

Abstract
Surgical resection is the effective treatment modality for hepatocellular carcinoma (HCC); however, rapid recurrence of the tumors are frequently observed even after apparently curative resection. The recurrence and prognostic assessment of patients with HCC after resection is an important clinical issue. We recently reported that aberrant expression of Aurora B is observed in primary HCC, and that it can be a predictive factor for HCC recurrence exceeding Milan criteria after curative hepatectomy. In this study we investigated the expression of the newly observed Aurora B splicing variant forms in HCC, and their roles in hepatocarcinogenisis. The expression of Aurora B and splicing variant forms were screened in 125 HCC patients (94 chronic hepatitis with cirrhosis background liver specimens), 18 metastatic liver cancer patients and 16 normal liver specimens by cDNA microarray, reverse transcription – polymerase chain reaction (RT–PCR) and Real time quentitative Reverse Transcription PCR (qRT‐PCR). The results showed that expression of Aurora B splicing variant 2 (AURKB‐Sv2) variant form was absent in normal liver and was higher in metastatic liver cancer than HCC. This aberrant expression was associated with the advanced stages of HCC (P < 0.01), correlated with a poor outcome (P = 0.008) and short disease‐free period (P = 0.018). Furthermore, AURKB‐Sv2 variant form is associated with a higher level of serum α‐fetoprotein, protein induced by vitamin K absence or antagonist‐II (PIVKAII), tumor capsular invasion, multiple tumor formation and at an age younger than those with other variant forms (P < 0.05). The results thus suggest that AURKB‐Sv2 variant form is more significantly associated with the advanced stages of HCC than others and is a marker of poor prognosis. Founded in the tumor capsular invasion and multiple tumor regions, suggests that this might play a role in enhancing multiple malignant tumor formation and recurrence of HCC in hepatocarcinogenesis. This is the first study to report clinicopathological significance of aberrant expression of AURKB‐Sv2 variant form in hepatocellular carcinoma. (Cancer Sci 2009; 100: 472–480)
Abbreviation:
- HCC
- AURKB‐WT
Aurora B kinase
Aurora B kinase splicing variants
cDNA microarray
Hepatocellular carcinoma (HCC) is one of the major causes of cancer death in the world.( 1 , 2 ) As a result of advances in the diagnosis and disease management of HCC, significant improvements in overall and disease‐free survival rates after resection of HCC have been achieved within the past decade.( 3 , 4 ) Surgical resection is the effective treatment modality for HCC; however, rapid recurrence of the tumors are observed frequently even after apparently curative resection.( 5 , 6 ) Even when curative resection is performed, a considerable number of patients develop intrahepatic and/or extrahepatic recurrence postoperatively.( 7 , 8 , 9 ) The recurrence and prognostic assessment of patients with HCC after resection is an important clinical issue. Hepatic recurrence has been classified as intrahepatic metastasis and multicentric recurrence and the long‐term outcomes are affected mainly by metastatic recurrence.( 10 , 11 )
The development of DNA microarray technology has enabled us to analyze genome‐wide profiles of gene expression specific to malignant tumors. Using this technology, we investigated the messenger RNA (mRNA) expression patterns in primary HCC, and clarified that the genes associated with cell cycle, especially Aurora kinase B (AURKB), were up‐regulated significantly in hepatocellular carcinoma in relation to the invasion of the portal vein and/or hepatic vein. We recently reported that Aurora B aberrant expression in primary HCC can be the predictive factor of HCC recurrence exceeding Milan criteria after curative hepatectomy.( 12 )
AURKB is known as an aberrantly expressed gene in many cancer and tumor cell lines.( 13 , 14 , 15 , 16 ) Aurora B exists in a complex with at least two other proteins, inner centromere protein (INCENP) and Survivin. Aurora B, INCENP, and Survivin are so‐called chromosomal passenger proteins and they associate with inner centromere regions during prophase, but subsequently relocalize to the midzone of the central spindle and concentrate at the midbody.( 17 , 18 , 19 , 20 ) Aurora B is localized on the controversy from prophase through the metaphase‐anaphase transition. Another chromosome passenger protein is INCENP, which tightly associates with Aurora B, probably regulating its activity. It is overexpressed throughout the cell cycle in cancer cells.( 21 , 22 , 23 ) Overexpression of Aurora B produces multinuclearity and induces aggressive metastasis, suggesting that overexpressed Aurora B has multiple functions in cancer development. Honda et al.( 24 ) mentioned about smaller immunoreactive protein of Aurora B represents either a cleavage product or a splicing variant of Aurora B. Sistayanarain et al.( 13 ) reported that Aurora B and its two variant forms are expressed in HCC (by studies with some limited samples by reverse transcription – polymerase chain reaction [RT‐PCR]). However, the roles of expression of Aurora B and variant forms in HCC and other adjacent tissues are unknown. In this study we aimed to investigate the expression of the newly observed Aurora B splicing variant forms in HCC, adjacent tissue and their roles in hepatocarcinogenisis.
Materials and Methods
Cancer cell lines, patients and tissue samples. The human HCC cell lines, PLC/PRF/5, SK‐Hep1, HepB3, were obtained from the American Type Culture Collection (Manassas, VA, USA), JHH4, JHH5, HLE, HepG2, Huh1, Huh6, Huh7 were obtained from the Human Science Research Resources Bank (Osaka Japan). All cell lines were maintained in Dulbecco's minimum essential medium (DMEM: Sigma, St Louis, MO, USA) containing nonessential amino acid (Invitrogen, Carlsbad, CA, USA) and 10% heat inactivated fetal bovine serum (FBS, JRH Bioscience, Lenexa, KS, USA) and was grown at 37°C in 5% CO2. Primary HCC and metastatic liver cancer tissues were obtained with informed consent from 125 patients and 18 patients, respectively, by surgical resection in the Department of Hepato‐Biliary‐Pancreatic Surgery at Tokyo Medical and Dental University Hospital between November 2005 and May 2008. This research project was approved by the local ethical committee and all samples were obtained with the patient's informed consent. A part of the resected sample was fixed in formalin and embedded in paraffin for histological diagnosis and all tissues were snap frozen in liquid nitrogen and then stored at –80°C for RNA analysis. Sections were obtained from each frozen sample before mRNA extraction. One section was stained with hematoxylin and eosin to verify the presence of viable tumor. Histological diagnosis was made when two pathologists specializing in liver disease reached the same conclusion. The patients consisted of 93 (74.4%) males and 32 (25.3%) females, 40–85 years old (mean 66.3). Other clinicopathological features are shown in Table 1.
Table 1.
Clinicopathological data for 125 primary HCC cases
| Clinicopathologic factor | Primiry HCC n = 125 |
|---|---|
| Age(y, mean ± SD) | 66.3 ± 0.9 |
| Gender (male:female) | 93:32 |
| Hepatitis virus (HBV:HCV:non‐B‐non‐C) | 26:66:33 |
| AST (IU/L, mean ± SD) | 61.4 ± 11.1 |
| ALT (IU/L, mean ± SD) | 54.8 ± 7.4 |
| Plt (×109L, mean ± SD) | 14.7 ± 0.7 |
| ICG‐R15(%, mean ± SD) | 19.7 ± 1.1 |
| PT% (mean ± SD) | 83.8 ± 1.1 |
| Total bilirubin (mg/dL, mean ± SD) | 0.9 ± 0.04 |
| Alb (g/dL, mean ± SD) | 3.9 ± 0.04 |
| AFP (>100 ng/mL) | 46:79 |
| AFP (>400 ng/mL) | 29:96 |
| PIVKA‐II (>40mAU/mL) | 83:42 |
| PIVKA‐II (>100mAU/mL) | 63:62 |
| Number of tumors (A:B:C:D) | 91:26:2:6 |
| Tumor size (A:B:C) | 14:72:39 |
| Tumor size (>5.1 cm) | 39:86 |
| Histological differentiation(well:mod:poor) | 22:53:50 |
| Histological structure(trab:pseud:comp:scir) | 95:8:21:1 |
| Growth pattern(expansive:invasive) | 108:17 |
| Capsular formation (+:) | 100:25 |
| Capsular invasion (+:) | 81:44 |
| Portal vein invasion (+:) | 56:69 |
| Hepatic vein invasion (+:) | 16:109 |
| Stages (I:II:III:IV) | 6:58:45:16 |
NOTE: AST: aspartate amino transferase, ALT: alanine aminotransferase, PLT: platelet, ICG‐R15: indocyanine green retention rate at 15 min, PT%: prothrombin time, Alb: albumin, AFP: α‐fetoprotein, PIVKAII, protein induced by vitamin K absence or antagonists II. Number of tumors (A:B:C:D); A = 1; B = 2; C = 3; D = 4, tumor size (A: B:C); A: = 2.0; B: = 0.2.1, = 5.0; C: = 5.1, trab: trabecular type, pseud: pseundoglandular type, comp: compact type, scir: srirrhous type, positive: +, negative: –.
RNA isolation, complementary RNA (cRNA) preparation and microarray analysis. One hundred and twenty‐five primary HCC and correlated background specimens were obtained from surgically resected materials. Total RNA was extracted from tissue specimens and human HCC cell lines using RNeasy Mini kit (Qiagen, Hilden, Germany) and treated with RNase‐free DNase according to the manufacturer's instructions. Integrity of obtained RNA was assessed using Agilent Bioanalyzer RNA 6000 Nano Assay (Agilent Technologies, Palo Alto, CA, USA). All samples had RNA Integrity Number (RIN) > 5.0. Using 2 µg of total tissue RNA specimens, cRNA was prepared using one‐cycle target labeling and control reagents by Affymetrix, P/N 900493 (Affymetrix, Santa Clara, CA, USA). Hybridization and signal detection of HG‐U133 plus 2.0 arrays (Affymetrix) were performed following the manufacturer's instruction.
Western blot analysis. The cells were maintained in continuous monolayer cultures at 37°C and 5% CO2, expanded up to 70–80% confluence and then employed for the experiments, as described below. For Western blotting, cellular proteins were solubilized in 2× gel sample buffer, boiled for 5 min, and resolved by 10% sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS‐PAGE). Proteins were then transferred to nitrocellulose membranes, which were blocked in TBST (50 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween‐20) plus 5% non‐fat dried milk and incubated with the primary antibodies antihuman Aurora B (1 : 1000; Abcam, Cambridge, UK, catalog no. ab2254) in TBST plus 1% non‐fat dried milk. Secondary antibodies were examined using the ECL Western Blotting Detection System (GE Healthcare, Buckinghamshire, UK). The expression ratio of Aurora B to the control was examined using Multi‐Gage software (FUJIFILM, Tokyo, Japan).
TaqMan® MGB probe real‐time PCR. Two micrograms of tissue RNA were reverse transcribed to cDNA with High‐Capacity cDNA Reverse Transcription System (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using the TaqMan Fast Universal PCR master mix 2x (Applied Biosystems), TaqMan Gene Expression Assays AURKB (Hs00177782_m1, Context Sequence GCCGACAGACGGCTCCATCTGGCCT) for AURKB. Triplicate 20 µL RT‐PCR reactions for each sample contained 10 µL of AB TaqMan Fast Universal PCR Master Mix, 1 µL of the relevant 20× assay, 1 µL of target cDNA and dH2O under following conditions: one cycle of 20 sec at 95°C, 40 cycles of 3 sec at 95°C, 30 sec at 60°C. Data was analyzed using the comparative relative quantification method and samples were normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH).
Quantitative real time RT‐PCR. Two micrograms of cells and tissues RNA were reverse‐transcribed to cDNA with High‐Capacity cDNA Reverse Transcription System (Applied Biosytems). Quantitative PCR was performed using the SYBR Green Supermix (Bio‐Rad Laboratories, Hercules, CA, USA) on the AB 7500 Fast Real Time PCR System (Applied Biosystems) with 30‐mer sense (5′‐GAGAGTGCATCACACAACGAGACCTATCGC‐3′) and antisense (5′‐AGAAAACAGATAAGGGAACAGTTAGGGATC‐3′) for AURKB‐WT (Aurora B‐wild type) primers, with 30‐mer sense (5′‐CGGCACTTCACAATTGATGACTTTGAGATT‐3′) and antisense (5′‐TTATCAACATCTCTGCGTCCTACAACCCTA‐3′) for AURKB‐Sv1 (Aurora B‐splicing variant 1) primers, with 20‐mer sense (5′‐ATCTTAACCAGGCGGCACTT‐3′) and antisense (5′‐ACTCCTCCATGATTGCAGGT‐3′) for AURKB‐Sv2 (Aurora B‐splicing variant 2) primers under the following conditions: 2 min at 50°C, 10 min at 95°C one cycle; 40 cycles of 15 s at 95°C, 60 s at 60°C. Immediately after the amplification, melt curve protocols were performed to ensure primer‐dimers and other non‐specific products had been minimized or eliminated. GAPDH transcript was tested as an endogenous reference to calculate the relative expression levels of target genes according to Applied Biosystems instructions. The PCR reactions were separated by gel electrophoresis and the DNA bands were visualized under ultraviolet light for photographing.
Normalization and statistical analysis of microarray data. Obtained 80 microarray datasets were normalized using the robust multiarray average (RMA) method (R 2.4.1 statistical software together with BioConductor package).( 25 ) Estimated gene expression levels were log2‐transformed, and 62 control probe sets were removed for further analysis. Out of 80 patients, 23 showed expression of AURKB‐SV2. To identify genes associated with the expression of AURKB‐SV2, Wilcoxon rank‐sum test was performed to estimate the significance of gene expression differences between AURKB‐SV2 (+) and (–) groups for each 54 613 probe sets. Obtained P‐values from the multiple hypothetical testing were adjusted by the false discovery rate (FDR), and probe sets with P < 0.0005 (FDR < 0.266) were considered for further analysis. Hierarchical clustering with the selected probe sets was performed on R software using Pearson's correlation coefficient as a similarity index and complete linkage method for agglomeration. For visualization, the expression levels were standardized by z‐scores (mean = 0 and variance = 1) for each probe set.
Statistical analysis of qRT‐PCR. A quantitative analysis of specific mRNA expression was performed by qRT‐PCR using the Applied Biosystems 7500 Fast Real‐Time PCR System. CT values were calculated using the 7500 SDS software. For each sample, expressions of AURKB gene and splicing variant forms were normalized with expression of control gene and fold difference between tumor and normal tissue calculated using the ??CT method (Applied Biosystems User Bulletin #2, 1997).
Statistical analysis of clinicopathogical correlation. Student's t‐test was used to analyze the differences in age of patients. Fisher's exact test was used to compare the categorical data between groups. Wilcoxon rank sum test was used to analyze the non‐categorical data. The overall survival curve and disease‐free survival rate was calculated by the Kaplan‐Meier method and rates are reported with 95% confidence intervals. Differences were tested for significance using the log‐rank test. The overall survival rate was measured from the date of resection until the date when the recurrence of HCC was detected or when the patient died. P < 0.05 was deemed to be statistically significant.
Results
AURKB overexpressed in tumor than adjacent‐non‐tumor and vascular invasion cases than‐non‐vascular invasion cases in hepatocellular carcinoma. From Western blotting analysis, cultured human HCC cell lines were found to express Aurora B protein (41 kDa) Fig. 1(a). To assess the validity of our hybridization results, we examined the expressed gene using TaqMan® MGB probe Gene Expression Assays for AURKB, quantitative PCR was performed using the TaqMan Fast Universal PCR master mix 2× (Applied Biosystems). Strong correlation between PCR expression data and microarray values was found (Fig. 1b,c). All changes determined by RT‐PCR were statistically significant (P < 0.01) and consistent with the direction of change reported by the microarray analysis.
Figure 1.
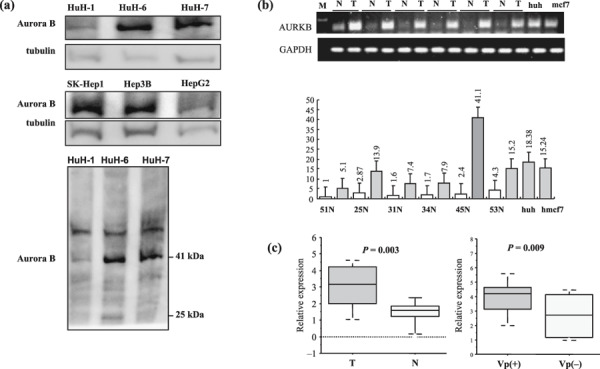
Expression of Aurora kinase B (AURKB) in human hepatocellular carcinoma (HCC) cell lines and HCC patients. (a) Protein levels of Aurora B and alpha‐tubulin (control) in HCC cell lines were examined using standard Western blot analysis on 10% sodium dodecyl sulfate – polyacrylamide gel electrophoresis. All cell lines indicated Aurora B (41 kDa). (b) TaqMan® MGB probe Gene Expression Assays for AURKB, Quantitative polymerase change reaction (PCR) was performed using the TaqMan Fast Universal PCR master mix 2× and normalized for sample variation by glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). (c) All changes determined by reverse transcription – PCR were statistically significant and consistent with the direction of change reported by the microarray analysis. The relative expression P‐values were tested for significance using Wilcoxon rank sum test. (N; non‐tumor, T; tumor, VP (+); portal vein positive, VP (–); portal vein negative).
Detection of AURKB‐WT and splicing variant forms in human HCC cell lines, HCC cases, metastatic liver cancer cases and normal liver specimens. In order to analyze the expression of AURKB and alternative splicing variant forms in human HCC cell lines, cancer and the related adjacent tissue were screened by RT‐PCR and qRT‐PCR. As with eight kinds of cell lines, 125 HCC cases, of which we found 94 chronic hepatitis and cirrhosis background liver specimens, 18 metastatic liver cancer cases and 16 normal liver specimens were screened by RT‐PCR and qRT‐PCR. Schematic illustration of the AURKB and alternative splicing variants are shown in Fig. 2(a). qRT‐PCR using the Applied Biosystems 7500 Fast Real‐Time PCR System is shown in Fig. 2(b,d). We found that the AURKB‐WT, AURKB‐Sv1 and AURKB‐Sv2 forms of mRNA were expressed in all cell lines and the relative expression ratios are shown in Fig. 2(c). AURKB‐WT mRNA was aberrantly expressed in all primary HCC tumor specimens (125 of 125), chronic hepatitis and cirrhosis background liver specimens (94 of 94), metastasis liver cancer cases (18 of 18) and normal liver specimens (16 of 16). AURKB‐Sv1 variant form was also up‐regulated at 100%, 97.8%, 88.9% and 25%, respectively. However, only 33% of primary HCC cases (42 of 125), 15% of chronic hepatitis and cirrhosis background liver specimens (15 of 94), 61% of metastasis liver cancer cases (11 of 18), 0% of normal liver specimens (0 of 16) had up‐regulated AURKB‐Sv2 variant form. The results are shown in Table 2 and the relative expression ratio is shown in Fig. 2(e).
Figure 2.
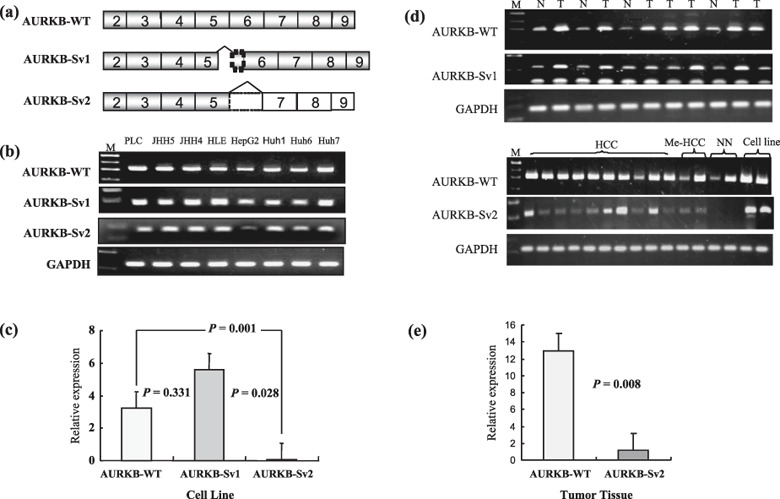
Expression of Aurora kinase B (AURKB) and splicing variant forms in human hepatocellular carcinoma (HCC) cell lines and in human cancer tissue specimens. (a) Schematic illustration of the AURKB alternative splicing coding messenger RNA (mRNA). The boxes indicate the exons numbered on Ensemble (access ENST380101). The gray boxes are coding regions and the white boxes are not translated due to frame shift. The box with dotted line is 47 bp of intron 5–6 retained in the variant AURKB splicing variant 1 (AURKB‐Sv1) and AURKB‐Sv2 in which the entire sequence is missing from exon 6. (b) Expression of AURKB and alternative splicing variant forms in human HCC cell lines. Relative AURKB‐WT, AURKB‐Sv1, AURKB‐Sv2 mRNA levels cell lines were evaluated by quantitative reverse transcription – polymerase chain reaction (qRT‐PCR). (c) Relative expression ratio for all cell lines expressed AURKB and variant forms. P‐values were evaluated by Student's t‐test. (d) Expression of AURKB and alternative splicing variant forms in HCC, related adjacent tissue, metastasis liver cancer cases and normal liver specimens. qRT‐PCR using the Applied Biosystems 7500 Fast real‐time PCR System. CT values were calculated using the 7500 SDS software. The relative expression P‐values were tested for significance using Student's t‐test.
Table 2.
Expression of Aurora kinase B (AURKB) and variant forms in HCC, adjacent tissue, metastatic cancer and normal liver specimens
| Specimens | AURKB‐WT | AURKB‐Sv1 | AURKB‐Sv2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Positive | Negative | % | Positive | Negative | % | Positive | Negative | % | |
| HCC | 125 | 0 | 100% | 125 | 0 | 100% | 42 | 83 | 33.6% |
| CH + LC | 94 | 0 | 100% | 92 | 2 | 97.8% | 15 | 79 | 15.9% |
| Me‐HCC | 18 | 0 | 100% | 16 | 2 | 88.9% | 11 | 7 | 61.1% |
| Normal liver | 16 | 0 | 100% | 4 | 12 | 25% | 0 | 16 | 0.0% |
NOTE: HCC: hepatocellular carcinoma, CH: chironic hepatitis, LC: liver cirrhosis; Me‐HCC: metastasis liver cancer, WT: wild type; Sv1: splicing variant 1; Sv2: splicing variant 2.
Expression of AURKB‐Sv2 variant form and clinicopathologic features. We detected AURKB‐Sv2 variant form in 33% of primary hepatocellular cases (42 of 125). AURKB‐WT and AURKB‐Sv1 were also detected in all positive cases. We studied the relationship between the expression of these mRNA (regardless of their variant forms) and clinicopathologic factors of HCC. As shown in Table 3 AURKB‐Sv2 was associated with the advanced stages of HCC more significantly than AURKB‐WT form (P = 0.008). The expression of AURKB‐Sv2 variant form was furthermore associated with a higher level of serum α‐fetoprotein (AFP) (P = 0.025), protein induced by vitamin K absence or antagonists‐II (PIVKAII) (P = 0.037), tumor capsular invasion (P = 0.029), histological differentiation (P = 0.044) and multiple tumor formation (P = 0.017). The AURKB‐Sv2 variant patients were of an age younger than those with other variant forms (P = 0.030). However, the expression of AURKB‐Sv2 variant did not show association with hepatitis B and hepatitis C virus infection, the histological structure patterns, tumor capsular formation, growth pattern, vascular invasion or other factors.
Table 3.
Expression of Aurora kinase B splicing variant 2 (AURKB‐Sv2) variant and clinicopathological findings in hepatocellular carcinoma
| Clinicopathologic factor | AURKB (Wt/Sv2) | ||
|---|---|---|---|
| +/+ (n = 42) | +/– (n = 83) | P‐value | |
| Age(y, mean ± SD) | 63.6 ± 1.6 | 67.7 ± 1.1 | 0.030* |
| Gender (Male:Female) | 34:8 | 59:24 | 0.282 |
| Hepatitis virus positive (HBV:HCV) | 8:24 | 18:42 | 0.808 |
| AFP(>100 ng/mL) | 23:19 | 23:60 | 0.006* |
| AFP (>400 ng/mL) | 15:27 | 14:69 | 0.025* |
| PIVKA‐II(>40mAU/mL) | 31:11 | 52:31 | 0.235 |
| PIVKA‐II(>100mAU/mL) | 27:15 | 36:47 | 0.037* |
| Solitary or multiple | 24:18 | 67:16 | 0.019* |
| Tumor size (>5.1 cm) | 10:32 | 29:54 | 0.227 |
| Histological differentiation(well:mod:poor) | 3:23:16 | 19:30:34 | 0.044* |
| Histological structure(trab:pseud:comp:scir) | 32:2:8:0 | 63:6:13:1 | 0.811 |
| Growth pattern(expansive:invasive) | 36:6 | 72:11 | 0.784 |
| Capsular formation (+:) | 32:10 | 68:15 | 0.453 |
| Capsular invasion (+:) | 33:9 | 48:35 | 0.029* |
| Portal vein invasion (+:) | 19:23 | 37:46 | 0.944 |
| Hepatic vein invasion (+:) | 9:33 | 7:76 | 0.050 |
| Stages (I + II:III + IV) | 14:28 | 50:33 | 0.008* |
NOTE: AFP: α‐fetoprotein, PIVKAII, protein induced by vitamin K absence or antagonists II.
trab: trabecular type, pseud: pseundoglandular type, comp: compact type, scir: srirrhous type, positive: +, negative: –, *Statistically significant.
Expression of AURKB‐Sv2 variant form and patient survival. We were able to follow the postoperative course of 110 patients. The follow‐up period until death or the end point of this study was 4–959 days (mean 276 days, median 206 days). Eighty patients survived without recurrence of HCC within the follow‐up period, and the mean survival (median, 193 days) was 276 days. Fifty‐five patients lived longer than 1 year after the operation. The mean survival period was 391 days (median; 389 days). Figure 3(a) shows the overall survival rate (110 patients) and Fig. 3(b) shows disease‐free survival rate (80 patients) of AURKB‐Sv2 variant +/– HCC patients. There was a significant difference in the overall survival rate (P = 0.008) and disease‐free survival rate (P = 0.018) between AURKB‐Sv2 positive patients and negative patients.
Figure 3.

Kaplan‐Meier method analysis survival curves for primary hepatocellular carcinoma (HCC) with Aurora kinase B splicing variant 2 (AURKB‐Sv2) (+/–) cases. Differences were tested for significance using the log‐rank test. All of the microarray‐examined patients with HCC classified into AURKB‐Sv2 (+) group (n = 37, n = 29) and AURKB‐Sv2 (–) group (n = 73, n = 51) after curative resection (P = 0.008) (P = 0.018). (a) Cumulative overall survival curves (110 patients). (b) Disease‐free survival curves (80 patients). (c) Hierarchical clustering of gene expression profiles of selected 93 probe sets obtained from 80 HCC patients: AURKB‐SV2 (+) (denoted as red in vertical side bar), and AURKB‐Sv2 (–) (denoted as black). Dendrograms show the classification determined by hierarchical clustering analysis. Red and green colors indicate relative overexpression and underexpression, respectively.
Gene selection and hierarchical clustering. By evaluating gene expression changes between AURKB‐SV2 (+) and (–) groups, 93 probe sets that satisfied P < 0.0005 (FDR < 0.266) by Wilcoxon rank sum test were identified as differently expressed genes (Supporting Information Table S1). Table 4 shows up‐ and down‐regulated genes in 80 primary HCC with AURKB Sv2 variant (+/–) (P < 0.0005, FC > 1.4). Figure 3(c) shows the hierarchical clustering results using selected probe sets. All patients in the left cluster were AURKB‐SV2 (–), and the majority in the right cluster was AURKB‐SV2 (+).
Table 4.
Up‐ and down‐regulated genes in 80 primary hepatocellular carcinomas with Aurora kinase B splicing variant 2 (AURKB‐Sv2) (+/–) (P < 0.0005, FC > 1.4). We used the expression data by Affymetrix HG‐U133 Plus 2.0 (54 675 probe sets)
| Gene symbol | Up‐regulated gene in AURKB‐Sv2(+) | |||
|---|---|---|---|---|
| Title | P‐value | FC | FDR | |
| 1559213_at | Homo sapiens, clone IMAGE:5394246, mRNA | 4.77E‐04 | 5.34 | 2.74E‐01 |
| SSX1 | synovial sarcoma, X breakpoint 1 | 3.83E‐04 | 4.51 | 2.74E‐01 |
| GPR88 | G protein‐coupled receptor 88 | 3.51E‐04 | 4.19 | 2.74E‐01 |
| COCH | coagulation factor C homolog, cochlin (Limulus polyphemus) | 7.33E‐05 | 3.47 | 2.16E‐01 |
| COCH | coagulation factor C homolog, cochlin (Limulus polyphemus) | 2.20E‐05 | 2.52 | 1.20E‐01 |
| MAGEA2///2B | melanoma antigen family A, 2///melanoma antigen family A, 2B | 1.59E‐04 | 2.32 | 2.35E‐01 |
| DSG2 | desmoglein 2 | 2.67E‐04 | 2.11 | 2.74E‐01 |
| LOC133874 | hypothetical gene LOC133874 | 9.88E‐05 | 1.92 | 2.16E‐01 |
| EPB41L2 | erythrocyte membrane protein band 4.1‐like 2 | 2.67E‐04 | 1.81 | 2.74E‐01 |
| TMEM48 | transmembrane protein 48 | 4.77E‐04 | 1.78 | 2.74E‐01 |
| CEACAM1 | carcinoembryonic antigen‐related cell adhesion molecule 1 (biliary glycoprotein) | 3.83E‐04 | 1.61 | 2.74E‐01 |
| PLXNC1 | plexin C1 | 3.67E‐04 | 1.59 | 2.74E‐01 |
| RAB3IP | RAB3A interacting protein (rabin3) | 2.93E‐04 | 1.59 | 2.74E‐01 |
| TMEM118 | transmembrane protein 118 | 1.26E‐04 | 1.55 | 2.30E‐01 |
| POT1 | POT1 protection of telomeres 1 homolog (S. pombe) | 3.51E‐04 | 1.53 | 2.74E‐01 |
| PSENEN | presenilin enhancer 2 homolog (C. elegans) | 1.58E‐05 | 1.52 | 1.20E‐01 |
| LOC642236 | similar to FRG1 protein (FSHD region gene 1 protein) | 5.21E‐04 | 1.52 | 2.74E‐01 |
| CKLF | chemokine‐like factor | 2.33E‐04 | 1.51 | 2.74E‐01 |
| MAPRE2 | microtubule‐associated protein, RP/EB family, member 2 | 2.03E‐04 | 1.49 | 2.52E‐01 |
| SSX3 | synovial sarcoma, X breakpoint 3 | 1.68E‐04 | 1.49 | 2.35E‐01 |
| CKLF | chemokine‐like factor | 1.94E‐04 | 1.46 | 2.46E‐01 |
| UROD | uroporphyrinogen decarboxylase | 1.26E‐04 | 1.44 | 2.30E‐01 |
| ULK4 | unc‐51‐like kinase 4 (C. elegans) | 2.93E‐04 | 1.42 | 2.74E‐01 |
| ERGIC3 | ERGIC and golgi 3 | 5.58E‐06 | 1.41 | 1.20E‐01 |
| ATP6V0B | ATPase, H + transporting, lysosomal 21 kDa, V0 subunit b | 1.68E‐04 | 1.41 | 2.35E‐01 |
| H3F3B | H3 histone, family 3B (H3.3B) | 4.19E‐04 | 1.40 | 2.74E‐01 |
| MED8 | mediator of RNA polymerase II transcription, subunit 8 homolog (S. cerevisiae) | 1.46E‐04 | 1.40 | 2.35E‐01 |
| AIP | aryl hydrocarbon receptor interacting protein | 1.46E‐04 | 1.40 | 2.35E‐01 |
| Gene symbol | Down‐Regulated Gene in AURKB‐Sv2(+) | |||
|---|---|---|---|---|
| Title | P‐value | FC | FDR | |
| 1562346_at | MRNA; cDNA DKFZp313F2234 (from clone DKFZp313F2234) | 3.21E‐04 | 0.89 | 2.74E‐01 |
| IL1RL1 | Interleukin 1 receptor‐like 1 | 4.01E‐04 | 0.89 | 2.74E‐01 |
| FUT6 | Fucosyltransferase 6 (alpha (1,3) fucosyltransferase) | 8.95E‐05 | 0.88 | 2.16E‐01 |
| IL28RA | interleukin 28 receptor, alpha (interferon, lambda receptor) | 3.06E‐04 | 0.88 | 2.74E‐01 |
| 1555488_at | NA | 5.21E‐04 | 0.88 | 2.74E‐01 |
| KCNAB3 | potassium voltage‐gated channel, shaker‐related subfamily, beta member 3 | 7.33E‐05 | 0.88 | 2.16E‐01 |
| DIXDC1 | DIX domain containing 1 | 1.41E‐05 | 0.87 | 1.20E‐01 |
| 1569912_at | Homo sapiens, clone IMAGE:5459012, mRNA | 1.74E‐06 | 0.87 | 9.52E‐02 |
| 236991_at | Transcribed locus | 1.20E‐04 | 0.87 | 2.30E‐01 |
| ZNF474 | zinc finger protein 474 | 9.33E‐05 | 0.87 | 2.16E‐01 |
| CCDC85A | Coiled‐coil domain containing 85A | 4.77E‐04 | 0.87 | 2.74E‐01 |
| BAI2 | brain‐specific angiogenesis inhibitor 2 | 2.55E‐04 | 0.87 | 2.74E‐01 |
| MDGA1 | MAM domain containing glycosylphosphatidylinositol anchor 1 | 1.46E‐04 | 0.87 | 2.35E‐01 |
| KIAA1787 | KIAA1787 protein | 5.44E‐04 | 0.86 | 2.74E‐01 |
| MORN1 | MORN repeat containing 1 | 4.99E‐04 | 0.86 | 2.74E‐01 |
| FAM41AY | family with sequence similarity 41, member A, Y‐linked | 8.04E‐05 | 0.86 | 2.16E‐01 |
| ITGA10 | integrin, alpha 10 | 5.99E‐05 | 0.85 | 2.16E‐01 |
| 236541_at | Transcribed locus | 4.99E‐04 | 0.84 | 2.74E‐01 |
| AMACR | alpha‐methylacyl‐CoA racemase | 1.21E‐04 | 0.84 | 2.30E‐01 |
| DYNLRB2 | dynein, light chain, roadblock‐type 2 | 4.57E‐04 | 0.84 | 2.74E‐01 |
| RBM25 | RNA binding motif protein 25 | 1.85E‐04 | 0.83 | 2.46E‐01 |
| 228525_at | Transcribed locus, strongly similar to XP_512572.1 similar to low density lipoprotein receptor‐related protein 3 [Pan troglodytes] | 5.21E‐04 | 0.81 | 2.74E‐01 |
| LOC338328 | high density lipoprotein‐binding protein | 3.83E‐04 | 0.79 | 2.74E‐01 |
| CYP46A1 | cytochrome P450, family 46, subfamily A, polypeptide 1 | 2.20E‐05 | 0.77 | 1.20E‐01 |
| RICS | Rho GTPase‐activating protein | 3.51E‐04 | 0.73 | 2.74E‐01 |
| EML1 | echinoderm microtubule associated protein like 1 | 5.44E‐04 | 0.73 | 2.74E‐01 |
| DGCR5 | DiGeorge syndrome critical region gene 5 (non‐coding) | 1.67E‐04 | 0.72 | 2.35E‐01 |
Discussion
We have previously identified AURKB as the only independent predictor of the aggressive recurrence of HCC.( 12 ) AURKB is a chromosomal passenger serine/threonine protein kinase that regulates accurate chromosomal segregation, cytokinesis, protein localization to the centromere and kinetochore, correct microtubule‐kinetochore attachments and regulation of the mitotic checkpoint.( 26 ) Our previous study revealed that AURKB expression was closely associated with genetic instability of HCC tumors. More importantly, AURKB has recently received increasing attention as an eligible target of molecular cancer therapy.( 27 ) AURKB‐targeted therapy might be a promising neoadjuvant approach for the occult vascular invasion of HCC.
DNA microarray technology enabled us to analyze the genome‐wide profile of gene expression specific to malignant tumor. Using this technology, we investigated the mRNA expression patterns in 80 cases of primary HCC, and clarified that the genes associated with cell cycle, especially AURKB, were significantly up‐regulated in HCC in relation to the invasion of the portal vein and/or hepatic vein. From Western blotting analysis, Aurora B protein was expressed in all cultured human HCC cell lines. To assess the validity of our microarray results, we examined by the RT‐PCR using TaqMan® MGB probe Gene Expression Assays for AURKB. A strong correlation between qRT‐PCR expression data and microarray values was found. AURKB was overexpressed in tumor more than adjacent non‐tumor specimens; overexpression was also observal in vascular invasion cases rather than non‐vascular invasion cases in HCC.
To confirm the analysis of Aurora B mRNA expression pattern in human HCC and adjacent tissue, we screened eight human HCC cell lines, 125 tumor specimens, 94 chronic hepatitis and cirrhosis background liver specimens, 18 metastatic liver cancer cases and 16 normal liver specimens (based on the sequencing) for different Aurora B variants including AURKB‐WT and two variant forms AURKB‐Sv1, AURKB‐Sv2 by RT‐PCR and qRT‐PCR. AURKB‐WT was expressed in all HCC tumor specimens, chronic hepatitis and cirrhosis background liver specimens, metastasis liver cancer and normal liver specimens. However, the expression of AURKB‐Sv1 variant form were 100%, 97.8%, 88.9% and 25%, respectively. In cases of the AURKB‐Sv2 variant form, 33.6%, 15.9%, 61.1%, 0% specimens only expressed, respectively. It would be a very interesting finding that the expression of AURKB‐Sv2 was absent in normal liver and was higher in metastatic liver cancer than HCC. Sistayanarain et al.( 13 ) reported that Aurora B and two variant forms were expressed in HCC, but this was studied only with some limited samples including 11 frozen tissues and six paraffin‐embedded tissues by RT‐PCR. In that study, they showed the two variants (B1 and B2) present in tumor specimens at 41.1%, 52.9%, respectively, but by the analysis of only 11 frozen tumor specimens showed 45% and 27.3%, respectively. In contrast, all specimens were fresh and frozen in our study. Co‐expression of AURKB‐WT or alternative variant AURKB‐Sv1 was detected in all positive HCC cases; AURKB‐Sv2 was detected in some limited cases. This result was similar to that which Sistayanarain et al. reported previously. Here, we also studied the significance of AURKB‐Sv2 variant form from a the clinical viewpoint, for the progression of hepatocellular carcinoma, and showed that expression of AURKB‐Sv2 form was associated with the advanced stage of HCC rather than others (P < 0.01). The AURKB‐Sv2 positive cases showed a higher level of serum α‐AFP and PIVKAII, poor differentiation cases, tumor capsular invasion, multiple tumor formation cases and at a younger age than with other variant forms (P < 0.05). Although this variant did not show the association with vascular invasion, we previously reported that the AURKB‐WT was more readilyassociated with vascular invasion and recurrence cases in HCC. Then we analyzed the overall survival rate for 110 patients and disease‐free survival rate for 80 AURKB‐Sv2 variant positive/negative HCC patients. Statistical significance was observable for poor survivals of AURKB‐Sv2 (+) cases.
We tried to detect AURKB‐Sv2 protein in tissue samples, but could not detect this. As the mRNA expression level of AURKB‐Sv2 was very low compared to the AURKB‐WT, we assume the protein could not be detected by our antibody.
This opens a question: what are the roles of AURKB splicing variants in hepatocarcinogenesis? Recently our research group predicted the 3D‐stuctures of splicing variants by their protein sequences (Kim Hyeryun, Y. Mahmut et al., data not published), using computer simulation models; molecular dynamics (MD) and root means square deviation (RMSD)–time plots. AURKB‐WT has α‐helix domain activated by INCEP. AURKB‐Sv1 lacks this domain because of its absence in some part of exon 5, but the model suggests that α‐helix was newly formed by intron 5‐6 and replaced the exon 5α‐helix, suggesting that the conformation direction was different from AURKB‐WT. This means that AURKB‐Sv1 has difficulty interacting with INCENP. On the other hand, AURKB‐Sv2 kept this domain, but lacked many parts of kinase domain by truncation. This suggests that if it has no kinase activity, it is possible to compete with normal AURKB in a dominant negative manner. However, we lack the functional analysis data and further experiments are necessary to elucidate this point.
Criteria of AURKB‐SV2 (+)/(–) groups used for Wilcoxon rank sum test were identified as differently revealed AURKB‐Sv2 associated up‐ and down‐regulated genes (Table 4). Several genes (CEACAM1, SSX1, SSX3 and MAGEA) are up‐regulated in AURKB‐Sv2 (+) cases. Such a cell adhesion molecule CEACAM1 (Carcinoembryonic Antigen‐Related Cell Adhesion Molecule 1) gene encodes a member of the carcinoembryonic antigen (CEA) gene family, which belongs to the immunoglobulin super family. Several studies showed that multiple cellular activities have been attributed to the encoded protein, including roles in the differentiation and arrangement of tissue three‐dimensional structure, angiogenesis, apoptosis, tumor suppression, metastasis, and the modulation of innate and adaptive immune responses, was down‐regulated in several types of human cancers, including prostate, colorectal and breast cancers. Thies et al.( 28 ) showed that expression of CEACAM1 in primary tumors in melanoma patients is associated with the subsequent development of metastatic disease. Hokari et al.( 29 ) analyzed several hepatoma cell lines and found that CEACAM1 was only expressed in HepG2 cells and the cells were treated with small interfering RNA targeted against CEACAM1, the growth rate in monolayer culture was increased. In contrast, when HepG2 cells were cultured in suspension, inhibition of CEACAM1 expression significantly decreased the growth rate, suggesting a role for CEACAM1 on hepatocarcinogenesis, by showing that CEACAM1 acts as a tumor suppressor in HepG2 cells in anchorage‐dependent growth conditions, while in anchorage‐independent growth conditions, it augments cell proliferation by potentiating the cell‐cell attachment. SSX1 and SSX3 belong to the SSX family whose transcriptional repressor and cellular immune responses in cancer patients, overexpress in synovial sarcomas and are potentially useful targets in cancer vaccine‐based immunotherapy. MAGEA (melanoma‐associated antigen gene) belongs to the chromosome X‐clustered cancer/testis antigens that normally express in the human germ line and are overexpressed in various tumor types. Monte et al.( 30 ) showed that MAGEA tumor antigen target p53 transactivation functions through histone deacetylase recruitment and confers resistance to chemotherapeutic agents. Tsai et al.( 31 ) reported that differential expression profile analysis of lung cancer suggests MAGE gene is not only for immunotherapy, but also valuable markers for further diagnosis and prognosis. In constant, several genes (BAI2, CYP46A1 and DYNLRB2) are down‐regulated in AURKB‐Sv2 (+) cases. BAI2 (brain‐specific angiogenesis inhibitor 2) a p53‐target gene, encodes brain‐specific angiogenesis inhibitor, a seven‐span transmembrane protein, and is thought to be a member of the secretin receptor family, and plays a role in angiogenesis. BAI2 expression decreased after hypoxia and preceded the increased expression of vascular endothelial growth factor.( 32 ) CYP46A1 (cytochrome P450, family 46, subfamily A, polypeptide 1) gene encodes a member of the cytochrome P450 super family of enzymes. The cytochrome P450 proteins are mono‐oxygenases which catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids and other lipids. DYNLRB2 (dynein, light chain, roadblock‐type 2) is member of an ancient dynein light chain protein family, conserved in nematode, fruit fly, mouse and rat. The expression of DNLC2B was generally high compared with that of DNLC2A except in the liver. Jiang et al.( 33 ) analyzed 68 hepatocellular carcinoma tissue samples, suggesting that down‐regulation of DNLC2B and up‐regulation of DNLC2A genes might be involved in tumor progression.
In conclusion, an abnormal expression of Aurora B and alterative splicing variants was found to be involved in hepatocarcinogenesis. The AURKB‐Sv2 variant form is more significantly associated with advanced stages of HCC than others. It is suggested to be a marker of poor prognosis. Founded in the tumor capsular invasion and multiple tumor regions, suggests that this might play a role in enhancing multiple malignant tumor formation and recurrence of HCC in hepatocarcinogenesis. Associated up‐ and down‐regulated genes were identified by comparison of cases where AURKB‐Sv2 differentially expressed or not. Further experiments are necessary to elucidate the functional properties of this variant in HCC.
Supporting information
Table S1. 93 probe sets identified as differently expressed gene that satisfied false discovery rate (FDR) < 5% (P < 0.0005) with Wilcoxon rank sum test between AURKB‐Sv2(+) and AURKB‐Sv(–) group.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported by Special Coordination Funds for Promoting Science and Technology (Japan Science & Technology Agency), and a Grant‐in‐Aid from Ministry of Education, Culture, Sports, Science & Technology of Japan.
References
- 1. Yip D, Findlay M, Boyer M, Tattersall MH. Hepatocellular carcinoma in central Sydney: a 10‐year review of patients seen in a medical oncology department. World J Gastroenterol 1999; 5: 483–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schmid R. Prospect of gastroenterology and hepatology in the next century. World J Gastroenterol 1999; 5: 185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poon RT, Fan ST, Lo CM et al . Improving survival results after resection of hepatocellular carcinoma: a prospective study of 377 patients over 10 years. Ann Surg 2001; 234: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parks RW, Garden OJ. Liver resection for cancer. World J Gastroenterol 2001; 7: 766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arii S, Yamaoka Y, Futagawa S et al . Results of surgical and nonsurgical treatment for small‐sized hepatocellular carcinomas: a retrospective and nationwide survey in Japan. The Liver Cancer Study Group Japan Hepatol 2000; 32: 1224–9. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka S, Noguchi N, Ochiai T et al . Outcomes and recurrence of initially resection of hepatocellular carcinoma meeting Milan Criteria: Raionale for partial hepatectomy as first strategy. J Am Coll Surg 2007; 204: 1–6. [DOI] [PubMed] [Google Scholar]
- 7. Paquet KJ, Lazar A, Heine WP, Jachmann‐Jahn V. Small unilocular hepatocellular carcinoma (0 < 5 cm) in patients with liver cirrhosis. Early diagnosis, surgical indications, resection and prognosis. Zentralbl Chir 2000; 125: 629–36. [PubMed] [Google Scholar]
- 8. Rabe C, Pilz T, Klostermann C et al . Clinical characteristics and outcome of a cohort of 101 patients with hepatocellular carcinoma. World J Gastroenterol 2001; 7: 208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou XD, Tang ZY, Yang BH et al . Experience of 1000 patients who underwent hepatectomy for small hepatocellular carcinoma. Cancer 2001; 91: 1479–86. [DOI] [PubMed] [Google Scholar]
- 10. Poon RT, Fan ST, Ng IO, Lo CM, Liu CL, Wong J. Different risk factors and prognosis for early and late intrahepatic recurrence after resection of hepatocellular carcinoma. Cancer 2000; 89: 500–7. [PubMed] [Google Scholar]
- 11. Kumada T, Nakano S, Takeda I et al . Patterns of recurrence after initial treatment in patients with small hepatocellular carcinoma. Hepatology 1997; 25: 87–92. [DOI] [PubMed] [Google Scholar]
- 12. Tanaka S, Arii S, Yasen M et al . Aurora kinase B is a predictive factor for aggressive recurrence of hepatocellular carcinoma after curative hepatectomy. Br J Surg 2008; 95: 611–19. [DOI] [PubMed] [Google Scholar]
- 13. Sistayanarain A, Tsuneyama K, Zheng H et al . Expression of Aurora‐B kinase and phosphorylated histone H3 in hepatocellular carcinoma. Anticancer Res 2006; 26: 3585–94. [PubMed] [Google Scholar]
- 14. Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer: a coincidence or a real link? Trends Cell Biol 2005; 15: 241–7. [DOI] [PubMed] [Google Scholar]
- 15. Klein A, Reichardt W, Jung V et al . Overexpression and amplification of STK15 in human gliomas. Int J Oncol 2004; 25: 1789–94. [PubMed] [Google Scholar]
- 16. Sorrentino R, Libertini S, Pallante PL et al . Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J Clin Endocrinol Metab 2005; 90: 928–35. [DOI] [PubMed] [Google Scholar]
- 17. Adams RR, Wheatley SP, Gouldsworthy AM et al . INCENP binds the Aurora‐related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr Biol 2000; 10: 1075–8. [DOI] [PubMed] [Google Scholar]
- 18. Kaitna S, Mendoza M, Jantsch‐Plunger V, Glotzer M. Incenp and an aurora‐like kinase form a complex essential for chromosome segregation and efficient completion of cytokinesis. Curr Biol 2000; 10: 1172–81. [DOI] [PubMed] [Google Scholar]
- 19. Wheatley SP, Carvalho A, Vagnarelli P, Earnshaw WC. INCENP is required for proper targeting of survivin to the centromeres and the anaphase spindle during mitosis. Curr Biol 2001; 11: 886–90. [DOI] [PubMed] [Google Scholar]
- 20. Adams RR, Carmena M, Earnshaw WC. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol 2001; 11: 49–54. [DOI] [PubMed] [Google Scholar]
- 21. Adams RR, Maiato H, Earnshaw WC, Carmena M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol 2001; 153: 865–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sessa F, Mapelli M, Ciferri C et al . Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell 2005; 29: 379–91. [DOI] [PubMed] [Google Scholar]
- 23. Kaitna S, Pasierbek P, Jantsch M, Loidl J, Glotzer M. The aurora B kinase AIR‐2 regulates kinetochores during mitosis and is required for separation of homologous chromosomes during meiosis. Curr Biol 2002; 12: 798–812. [DOI] [PubMed] [Google Scholar]
- 24. Reico H, Roman K, Erich A. Nigg, exploring the functional interactions between Aurora B, INCENP, and Survivin in mitosis. Mol Cell Biol 2003; 14: 3325–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Irizarry RA, Hobbs B, Collin F et al . Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003; 4: 249–64. [DOI] [PubMed] [Google Scholar]
- 26. Vader G, Medema RH, Lens SM. The chromosomal passenger complex: guiding Aurora‐B through mitosis. J Cell Biol 2006; 173: 833–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Girdler F, Gascoigne KE, Eyers PA et al . Validating Aurora B as an anti‐cancer drug target. J Cell Sci 2006; 119: 3664–75. [DOI] [PubMed] [Google Scholar]
- 28. Thies A, Moll I, Berger J et al . CEACAM1 expression in cutaneous malignant melanoma predicts the development of metastatic disease. J Clin Oncol 2002; 20: 2530–6. [DOI] [PubMed] [Google Scholar]
- 29. Hokari M, Matsuda Y, Wakai T et al . Tumor suppressor carcinoembryonic antigen‐related cell adhesion molecule 1 potentates the anchorage‐independent growth of human hepatoma HepG2 cells. Life Sci 2007; 81: 336–45. [DOI] [PubMed] [Google Scholar]
- 30. Monte M, Simonatto M, Peche LY et al . MAGE‐A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci USA 2006; 103: 11160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsai JR, Chong IW, Chen YH et al . Differential expression profile of MAGE family in non‐small‐cell lung cancer. Lung Cancer 2007; 56: 185–92. [DOI] [PubMed] [Google Scholar]
- 32. Kee HJ, Koh JT, Kim MY et al . Expression of brain‐specific angiogenesis inhibitor 2 (BAI2) in normal and ischemic brain: involvement of BAI2 in the ischemia‐induced brain angiogenesis. J Cereb Blood Flow Metab 2002; 22: 1054–67. [DOI] [PubMed] [Google Scholar]
- 33. Jiang J, Yu L, Huang X et al . Identification of two novel human dynein light chain genes, DNLC2A and DNLC2B, and their expression changes in hepatocellular carcinoma tissues from 68 Chinese patients. Gene 2001; 281: 103–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. 93 probe sets identified as differently expressed gene that satisfied false discovery rate (FDR) < 5% (P < 0.0005) with Wilcoxon rank sum test between AURKB‐Sv2(+) and AURKB‐Sv(–) group.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item