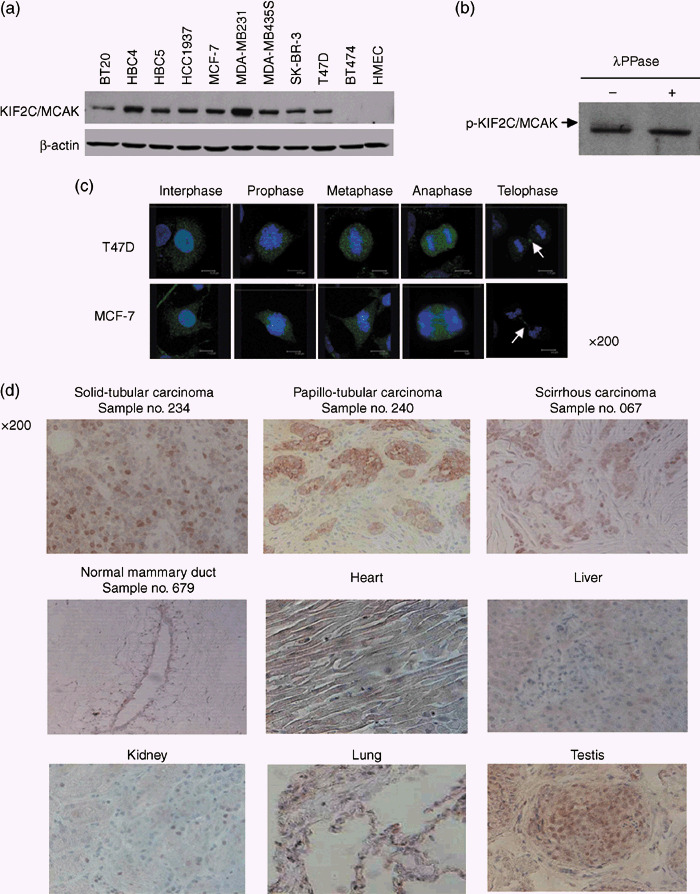
Expression of kinesin family member 2C (KIF2C)/mitotic centromere‐associated kinesin (MCAK) in breast cancer cell lines and tissue sections. (a) Expression of endogenous KIF2C/MCAK protein in 10 breast cancer cell lines (BT‐20, HBC4, HBC5, HCC1937, MCF‐7, MDA‐MB‐231, MDA‐MB‐435S, SK‐BR‐3, T47D, and BT474) in comparison with the human mammalian epithelial cell line by western blot analysis using affinity‐purified anti‐KIF2C/MCAK polyclonal antibody. (b) Phosphorylation of KIF2C/MCAK protein in T47D cells. The cell lysates were incubated with or without lambda phosphatase for 2 h at 30°C. A phosphorylated KIF2C/MCAK protein was observed as a slowly migrating band, indicated by an arrow. (c) Subcellular localization of endogenous KIF2C/MCAK protein in breast cancer cells at various cell‐cycle points. T47D (upper panels) and MCF‐7 (lower panels) cells were stained immunocytochemically using affinity‐purified anti‐KIF2C/MCAK polyclonal antibody (green), and 4′,6′‐diamidine‐2′‐phenylindole dihydrochloride (DAPI) (blue). The arrows indicate midbody in telophase cells. (d) Representative images of immunohistochemical staining of KIF2C/MCAK in breast cancer and normal tissue sections (normal mammary gland, heart, liver, kidney, lung, and testis). Endogenous KIF2C/MCAK protein was stained using anti‐KIF2C/MCAK polyclonal antibody. The expression was hardly detectable in normal breast tissue (sample no. 679) or any of the other normal human tissues (heart, liver, kidney, and lung), but cancer cells were stained strongly in all cancer tissue sections investigated; solid‐tubular (sample no. 234), papillotubular (sample no. 240), and scirrhous (sample no. 067) carcinomas as well as testis. Original magnification ×200.
To characterize the biological role of KIF2C/MCAK, we first examined the subcellular localization of endogenous KIF2C/MCAK protein in the breast cancer cell lines MCF‐7 and T47D by immunocytochemical staining using affinity‐purified anti‐KIF2C/MCAK polyclonal antibody. We found it in the nucleus and cytoplasm of both breast cancer cell lines at interphase (Fig. 2c). However, in prophase, it was localized on the chromosomes, especially the centomeres and kinetochores. Finally, this protein accumulated at the spindle midzone and midbody during anaphase to telophase (Fig. 2c).
Furthermore, we carried out immunohistochemical staining with anti‐KIF2C/MCAK antibody and observed its strong staining in the cytoplasm and nuclei of three different histological subtypes of breast cancer: solid‐tubular carcinoma (no. 234), papillotubular carcinoma (no. 240), and scirrhous carcinoma (no. 067) (Fig. 2d, upper panels). However, its expression was hardly detectable in normal mammary ductal cells (no. 679) (Fig. 2d, middle left panel). Furthermore, in concordance with the results of northern blot analysis, its expression was detected in the testis, whereas no expression was observed in the heart, liver, kidney, or lung (Fig. 2b, middle and lower panels).
Cell cycle‐dependent expression of KIF2C/MCAK. KIF2C/MCAK was suggested to be a mitotic protein,( 14 ) but little was known about the mechanism of how it functions during mitosis in breast cancer cells. Hence, we investigated its relation to cell‐cycle progression. We carried out FACS and western blot analyses using HBC5 cells after synchronization of the cell cycle by aphidicolin treatment. When a large proportion of the cells were considered to be in G2/M phase (at 8–12 h after release from cell‐cycle arrest) (Fig. 3a), western blot analysis detected an extra band of higher molecular weight KIF2C/MCAK, suggesting an important role for phosphorylated KIF2C/MCAK in mitosis (Fig. 3b).
Figure 3.
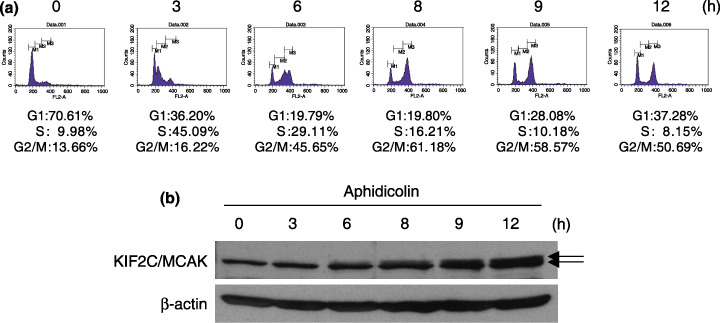
Cell cycle‐dependent expression of the kinesin family member 2C (KIF2C)/mitotic centromere‐associated kinesin (MCAK) protein. (a) Fluorescence‐activated cell sorting analysis of T47D cells collected every 3 h from 0 to 12 h after synchronization of the cell cycle. (b) Western blot analysis of KIF2C/MCAK during mitosis in T47D cells. KIF2C/MCAK was upregulated and phosphorylated at 6–12 h after release from cell‐cycle arrest when the majority of cells were at G2/M phase.
Growth‐inhibitory effects by siRNA to KIF2C/MCAK. To assess the growth‐promoting role of KIF2C/MCAK, we knocked down the expression of endogenous KIF2C/MACK in the breast cancer cell lines T47D and HBC5, in which KIF2C/MCAK was overexpressed, by means of the mammalian vector‐based RNA interference technique. Semiquantitative RT‐PCR analysis detected a significant knockdown effect on KIF2C/MCA expression in the cells transfected with psiU6BX‐KIF2C/MCAK‐specific siRNA (Si‐#2), compared with Mock, Si‐#1, or Si‐#3 (Fig. 4a). We then carried out MTT and colony‐formation assays (Fig. 4b,c), and found that introduction of the Si‐#2 construct remarkably suppressed the growth of both T47D and HBC5 cells (T47D: Si‐#2, P = 0.00011; HBC5: Si‐#2, P = 0.00105, Student's t‐test), concordantly with the knockdown effect. Furthermore, we confirmed a reduction in KIF2C/MCAK expression with KIF2C/MCAK‐siRNA (Si‐#2) at the protein level (Fig. 4d, third panel). To exclude the possibility of an off‐target effect by KIF2C/MCAK‐siRNA (Si‐#2), we generated two mismatched siRNA constructs, each of which contained three substituted nucleotides in Si‐#2 (si‐m#1 and si‐m#2), and found that these mismatched constructs had no suppressive effect on KIF2C/MCAK expression at the transcriptional or protein levels (Fig. 4d, first and third panels), as well as on the growth of HBC5 cells (Fig. 4e,f). Furthermore, we examined the morphological changes in HBC5 cells transfected with KIF2C/MCAK‐specific siRNA (Si‐#2) by microscopy and immunocytochemical staining analysis, and found a significant increase in multinucleated cells at 5 days after transfection (Fig. 4g,h). These findings indicate that the absence of KIF2C/MCAK caused the failure of cytokinesis, resulted in the formation of multinucleated cells, and then induced cell death. Together, we suggest that KIF2C/MCAK was likely to have a critical role in the cell growth of these breast cancer cells.
Figure 4.
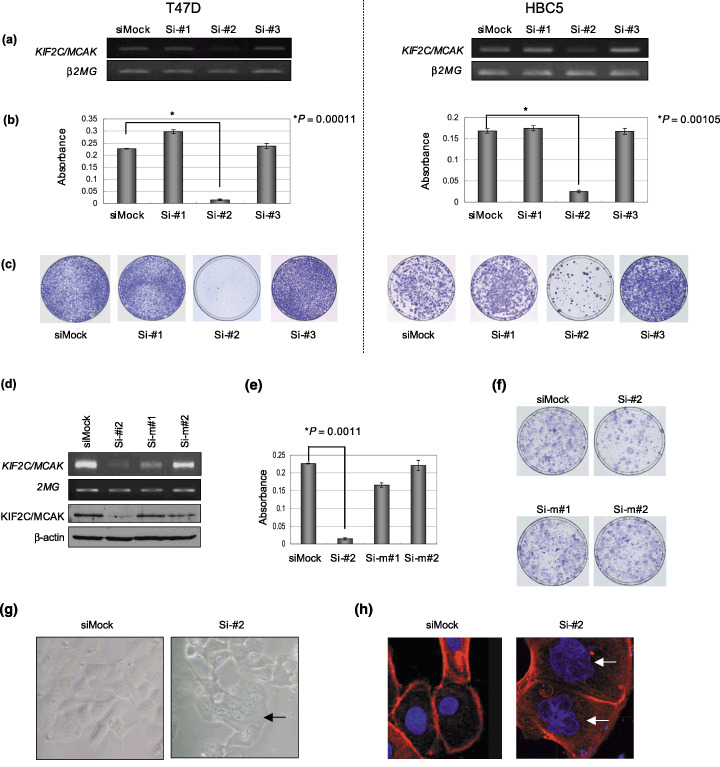
Growth‐inhibitory effects of kinesin family member 2C (KIF2C)/mitotic centromere‐associated kinesin (MCAK) small interfering RNA (siRNA) on breast cancer cells. (a) Semiquantitative reverse transcription–polymerase chain reaction (RT‐PCR) showing suppression of endogenous expression of KIF2C/MCAK by KIF2C/MCAK‐specific siRNA (Si‐#1, Si‐#2, and Si‐#3) in breast cancer cell lines, T47D (left panels), and HBC5 (right panels). Mock‐siRNA (siMock) was used as a control. β2‐Microgloblin (β2MG) was used as a quantitative control for RT‐PCR. (b) The 3‐(4,5‐dimethylthiaxol‐2‐yl)‐2,5‐diphenyltetraozolium bromide (MTT) assay demonstrated a decrease in the numbers of colonies by knockdown of KIF2C/MCAK in T47D (left panel; Si‐#2 P = 0.00011; unpaired t‐test) and HBC5 (right panel; Si‐#2 P = 0.00105; unpaired t‐test) cells. (c) The colony‐formation assay demonstrated a decrease in the numbers of colonies by knockdown of KIF2C/MCAK in T47D (left panels) and HBC5 (right panels) cells. (d) KIF2C/MCAK‐siRNA (Si‐#2) reduced the expression of KIF2C at the transcriptional and protein levels in HBC5 cells, whereas KIF2C/MCAK‐siRNA‐mismatch (Si‐m#1 and Si‐m#2) as well as siMock did not reduce KIF2C/MCAK expression by semiquantitative RT‐PCR (first and second panels) and western blot (third and forth panels) analyses (e) by MTT assay (Si‐#2, P = 0.0011; unpaired t‐test) and (f) by colony‐formation assay. (g) Morphological changes in HBC5 cells transfected with si‐KIF2C/MCAK observed by microscopy. siMock was used as control siRNA. The arrows indicate the midbodies of cells during cytokinesis (right panel). (h) Morphological changes in HBC5 cells transfected with si‐KIF2C/MCAK observed by immunocytochemical staining analysis with 4′,6′‐diamidine‐2′‐phenylindole dihydrochloride (DAPI) and phalloidin to distinguish the nucleus from the cytoplasm. siMock was used as control siRNA. The arrows indicate the multinucleation in KIF2C/MCAK‐depleted cells (right panel).
Identification of KIF2C/MCAK as a potential p53‐suppressive gene. We previously screened for genes that are regulated by p53 through comprehensive cDNA microarray analysis using mRNA isolated from p53‐mutant U373MG (p53‐null) cells infected with either adenovirus‐expressed (Ad)‐p53 or Ad‐lacZ.( 18 ) Through this analysis, we identified KIF2C/MCAK to be significantly downregulated by ectopic introduction of wild‐type p53. To verify whether KIF2C/MCAK is regulated by p53, we carried out semiquantitative RT‐PCR and western blot analyses, and confirmed that its expression was downregulated by the introduction of wild‐type p53 in a time‐dependent manner in U373MG cells at both the transcriptional and protein levels (Fig. 5a,b). In addition, we also confirmed its negative regulation by the introduction of wild‐type p53 into p53‐null H1299 lung carcinoma cell lines (data not shown). To further clarify the mechanism of its transcriptional repression by p53, we carried out a reporter assay with a heterologous luciferase gene fused to the DNA fragment corresponding to the KIF2C/MCAK promoter sequence (–598 to +53 to the transcriptional initiation site) into a pGL3‐basic vector (pKIF2C/MCAK‐Luc). Fig. 5c shows that the luciferase activity of pKIF2C/MCAK‐Luc (KIF2C/MCAK) was significantly suppressed by the introduction of wild‐type p53 vector into U373MG cells, but not suppressed by cotransfection with either mutant p53 or mock vector. Taken together, these findings support the p53‐mediated downregulation of KIF2C/MCAK transcription, although the direct or indirect effect of the p53 protein is unsolved.
Figure 5.
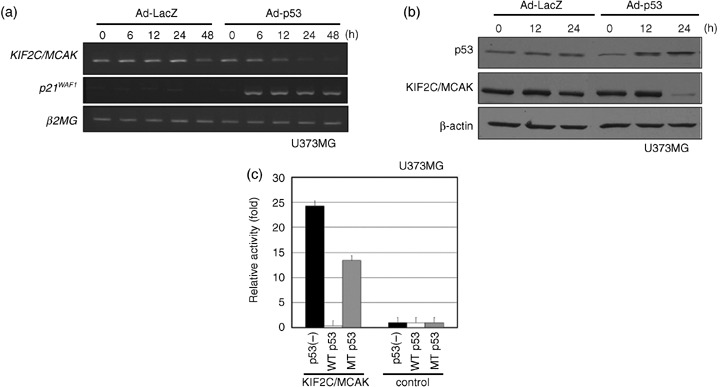
Repression of kinesin family member 2C (KIF2C)/mitotic centromere‐associated kinesin (MCAK) expression after infection with Ad‐p53. (a) Semiquantitative reverse transcription–polymerase chain reaction analysis for KIF2C/MCAK mRNA expression in U373MG cells at the indicated times (0–48 h) after infection with Ad‐p53 or Ad‐LacZ. β2‐Microgloblin (β2MG) served as a loading control. (b) Western blot analysis for KIF2C/MCAK protein in U373MG cells at the indicated times (0–24 h) after infection with Ad‐p53 or Ad‐LacZ. β‐Actin served as a loading control. (c) Luciferase assay. Luciferase activity is indicated relative to the activity of the pGL3‐promotor vector without the promoter region sequence.
p53‐dependent repression of KIF2C/MCAK following DNA damage. To further investigate the association between endogenous KIF2C/MCAK expression and p53 expression, HCT116 (p53 +/+) cells with wild‐type p53 or HCT116 (p53 −/–) cells without wild‐type p53 were exposed to adriamycin, and western blot analysis was carried out. Endogenous KIF2C/MCAK expression was significantly reduced in HCT116 (p53 +/+) cells at the protein level in a dose‐dependent manner (Fig. 6a), whereas its expression was not reduced in HCT116 (p53 −/–) cells, indicating that KIF2C/MCAK expression is regulated in a p53‐dependent manner in response to DNA damage.
Figure 6.
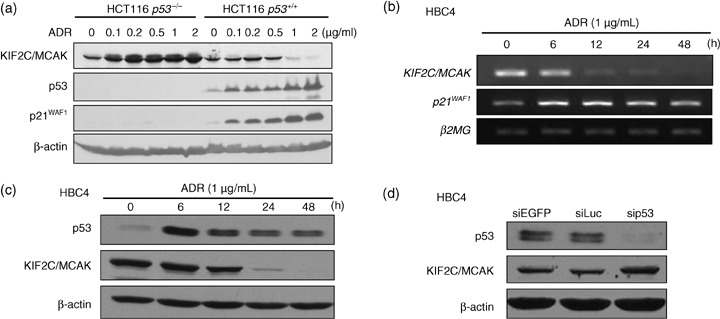
Downregulation of endogenous kinesin family member 2C (KIF2C)/mitotic centromere‐associated kinesin (MCAK) expression. (a) p53‐dependent repression of endogenous KIF2C/MCAK protein in p53 +/+ and p53 −/– ; HCT116 cells after DNA damage by treatment of various concentration of adriamycin (ADR; 0, 0.1, 0.2, 0.5, 1 and 2 µg/ml). β‐Actin served as a loading control for western blotting analysis. (b,c) p53‐dependent repression of endogenous KIF2C/MCAK transcription and protein in the breast cancer cell line HBC4 (wild‐type p53) after treatment with 1 µg/mL adriamycin. β2‐Microgloblin (β2MG) and β‐actin served as loading controls for reverse transcription–polymerase chain reaction and western blot analyses, respectively. (d) Increased expression of KIF2C/MCAK after p53 siRNA (sip53) treatment in HBC4 cells. The cells were transfected with sip53 and siEGFP or siLuc (negative controls). β‐Actin served as a loading control.
Furthermore, we examined the effect of p53 on the expression of endogenous KIF2C/MCAK in breast cancer cells. HBC4 breast cancer cells with wild‐type p53 were exposed to adriamycin. We found that the expression of KIF2C/MCAK was drastically suppressed in HBC4 cells at the transcriptional and protein levels at 12 and 24 h after the adriamycin treatment (Fig. 6b,c). In the cells, expression of p53 and p21WAF1 was induced as early as 6 h after the adriamycin treatment. To further confirm p53‐dependent KIF2C/MCAK repression, we applied siRNA treatment (sip53) and found that depletion of the p53 protein caused upregulation of the KIF2C/MCAK protein in HBC4 cells (Fig. 6d). Together, these finding suggest that downregulation of KIF2C/MCAK is specifically dependent on p53 protein accumulation in breast cancer cells.
Discussion
Significant advances in the development of molecular‐targeting drugs for cancer therapy have been achieved in the last two decades. However, the proportion of patients showing good response to presently available treatments is still very limited and a subset of patients suffers from severe adverse reactions without any benefit.( 2 ) Hence, the development of new anticancer agents that are highly specific to malignant cells and have a minimum risk of adverse reactions is needed. Through detailed expression‐profile analysis of clinical breast cancers, we identified KIF2C/MCAK to be significantly upregulated in the great majority of clinical breast cancer cases. In the present study, we reported the biological significance of KIF2C/MCAK in mammary carcinogenesis and the regulation of its expression through exogenous and endogenous p53 induction. Northern blot, western blot, and immunohistochemical analyses clearly demonstrated overexpression of KIF2C/MCAK in breast cancer cells, and its undetectable level of expression in normal human tissues except the testis, indicating that KIF2C/MCAK is a typical cancer–testis antigen. Furthermore, depletion of KIF2C/MCAK expression by means of siRNA treatment drastically suppressed the growth of breast cancer cells. However, because the introduction of KIF2C/MCAK into NIH3T3 cells could not enhance the growth of the cells (data not shown), we assume that KIF2C/MCAK is essential for the survival of breast cancer cells, but KIF2C/MCAK alone may not be sufficient for the transforming activity.
KIF2C/MCAK, a member of the kinesin‐13 family of kinesin‐related proteins, is a microtubule depolymerase that is necessary to ensure proper kinetocore–microtubule attachment during spindle formation.( 14 , 15 , 16 , 17 , 18 ) We previously demonstrated that KIF2C/MCAK plays an important role in breast carcinogenesis through its interaction with PRC1.( 4 ) We further demonstrated here that knockdown of KIF2C/MCAK expression with KIF2C/MCAK‐specific siRNA led to a failure to complete cytokinesis, as shown in Fig. 4g. Furthermore, we here showed that KIF2C/MCAK expression was downregulated directly or indirectly by p53. In addition, we examined the expression levels of KIF2C/MCAK in breast cancer cell lines, shown in Fig. 1b, by real‐time PCR, and found that its expression is relatively higher in p53 mutant cell lines compared with p53 wild‐type cell lines (data not shown), although its expression and p53 mutation were not completely concordant. The p53 tumor suppressor gene was shown to be mutated or inactivated in the majority of human cancers, including breast cancers.( 21 ) Because our findings demonstrated that KIF2C/MCAK was negatively regulated by p53 and significantly upregulated in breast cancer cases, we suggest that inactivation of p53 might be, in part, responsible for the enhanced expression of KIF2C/MCAK.
In conclusion, our findings clearly suggest that KIF2C/MCAK is overexpressed in breast cancer cells, and is likely to play a significant role in cytokinesis in these cells. Furthermore, we found that downregulation of KIF2C/MCAK by treatment with siRNA significantly suppresses the growth of breast cancer cells, indicating its crucial role in the growth of breast cancer cells. Moreover, we showed that KIF2C/MCAK is regulated directly or indirectly by p53. Taken together, our data should provide a new insight to a better understanding of mammary carcinogenesis, and that KIF2C/MCAK is a promising molecular target for the development of novel anticancer drugs. Furthermore, it is notable that our cDNA microarray data identified upregulation of KIF2C/MCAK in many types of clinical cancers, including bladder cancer, cholangiocarcinoma, esophagus, lung cancers, pancreatic cancer, and soft tissue tumors, as well as breast cancers (data not shown). These results show that this gene should serve as a valuable target for the development of anticancer agents for a wide range of human cancers.
Acknowledgments
We gratefully thank Dr Akira Togashi for western blot analysis in the breast cancer cell lines, Dr Mutsuo Furihata for evaluation of immunohistochemical staining analysis as a pathologist, and Ms Kie Naito, Ms Kyoko Kijima, Ms Akiko Konuma, and Ms Yoshiko Fujisawa for their technical assistance.
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Navolanic PM, McCubrey JA. Pharmacological breast cancer therapy (review). Int J Oncol 2005; 27: 1341–4. [PubMed] [Google Scholar]
- 3. Park JH, Lin ML, Nishidate T, Nakamura Y, Katagiri T. PDZ‐binding kinase/T‐LAK cell‐originated protein kinase, a putative cancer/testis antigen having an oncogenic activity in breast cancer. Cancer Res 2006; 66: 9186–95. [DOI] [PubMed] [Google Scholar]
- 4. Shimo A, Nishidate T, Ohta T, Fukuda M, Nakamura Y, Katagiri T. Elevated expression of PRC1, protein regulator of cytokinesis 1, involved in the growth of breast cancer cells. Cancer Sci 2007; 98: 174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin ML, Park JH, Nishidate T, Nakamura Y, Katagiri T. MELK, maternal embryonic leucine zipper kinase, involved in mammary carcinogenesis through interaction with Bcl‐G, a pro‐apoptotic member of Bcl‐2 family. Breast Cancer Res 2007; 9: R17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kanehira M, Katagiri T, Shimo A, Takata R et al . Oncogenic role of MPHOSPH1, a cancer–testis antigen specific to human bladder cancer. Cancer Res 2007; 67: 3276–85. [DOI] [PubMed] [Google Scholar]
- 7. Kanehira M, Harada Y, Takata R, Shuin T et al . Involvement of upregulation of DEPDC1 (DEP domain containing 1) in bladder carcinogenesis. Oncogene 2007; 26: 6448–55. [DOI] [PubMed] [Google Scholar]
- 8. Nagayama S, Iiizumi M, Katagiri T, Toguchida J, Nakamura Y. Identification of PDZK4, a novel human gene with PDZ domains, that is upregulated in synovial sarcomas. Oncogene 2004; 23: 5551–7. [DOI] [PubMed] [Google Scholar]
- 9. Nagayama S, Fukukawa C, Katagiri T et al . Therapeutic potential of antibodies against FZD 10, a cell‐surface protein, for synovial sarcomas. Oncogene 2005; 24: 6201–12. [DOI] [PubMed] [Google Scholar]
- 10. Okada K, Hirota E, Mizutani Y et al . Oncogenic role of NALP7 in testicular seminomas. Cancer Sci 2004; 95: 949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Togashi A, Katagiri T, Ashida S et al . Hypoxia‐inducible protein 2 (HIG2), a novel diagnostic marker for renal cell carcinoma and potential target for molecular therapy. Cancer Res 2005; 65: 4817–26. [DOI] [PubMed] [Google Scholar]
- 12. Hirota E, Yan L, Tsunoda T et al . Genome‐wide gene expression profiles of clear cell renal cell carcinoma: Identification of molecular targets for treatment of renal cell carcinoma. Int J Oncol 2006; 29: 799–827. [PubMed] [Google Scholar]
- 13. Nishidate T, Katagiri T, Lin ML et al . Genome‐wide gene‐expression profiles of breast‐cancer cells purified with laser microbeam microdissection: identification of genes associated with progression and metastasis. Int J Oncol 2004; 25: 797–819. [PubMed] [Google Scholar]
- 14. Wordeman L, Mitchison TJ. Identification and partial characterization of mitotic centromere‐associated kinesin, a kinesin‐related protein that associates with centromeres during mitosis. J Cell Biol 1995; 128: 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vale RD, Fletterick RJ. The design plan of kinesin motors. Annu Rev Cell Dev Biol 1997; 13: 745–77. [DOI] [PubMed] [Google Scholar]
- 16. Lawrence CJ, Dawe RK, Christie KR et al . A standardized kinesin nomenclature. J Cell Biol 2004; 167: 19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrews PD, Ovechkina Y, Morrice N et al . Aurora B regulates MCAK at the mitotic centromere. Dev Cell 2004; 6: 253–68. [DOI] [PubMed] [Google Scholar]
- 18. Lan W, Zhang X, Kline‐Smith SL et al . Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol 2004; 14: 273–86. [DOI] [PubMed] [Google Scholar]
- 19. Shimokawa T, Furukawa Y, Sakai M et al . Involvement of the FGF18 gene in colorectal carcinogenesis, as a novel downstream target of the β‐catenin/T‐cell factor complex. Cancer Res 2003; 63: 6116–20. [PubMed] [Google Scholar]
- 20. Mori T, Anazawa Y, Matsui K, Fukuda S, Nakamura Y, Arakawa H. Cyclin K as a direct transcriptional target of the p53 tumor suppressor. Neoplasia 2002; 4: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakamura Y. Isolation of p53‐target genes and their functional analysis. Cancer Sci 2004; 95: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]