

Abstract
The extracellular‐signal‐regulated kinase (ERK) mitogen‐activated protein (MAP) kinase signaling pathway plays an important role in various cellular responses, including cell proliferation, cell differentiation and cell survival. Recent studies have identified a number of Ras/ERK signaling‐related proteins, such as scaffold proteins and inhibitors. These proteins modulate ERK signaling and thereby could give variations in ERK signaling outputs that regulate cell fate decisions. Here we focus on the role of ERK signaling in cell cycle progression from G0/G1 to S phase and cancer. (Cancer Sci 2006; 97: 697–702)
The Ras/extracellular‐signal‐regulated kinase (ERK) mitogen‐activated protein (MAP) kinase signaling pathway is among the key mechanisms that transmit signals from the cell surface to the nucleus.( 1 , 2 , 3 , 4 , 5 , 6 , 7 ) A wide variety of extracellular stimuli induce sequential activation of three protein kinases, Raf, MEK and ERK, in the Ras/ERK signaling pathway (Fig. 1). ERK is a highly conserved serine/threonine kinase activated by MEK via phosphorylation on both threonine and tyrosine residues in the TEY sequence. Activated ERK phosphorylates both cytoplasmic and nuclear substrates, including many enzymes, cytoskeletal proteins and transcription factors. Recent studies have identified a number of Ras/ERK signaling‐related proteins, such as scaffold proteins and inhibitor proteins of this pathway. These proteins provide variations in ERK signaling by modulating the duration, magnitude and subcellular compartmentalization of ERK activity.( 3 , 8 ) Accumulating evidence suggests that such differences in ERK activity generate variations in signaling outputs that regulate cell fate decisions. Moreover, crosstalk with other pathways could also be crucial for determining signaling specificity. The Ras/ERK signaling pathway is known to regulate various cellular responses and, in particular, its role in cell cycle progression in G1 phase and cell proliferation is well established.( 9 , 10 , 11 , 12 ) In addition, the pathway is activated constitutively in many types of cancer. Here we discuss recent findings, focusing on ERK signaling‐mediated normal cell cycle progression and malignant transformation.
Figure 1.
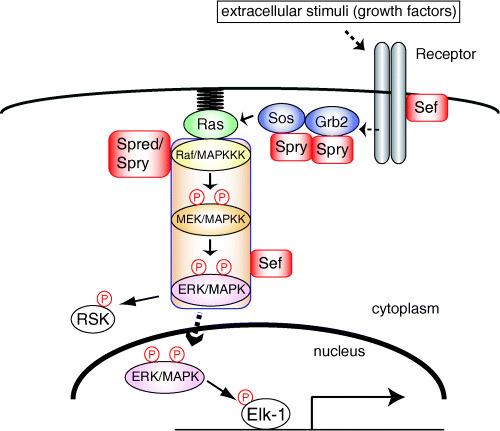
The Ras/extracellular‐signal‐regulated kinase (ERK) signaling pathway and its regulators. Extracellular stimuli induce the dimerization and activation of receptor tyrosine kinases (RTK). Activated RTK phosphorylate themselves or adaptor proteins, such as FRS2 and Shp2, on their tyrosine residues. Other adaptor proteins, such as Grb2, bind to phosphorylated tyrosine residues through their SH2 domain. The Grb2/Sos complex then translocates to the plasma membrane, where the guanine nucleotide exchange factor Sos activates Ras. Activated Ras induces activation of the ERK cascade composed of three protein kinases: Raf, MEK and ERK. Activated ERK translocates to the nucleus, in which ERK phosphorylates nuclear substrates, such as Elk‐1, while part of activated ERK remains in the cytoplasm and phosphorylates cytoplasmic ERK targets, such as RSK. Several regulators of the ERK signaling pathway, such as Sprouty (Spry), bind to and interfere with components of this pathway.
ERK signaling regulators
Recent studies have identified several negative regulators of the ERK signaling pathway and their action mechanisms have been analyzed. Among them, Sprouty, Spred and Sef were found to act as conserved inhibitors of the ERK signaling pathway.( 13 , 14 , 15 , 16 , 17 ) More recent reports have demonstrated the detailed molecular mechanisms of action of these regulators (Fig. 1). Spred inhibits the ERK signaling pathway at the level of Raf by binding to Ras and Raf.( 14 ) The inhibitory mechanisms of Sprouty and Sef have been controversial. Targets of Sprouty in the ERK signaling pathway are suggested to be Grb2,( 18 , 19 ) Sos( 19 ) and Raf1.( 20 ) Hanafusa et al. showed that Sprouty1 and 2 become phosphorylated on a conserved tyrosine residue (Y53 in Sprouty1 and Y55 in Sprouty2) in their amino‐terminal domain upon growth factor stimulation, and become bound to Grb2.( 18 ) This binding prevents Grb2 from binding to either tyrosine‐phosphorylated adaptor proteins or receptors, resulting in the inhibition of Ras/ERK signaling. This conserved tyrosine residue of Sprouty1 and 2 could be phosphorylated by Src family kinases and dephosphorylated by Shp2.( 21 , 22 , 23 ) Other reports have shown that Sprouty2 becomes phosphorylated on the same conserved tyrosine residue upon epidermal growth factor (EGF) stimulation and binds to c‐Cbl, the E3 ubiquitin ligase for the EGF receptor.( 22 , 24 , 25 , 26 ) This association leads to polyubiquitylation and subsequent degradation of Sprouty2, instead of the EGF receptor. In this case, tyrosine phosphorylation of Sprouty2 results in enhancement of EGF signaling. However, in MEF cells, Sprouty2 does not enhance EGF signaling, but rather inhibits both fibroblast growth factor (FGF) and EGF signaling.( 22 ) The difference observed might result from the difference in cell type. Recently, DaSilva et al. reported that Sprouty2 is phosphorylated by MAP kinase‐interacting kinase 1 (Mnkl) on serines 112 and 121 upon growth factor (FGF or EGF) stimulation.( 27 ) This phosphorylation enhances the stability of Sprouty2 by interfering with tyrosine phosphorylation of Sprouty2 and binding to c‐Cbl. Thus, the function of Sprouty might be regulated by tyrosine and serine phosphorylation, which are both involved in Sprouty binding to other factors. Another group reported that the carboxy‐terminal cysteine‐rich domain of Sprouty4 binds to Raf1.( 20 ) This binding prevents VFGF/PKC/Raf kinase‐mediated activation of Raf1, but not Ras‐mediated activation of Raf1. As Sprouty4 does not become phosphorylated in response to growth factor stimulation,( 18 , 22 ) the action mechanism of Sprouty4 would be different from that of Sprouty1 and 2. Recently, Ozaki et al. showed functional cooperation among Sprouty isoforms.( 19 ) The four Sprouty isoforms have been shown to interact with each other through their carboxy‐terminal domains.( 18 , 19 ) Ozaki et al. reported that Sprouty1 and Sprouty4 bind specifically to Grb2 and Sos1, respectively, and thus the Sprouty1/Sprouty4 heterooligomer prevents the Grb2/Sos1 complex from binding to FRS2, exhibiting the most potent inhibitory activity among hetrooligomers of Sprouty proteins. These findings increased our understanding of the Sprouty mechanisms of action. As described above, the conserved tyrosine residue of Sprouty is important for its inhibitory activity. The Sprouty1, 2 and 4 mutants, in which this conserved tyrosine residue is mutated (Sprouty YF), act as dominant‐negative forms.( 18 , 28 ) Hanafusa et al. have found that Sprouty controls the duration of ERK activity and thus acts as a temporal regulator of ERK signaling.( 18 )
The targets of Sef in the ERK signaling pathway are reported to be either the FGF receptor or MEK.( 29 ) Torii et al. have shown that Sef on the Golgi apparatus or plasma membrane regions binds to the MEK–ERK complex, blocks dissociation of the MEK–ERK complex, and thus inhibits ERK nuclear translocation. As a result, Sef inhibits phosphorylation of nuclear ERK substrates, such as Elk‐1.( 30 ) Interestingly, Sef does not inhibit phosphorylation of cytoplasmic ERK substrates, such as RSK2. Thus Sef provides spatial control for Ras/ERK signaling. In summary, these regulatory proteins could modulate the duration, magnitude and subcellular compartmentalization of ERK activity.
ERK signaling and cell cycle progression
Activation of the Ras/ERK signaling pathway in cell cycle progression from G0/G1 to S phase has been linked to cyclin D induction and consequent retinoblastoma (Rb) phosphorylation.( 9 , 10 , 11 , 12 , 31 , 32 ) Extracellular stimuli, such as growth factors, induce phosphorylation and activation of ERK. Activated ERK then translocates from the cytoplasm to the nucleus, where ERK phosphorylates and activates several nuclear ERK targets, including transcription factors such as Elk‐1. Consequently, ERK induces the expression of the immediate early genes, such as c‐fos. The expression of the immediate early genes has been implicated in regulating subsequent induction of the delayed early genes, including a first class of G1 cyclins, cyclin D. Upregulation of cyclin D expression results in upregulation of the cyclin D–CDK4/6 complex. Activation of cyclin D–CDK4/6 kinase activity leads to phosphorylation and inactivation of Rb, which then activates the E2F family of transcription factors and induces expression of target genes, including a second class of G1 cyclins, cyclin E. Activation of cyclin E–CDK2 kinase activity leads to further phosphorylation and inactivation of Rb, thus further enhancing the activity of the E2F family. This positive feedback leads to the synthesis of proteins required for S phase entry (Fig. 2).( 33 , 34 ) Therefore, in response to growth factor stimulation, ERK triggers these sequential events, including sequential induction of a number of genes, and thereby causes S phase entry.
Figure 2.
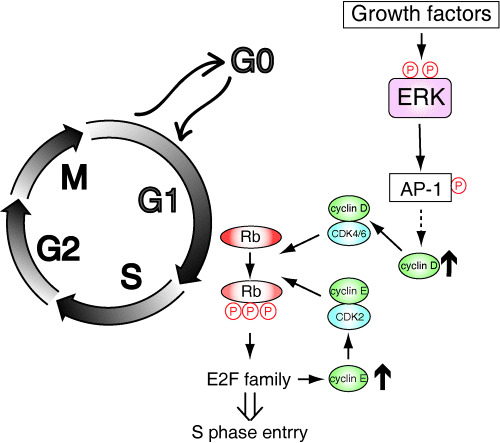
Regulation of G1 cell cycle progression through ERK signaling. In response to growth factor stimulation, ERK is phosphorylated and activated. Activated ERK causes enhanced expression of immediate early genes, including the AP‐1 protein family. Subsequently, delayed early genes, including cyclin D, are induced. The Cyclin D–CDK4/6 complex then initiates retinoblastoma (Rb) phosphorylation, which activates the E2F family of transcription factors and induces expression of target genes, including cyclin E. The cyclin E–CDK2 complex further phosphorylates Rb and thus activates the E2F family. This positive feedback loop drives the cells to S phase entry.
It has been suggested that sustained ERK activation, but not transient activation, is necessary for inducing S phase entry of quiescent fibroblastic cells.( 7 , 35 , 36 , 37 ) The ERK activity must be sustained until approximately 2 or 3 h before the onset of S phase.( 38 ) Thus, the duration of ERK activity is a key factor for ensuring G1 phase progression. How does sustained ERK activation elicit the appropriate cellular responses? Recently, Murphy et al. have provided some clues to this question.( 39 , 40 ) Both transient and sustained ERK activation induce expression of immediate early genes, such as Fos, Jun, Myc and Egr‐1. However, only sustained ERK activation induces sustained phosphorylation of immediate early gene products, which leads to their stabilization and activation, resulting in appropriate gene expression. Thus, sustained, but not transient, ERK activation can induce cyclin D expression several hours after growth factor stimulation.( 37 , 41 , 42 ) More recently, Yamamoto et al. have provided another clue to this question.( 39 ) Genome‐wide analyses of transcriptional programs in cell cycle progression from G0/G1 to S phase have shown that there are not only ERK‐dependent upregulated genes but also ERK‐dependent downregulated genes. The expression level of almost all of these ERK‐dependent downregulated genes is maintained at a lower level throughout G1 phase, and the decreased expression levels return to the original levels rapidly if ERK inactivation occurs. Remarkably, these ERK‐dependent downregulated genes include known and hitherto unknown antiproliferative genes, suggesting that continuous ERK activation downregulates antiproliferative genes until the onset of S phase to allow successful G1 phase progression (Fig. 3). The cells receive not only mitogenic signals but also diverse stimuli such as environmental stresses that induce transient ERK activation. As transient ERK activation does not induce sustained downregulation of antiproliferative genes, these inappropriate stimuli do not cause cell proliferation. Thus, this mechanism may work as a fail‐safe mechanism, which prevents inappropriate stimuli from causing cell cycle progression.
Figure 3.
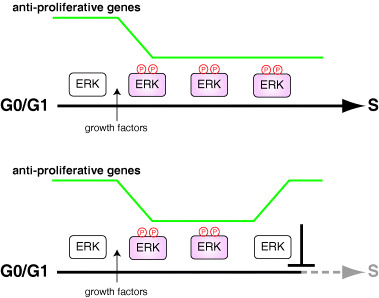
Role of ERK‐dependent downregulation of antiproliferative genes in G1 phase progression. Sustained activation of ERK induces and maintains decreased expression levels of antiproliferative genes, and thus causes S phase entry (upper). Cessation of ERK activation even at mid or late G1 leads to a rapid increase in the antiproliferative genes, and thus inhibits S phase entry (lower).
Among the antiproliferative genes that are downregulated in an ERK‐dependent manner, Tob1 and JunD have been relatively well characterized.( 43 , 44 , 45 ) Tob1 has been shown to regulate cyclin D1 expression negatively by recruiting histone deacetylase to the cyclin D1 promoter.( 46 ) JunD has also been shown to inhibit cyclin D1 expression, and it may exert this effect through antagonizing the function of c‐Jun.( 47 ) In contrast, the other antiproliferative genes might target molecules other than cyclin D to inhibit cell cycle progression. In fact, recent studies have shown that quiescent fibroblasts lacking D‐type cyclins or D‐type cyclin‐dependent kinases are able to enter S phase in response to mitogenic stimulation.( 48 , 49 ) In addition, Dekanty et al. have shown that when leukemia inhibitory factor induces DNA synthesis in Swiss 3T3 cells, ERK activity is required for their mitogenic responses, which are independent of cyclin D1 expression.( 50 ) Detailed functional analysis of these ERK‐dependent downregulated genes and identification of their target molecules will provide new insights into G1 phase progression.
PI3K/Akt signaling and cell cycle progression
The lipid kinase phosphatidylinositol 3‐kinase (PI3K) and its downstream protein kinase Akt, also known as PKB, are involved in growth factor‐stimulated cell cycle progression from G0/G1 to S phase.( 9 , 10 , 11 , 12 , 51 , 52 ) PI3K translocates to the cell membrane and binds to receptor tyrosine kinases or Ras. At the membrane, PI3K generates phosphatidylinositol‐3,4,5‐triphosphate (PIP3). Akt then translocates to the PIP3‐rich membrane and is phosphorylated by protein kinases, such as PDK1 and PDK2, and becomes activated. Activated Akt phosphorylates various targets, such as Bad, and has a strong anti‐apoptotic function, thus playing an important role in cell survival signaling.( 51 , 52 ) PI3K/Akt signaling has also been implicated in CDK activation. First, Akt phosphorylates and inhibits glycogen synthase kinase 3‐β (GSK3‐β), which phosphorylates and destabilizes cyclin D1 protein. Second, Akt phosphorylates and inhibits FOXO transcription factors, which can repress cyclin D1 expression and induce expression of p27 (kip1) and p21 (WAF), the inhibitors of G1 cyclin/CDK activity. Thus, PI3K/Akt signaling is involved in the regulation of cell cycle progression.
Activation of the ERK signaling pathway usually promotes cell cycle progression. However, in some cases it leads to cell cycle arrest. In fact, high level activation of ERK upregulates the activity of the CDK inhibitor p21 (WAF) through both transcriptional and post‐translational mechanisms.( 53 , 54 , 55 ) Interestingly, this ERK‐mediated cell cycle arrest can be bypassed by the PI3K/Akt signaling pathway.( 56 ) In addition, PI3K/Akt signaling would regulate ERK signaling, more directly. Recently, Hayashi et al. have shown that Centaurin‐α1 is a PI3K‐dependent activator of ERK signaling.( 57 ) Centaurin‐α1 is known as a PIP3‐binding protein that has two pleckstrin homology (PH) domains and a putative ADP ribosylation factor GTPase‐activation protein domain. Transient expression of Centaurin‐α1 is shown to induce activation of ERK, and this activation is dependent on PI3K and Ras. Taken together, PI3K/Akt signaling acts cooperatively with ERK signaling in regulating growth factor‐stimulated cell cycle progression.
ERK signaling in cancer
As mentioned above, both the ERK and PI3K signaling pathways are involved in G1 cell cycle progression. Thus, it is not surprising that several components of these signaling pathways are involved in carcinogensis.( 10 , 12 , 51 ) In fact, increased expression of EGF receptor is found in most carcinomas. Human EGF receptor‐2 (HER2)/neu expression is increased in breast cancers. Platelet‐derived growth factor (PDGF) expression is elevated in glioblastoma, and mutations in PDGF and kit receptors are found in gastrointestinal sarcomas. Ras activation by point mutations occurs in approximately 20% of human cancers. Three members of the Ras family, H‐Ras, K‐Ras and N‐Ras, are found to be activated in human cancers. Among them, K‐Ras is activated most frequently. B‐Raf is mutated and activated in approximately 7% of human cancers, particularly in melanomas. Mutations in B‐Raf occur in a very limited number of residues in the kinase domain, resulting in its constitutive kinase activation. One of these mutants is a V600E mutant. Recently, Solit et al. showed that B‐Raf mutant cancer cells have enhanced and selective sensitivity to MEK inhibition when compared to cells in which Ras is mutated.( 58 ) In the B‐Raf V600E mutant cells, MEK inhibition causes reduced cyclin D expression and Rb hypophosphorylation, resulting in G1 arrest. This finding suggests that there is a strong dependency on MEK activity in active B‐Raf‐mediated cancers.
Activating mutation or amplification of PI3K and Akt family members is found in carcinomas, especially in ovarian and breast cancers. The most significant activation of the PI3K/Akt signaling pathway in cancers is caused by a loss of PTEN, which acts as a PI3K signaling inhibitor. PTEN is depleted in approximately 30–40% of human cancers. Cyclin D1 expression is enhanced in 50% of breast cancers. As cyclin D1‐deficient mice are resistant to breast cancers induced by the HER2/neu and ras oncogenes, cyclin D1 should be required for mammary tumor formation by aberrant activation of the HER2/Ras pathway.( 59 ) Taken together, ERK and PI3K/Akt signaling pathways are involved in many types of cancer, suggesting that several components of these signaling pathways could be useful therapeutic targets in the treatment of human cancers.
ERK signaling regulators and cancer
Several regulators of ERK signaling, such as Sprouty, Spred and Sef, could control the duration, magnitude and/or subcellular compartmentalization of ERK activity. Therefore, these regulators may be involved in regulating cell cycle progression or carcinogensis. Indeed, recent reports have shown a correlation between expression of these regulators and cancer (Table 1).( 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 ) The expression of Sprouty isoforms or Sef is found to be downregulated in several cancers, including breast, prostate and liver cancers.( 61 , 62 , 63 , 64 , 65 , 66 ) Generally, the expression levels of Sprouty isoforms and Sef are positively regulated by ERK signaling, and thus these regulators act as a negative feedback regulator. In these cancers, expression of these factors is downregulated by several mechanisms, such as loss of heterozygosity, methylation of their promoter sites, and other unknown mechanisms. Moreover, Lo et al. reported that Sprouty2 YF, a dominant‐negative form of Sprouty2, induces enhanced cell proliferation, and injection of cells expressing Sprouty2 YF into nude mice causes a significantly larger tumor mass.( 62 ) On the other hand, two groups reported that expression of Sprouty2 is upregulated in melanoma cell lines with a B‐Raf V600E mutation or an N‐Ras activated mutation.( 67 , 68 ) Interestingly, Sprouty2 has an inhibitory effect on ERK signaling in melanoma cell lines with wild‐type B‐Raf, but not with B‐Raf mutations, especially exon 15 B‐Raf mutations such as V600E. In addition, it was shown that Sprouty2 and Sprouty4 bind to wild‐type B‐Raf, but not exon 15 B‐Raf mutants, although there is no evidence that binding of Sprouty to B‐Raf is required for the inhibitory activity of Sprouty in ERK signaling. Nevertheless, it is possible that, in these melanoma cells, increased levels of Sprouty fail to inhibit ERK signaling, as Sprouty proteins inhibit ERK signaling upstream of Ras. These reports point to the possibility that ERK signaling regulators, such as Sprouty and Sef, are involved in malignant transformation and cancer.
Table 1.
ERK signaling regulators and cancer
| Regulator | Action point | Cancer type | Gene expression | Reference |
|---|---|---|---|---|
| Sprouty1 | Grb2 | Breast, prostate | Downregulated | 60, 61 |
| Sprouty2 | Grb2, Raf, c‐Cbl | Breast, prostate, liver | Downregulated | 61, 62, 63 |
| Melanoma | Upregulated | 66, 67 | ||
| Sprouty4 | Sos, Raf | Prostate | Downregulated | 64 |
| Spred | Raf | – | – | – |
| Sef | FGF receptor, MEK/ERK | Prostate | Downregulated | 65 |
Conclusions
The ERK MAP kinase signaling pathway plays a pivotal role in various cellular responses, including cell proliferation, cell differentiation and cell survival. In addition, Ras/ERK signaling‐related proteins, such as Sprouty and Sef, are also important for the regulation of these cellular responses. In fact, deregulation of ERK signaling leads to inappropriate responses, perhaps through inappropriate gene expression. Further elucidation of the action mechanisms of ERK signaling regulators, their control mechanism, and their expression patterns will provide new insights into the signal transduction mechanisms underlying cell cycle progression and malignant transformation.
Acknowledgments
We thank members of our laboratory for their helpful discussion. The studies from our laboratory were supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to EN) and by a JSPS Research Fellowship for Young Scientists (to ST).
References
- 1. Sturgill TW, Wu J. Recent progress in characterization of protein kinase cascades for phosphorylation of ribosomal protein S6. Biochim Biophys Acta 1991; 1092: 350–7. [DOI] [PubMed] [Google Scholar]
- 2. Nishida E, Gotoh Y. The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem Sci 1993; 18: 128–31. [DOI] [PubMed] [Google Scholar]
- 3. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal‐regulated kinase activation. Cell 1995; 80: 179–85. [DOI] [PubMed] [Google Scholar]
- 4. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res 1998; 74: 49–139. [DOI] [PubMed] [Google Scholar]
- 5. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37–40. [DOI] [PubMed] [Google Scholar]
- 6. Pearson G, Robinson F, Beers Gibson T et al. Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22: 153–83. [DOI] [PubMed] [Google Scholar]
- 7. Pouyssegur J, Lenormand P. Fidelity and spatio‐temporal control in MAP kinase (ERKs) signalling. Eur J Biochem 2003; 270: 3291–9. [DOI] [PubMed] [Google Scholar]
- 8. Ebisuya M, Kondoh K, Nishida E. The duration, magnitude and compartmentalization of ERK MAP kinase activity: mechanisms for providing signaling specificity. J Cell Sci 2005; 118: 2997–3002. [DOI] [PubMed] [Google Scholar]
- 9. Sears RC, Nevins JR. Signaling networks that link cell proliferation and cell fate. J Biol Chem 2002; 277: 11 617–20. [DOI] [PubMed] [Google Scholar]
- 10. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3: 11–22. [DOI] [PubMed] [Google Scholar]
- 11. Coleman ML, Marshall CJ, Olson MF. RAS and RHO GTPases in G1‐phase cell‐cycle regulation. Nat Rev Mol Cell Biol 2004; 5: 355–66. [DOI] [PubMed] [Google Scholar]
- 12. Massague J. G1 cell‐cycle control and cancer. Nature 2004; 432: 298–306. [DOI] [PubMed] [Google Scholar]
- 13. Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell 1998; 92: 253–63. [DOI] [PubMed] [Google Scholar]
- 14. Wakioka T, Sasaki A, Kato R et al. Spred is a Sprouty‐related suppressor of Ras signalling. Nature 2001; 412: 647–51. [DOI] [PubMed] [Google Scholar]
- 15. Furthauer M, Lin W, Ang SL, Thisse B, Thisse C. Sef is a feedback‐induced antagonist of Ras/MAPK‐mediated FGF signalling. Nat Cell Biol 2002; 4: 170–4. [DOI] [PubMed] [Google Scholar]
- 16. Tsang M, Friesel R, Kudoh T, Dawid IB. Identification of Sef, a novel modulator of FGF signalling. Nat Cell Biol 2002; 4: 165–9. [DOI] [PubMed] [Google Scholar]
- 17. Kim HJ, Bar‐Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol 2004; 5: 441–50. [DOI] [PubMed] [Google Scholar]
- 18. Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol 2002; 4: 850–8. [DOI] [PubMed] [Google Scholar]
- 19. Ozaki K, Miyazaki S, Tanimura S, Kohno M. Efficient suppression of FGF‐2‐induced ERK activation by the cooperative interaction among mammalian Sprouty isoforms. J Cell Sci 2005; 118: 5861–71. [DOI] [PubMed] [Google Scholar]
- 20. Sasaki A, Taketomi T, Kato R et al. Mammalian Sprouty4 suppresses Ras‐independent ERK activation by binding to Raf1. Nat Cell Biol 2003; 5: 427–32. [DOI] [PubMed] [Google Scholar]
- 21. Hanafusa H, Torii S, Yasunaga T, Matsumoto K, Nishida E. Shp2, an SH2‐containing protein‐tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J Biol Chem 2004; 279: 22 992–5. [DOI] [PubMed] [Google Scholar]
- 22. Mason JM, Morrison DJ, Bassit B et al. Tyrosine phosphorylation of Sprouty proteins regulates their ability to inhibit growth factor signaling: a dual feedback loop. Mol Biol Cell 2004; 15: 2176–88. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Jarvis LA, Toering SJ, Simon MA, Krasnow MA, Smith‐Bolton RK. Sprouty proteins are in vivo targets of Corkscrew/SHP‐2 tyrosine phosphatases. Development 2006; 133: 1133–42. [DOI] [PubMed] [Google Scholar]
- 24. Fong CW, Leong HF, Wong ES, Lim J, Yusoff P, Guy GR. Tyrosine phosphorylation of Sprouty2 enhances its interaction with c‐Cbl and is crucial for its function. J Biol Chem 2003; 278: 33 456–64. [DOI] [PubMed] [Google Scholar]
- 25. Hall AB, Jura N, DaSilva J, Jang YJ, Gong D, Bar‐Sagi D. hSpry2 is targeted to the ubiquitin‐dependent proteasome pathway by c‐Cbl. Curr Biol 2003; 13: 308–14. [DOI] [PubMed] [Google Scholar]
- 26. Rubin C, Litvak V, Medvedovsky H, Zwang Y, Lev S, Yarden Y. Sprouty fine‐tunes EGF signaling through interlinked positive and negative feedback loops. Curr Biol 2003; 13: 297–307. [DOI] [PubMed] [Google Scholar]
- 27. DaSilva J, Xu L, Kim HJ, Miller WT, Bar‐Sagi D. Regulation of sprouty stability by Mnk1‐dependent phosphorylation. Mol Cell Biol 2006; 26: 1898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sasaki A, Taketomi T, Wakioka T, Kato R, Yoshimura A. Identification of a dominant negative mutant of Sprouty that potentiates fibroblast growth factor‐ but not epidermal growth factor‐induced ERK activation. J Biol Chem 2001; 276: 36 804–8. [DOI] [PubMed] [Google Scholar]
- 29. Torii S, Nakayama K, Yamamoto T, Nishida E. Regulatory mechanisms and function of ERK MAP kinases. J Biochem (Tokyo) 2004; 136: 557–61. [DOI] [PubMed] [Google Scholar]
- 30. Torii S, Kusakabe M, Yamamoto T, Maekawa M, Nishida E. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev Cell 2004; 7: 33–44. [DOI] [PubMed] [Google Scholar]
- 31. Lavoie JN, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 1996; 271: 20 608–16. [DOI] [PubMed] [Google Scholar]
- 32. Kerkhoff E, Rapp UR. Cell cycle targets of Ras/Raf signalling. Oncogene 1998; 17: 1457–62. [DOI] [PubMed] [Google Scholar]
- 33. Dyson N. The regulation of E2F by pRB‐family proteins. Genes Dev 1998; 12: 2245–62. [DOI] [PubMed] [Google Scholar]
- 34. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev 1999; 13: 1501–12. [DOI] [PubMed] [Google Scholar]
- 35. Meloche S, Seuwen K, Pages G, Pouyssegur J. Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol Endocrinol 1992; 6: 845–54. [DOI] [PubMed] [Google Scholar]
- 36. Cook SJ, McCormick F. Kinetic and biochemical correlation between sustained p44ERK1 (44 kDa extracellular signal‐regulated kinase 1) activation and lysophosphatidic acid‐stimulated DNA synthesis in Rat‐1 cells. Biochem J 1996; 320: 237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays 2000; 22: 818–26. [DOI] [PubMed] [Google Scholar]
- 38. Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates anti‐proliferative genes throughout G1 phase to allow cell cycle progression. Curr Biol (forthcoming). [DOI] [PubMed] [Google Scholar]
- 39. Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nature Cell Biol 2002; 4: 556–64. [DOI] [PubMed] [Google Scholar]
- 40. Murphy LO, MacKeigan JP, Blenis J. A network of immediate early gene products propagates subtle differences in mitogen‐activated protein kinase signal amplitude and duration. Mol Cell Biol 2004; 24: 144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular‐signal‐regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J 1997; 326: 61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Balmanno K, Cook SJ. Sustained MAP kinase activation is required for the expression of cyclin D1, 21Cip1 and a subset of AP‐1 proteins in CCL39 cells. Oncogene 1999; 18: 3085–97. [DOI] [PubMed] [Google Scholar]
- 43. Pfarr CM, Mechta F, Spyrou G, Lallemand D, Carillo S, Yaniv M. Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell 1994; 76: 747–60. [DOI] [PubMed] [Google Scholar]
- 44. Maekawa M, Nishida E, Tanoue T. Identification of the anti‐proliferative protein Tob as a MAPK substrate. J Biol Chem 2002; 277: 37 783–7. [DOI] [PubMed] [Google Scholar]
- 45. Suzuki T, K‐Tsuzuki J, Ajima R, Nakamura T, Yoshida Y, Yamamoto T. Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras‐mediated cell proliferation and transformation. Genes Dev 2002; 16: 1356–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshida Y, Nakamura T, Komoda M et al. Mice lacking a transcriptional corepressor Tob are predisposed to cancer. Genes Dev 2003; 17: 1201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weitzman JB, Fiette L, Matsuo K, Yaniv M. JunD protects cells from p53‐dependent senescence and apoptosis. Mol Cell 2000; 6: 1109–19. [DOI] [PubMed] [Google Scholar]
- 48. Kozar K, Ciemerych MA, Rebel VI et al. Mouse development and cell proliferation in the absence of D‐cyclins. Cell 2004; 118: 477–91. [DOI] [PubMed] [Google Scholar]
- 49. Malumbres M, Sotillo R, Santamaria D et al. Mammalian cells cycle without the D‐type cyclin‐dependent kinases Cdk4 and Cdk6. Cell 2004; 118: 493–504. [DOI] [PubMed] [Google Scholar]
- 50. Dekanty A, Sauane M, Cadenas B et al. Leukemia inhibitory factor induces DNA synthesis in Swiss mouse 3T3 cells independently of cyclin D1 expression through a mechanism involving MEK/ERK1/2 activation. J Biol Chem 2006; 281: 6136–43. [DOI] [PubMed] [Google Scholar]
- 51. Vivanco I, Sawyers CL. The phosphatidylinositol 3‐kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2: 489–501. [DOI] [PubMed] [Google Scholar]
- 52. Wymann MP, Marone R. Phosphoinositide 3‐kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol 2005; 17: 141–9. [DOI] [PubMed] [Google Scholar]
- 53. Sewing A, Wiseman B, Lloyd AC, Land H. High‐intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol 1997; 17: 5588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf‐induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol 1997; 17: 5598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coleman ML, Marshall CJ, Olson MF. Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1‐imposed block in proteasome‐mediated degradation. EMBO J 2003; 22: 2036–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mirza AM, Gysin S, Malek N, Nakayama K, Roberts JM, McMahon M. Cooperative regulation of the cell division cycle by the protein kinases RAF and AKT. Mol Cell Biol 2004; 24: 10 868–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hayashi H, Matsuzaki O, Muramatsu S et al. Centaurin‐alpha1 is a phosphatidylinositol 3‐kinase‐dependent activator of ERK1/2 mitogen‐activated protein kinases. J Biol Chem 2006; 281: 1332–7. [DOI] [PubMed] [Google Scholar]
- 58. Solit DB, Garraway LA, Pratilas CA et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006; 439: 358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001; 411: 1017–21. [DOI] [PubMed] [Google Scholar]
- 60. Lo TL, Fong CW, Yusoff P et al. Sprouty and cancer: The first terms report. Cancer Lett 2006; (forthcoming). [DOI] [PubMed] [Google Scholar]
- 61. Kwabi‐Addo B, Wang J, Erdem H et al. The expression of Sprouty1, an inhibitor of fibroblast growth factor signal transduction, is decreased in human prostate cancer. Cancer Res 2004; 64: 4728–35. [DOI] [PubMed] [Google Scholar]
- 62. Lo TL, Yusoff P, Fong CW et al. The ras/mitogen‐activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty 1 and sprouty 2 are deregulated in breast cancer. Cancer Res 2004; 64: 6127–36. [DOI] [PubMed] [Google Scholar]
- 63. McKie AB, Douglas DA, Olijslagers S et al. Epigenetic inactivation of the human sprouty2 (hSPRY2) homologue in prostate cancer. Oncogene 2005; 24: 2166–74. [DOI] [PubMed] [Google Scholar]
- 64. Fong CW, Chua MS, McKie AB et al. Sprouty2, an inhibitor of mitogen‐activated protein kinase signaling, is down‐regulated in hepatocellular carcinoma. Cancer Res 2006; 66: 2048–58. [DOI] [PubMed] [Google Scholar]
- 65. Wang J, Thompson B, Ren C, Ittmann M, Kwabi‐Addo B. Sprouty4, a suppressor of tumor cell motility, is downregulated by DNA methylation in human prostate cancer. Prostate 2006; 66: 613–24. [DOI] [PubMed] [Google Scholar]
- 66. Darby S, Sahadevan K, Khan MM, Robson CN, Leung HY, Gnanapragasam VJ. Loss of Sef (similar expression to FGF) expression is associated with high grade and metastatic prostate cancer. Oncogene 2006; (forthcoming). [DOI] [PubMed] [Google Scholar]
- 67. Tsavachidou D, Coleman ML, Athanasiadis G et al. SPRY2 is an inhibitor of the ras/extracellular signal‐regulated kinase pathway in melanocytes and melanoma cells with wild‐type BRAF but not with the V599E mutant. Cancer Res 2004; 64: 5556–9. [DOI] [PubMed] [Google Scholar]
- 68. Bloethner S, Chen B, Hemminki K et al. Effect of common B‐RAF and N‐RAS mutations on global gene expression in melanoma cell lines. Carcinogenesis 2005; 26: 1224–32. [DOI] [PubMed] [Google Scholar]