

Abstract
Aberrant reactivation of hedgehog (Hh) signaling has been described in a wide variety of human cancers and in cancer stem cells. However, the contribution of Hh signaling to leukemic cell regulation has remained unclear. In this study, we assessed the possibility that Hh pathway activation contributes to the survival and drug resistance of cluster of differentiation (CD)34+ leukemia cells. Hh signaling in leukemic cell lines and primary leukemic cells was screened by reverse transcription – polymerase chain reaction (RT‐PCR) and a Hh signaling reporter assay. We found that Hh signaling is active in several human acute myeloid leukemia (AML) cells, especially primary CD34+ leukemic cells and cytokine‐responsive CD34+ cell lines such as Kasumi‐1, Kasumi‐3 and TF‐1. These CD34+ cells express the downstream effectors glioma‐associated oncogene homolog (GLI)1 or GLI2, indicative of active Hh signaling. Moreover, inhibition of Hh signaling with the naturally derived Smoothened antagonist cyclopamine, endogenous Hh inhibitor hedgehog‐interacting protein or anti‐hedgehog neutralizing antibody induced apoptosis after 48 h of exposure, although these CD34+ cell lines exhibited resistance to cytarabine (Ara‐C). In contrast, cyclopamine failed to affect growth or survival in U937 and HL‐60 cell lines that lack expression of Hh receptor components, confirming that the effect of Hh inhibition is specific. Furthermore, combination with 10 µM cyclopamine significantly reduced drug resistance of CD34+ cell lines and primary CD34+ leukemic cells to Ara‐C. These results suggest that aberrant Hh pathway activation is a feature of some CD34+ myeloid leukemic cells and Hh inhibitors may have a therapeutic role in the treatment of AML. (Cancer Sci 2009; 100: 948–955)
The hedgehog (Hh) protein family is a group of secreted intercellular signaling molecules. Secreted Hh protein binds to a receptor, c, expressed on the plasma membrane of target cells. This interaction triggers the dissociation of Ptc from Smoothened (Smo), which is a constitutively active signal transducer.( 1 ) Hh signals are eventually transmitted to the transcription factors glioma‐associated oncogene homolog (GLI)1, GLI2 and GLI3, whose downstream signaling leads to entry of the cells into the cell cycle,( 2 ) inhibition of apoptosis,( 3 ) maintenance of self‐renewal of stem cells in various tissues,( 4 , 5 ) regulation of tissue stem cell differentiation,( 6 , 7 ) and modulation of tissue polarity.( 8 ) Aberrant reactivation of Hh signaling has been described in a wide variety of human cancers,( 9 , 10 , 11 , 12 , 13 , 14 , 15 ) and its role in normal stem cells suggest that pathway dysregulation contributes to oncogenesis and influences cell fate decisions in cancer stem cells.
In hematopoiesis, it was shown that Hh family members play an important role in regulation of stem/progenitor cell expansion in vitro and in vivo.( 16 ) In particular, we previously showed that Indian hedgehog (Ihh), but not Sonic hedgehog (Shh), and its receptor molecules are expressed in cord blood (CB) cluster of differentiation (CD)34+ cells. Further, human stromal cells and Ihh regulate proliferation of hematopoietic progenitor cells and short‐tem myeloid‐ and lymphoid‐repopulating cells.( 17 ) In contrast, the Hh inhibitor cyclopamine blocked differentiation of hematopoietic progenitor cells, especially erythroid progenitor cells.( 18 ) Furthermore, we have recently demonstrated that over‐expression of Ihh protein in mice inhibited proliferation and differentiation of splenic B lymphocytes and thymic T lymphocytes during BM hematopoiesis in vivo.( 19 ) Collectively, Ihh plays critical roles in regulation of differentiation and proliferation of myeloid‐ and lymphoid stem/progenitor cells.
Previously, the expression of Hh receptor Ptc, signal transducer Smo and transcription factor GLI1 was examined in several leukemic cell lines. For example, Ptc and Smo were expressed in Jurkat cells,( 18 ) and Shh and GLI1 were expressed in human promyelocytic leukemia (HL‐60) and KG‐1 cells.( 20 ) Moreover, these components of Hh signaling were detectable in some primary acute leukemic cells, but not acute lymphoblastic leukemic cells.( 20 ) However, it is uncertain whether the Hh signaling pathway was functional or not in previous studies. In addition, the role of Hh signaling in leukemic stem/progenitor cells, some of which expressed CD34 antigen,( 21 , 22 ) remained unclear.
In the present study, we evaluated the function of Hh signaling in acute leukemic cells including CD34+ cell lines using both reverse transcription – polymerase chain reaction (RT‐PCR) for Hh pathway components and a GLI‐responsive reporter assay. Furthermore, to gain insight into the role of Hh signaling in acute leukemic cell lines, we blocked the Hh signaling pathway with three types of Hh signaling inhibitors. We show here that Ihh signaling is essential for survival and drug resistance of cytokine‐responsive CD34+ leukemic cells.
Materials and Methods
Cell lines. Human myeloid leukemic cell lines NB4, HL60, U937 and KG1 were cultured in Roswell Park Memorial Institute (RPMI)1640 containing 10% heat‐inactivated fetal calf serum (FCS) (Gibco BRL, Rockville, MD, US), 2 mM/L L‐glutamine, 0.1% penicillin (100 U/mL) and streptomycin (100 mg/mL). CD34+ leukemic cell lines, such as Kasumi‐1, Kasumi‐3 and TF‐1 (American Type Culture Collection), were cultured in RPMI1640 containing 20% heat‐inactivated FCS, 2 mM/L L‐glutamine, 1 mM pyruvate, 0.1% penicillin (100 U/mL) and streptomycin (100 mg/mL). For long‐term culture of TF‐1, 10 U/mL interleukin‐3 (IL‐3, R & D Systems, Minneapolis, MA, US) was added to the complete medium described above.
Separation of primary CD34+ leukemic cells. Primary CD34+ leukemic cells were obtained from patients suffering acute myeloid leukemia (AML) by BM aspiration after obtaining informed consent. The study was approved by the Sapporo Medical University institutional review board. Low‐density (<1.077 g/mL) mononuclear cells were separated by Histopaque®‐1077 (Sigma‐Aldrich, Tokyo, Japan). CD34+ cell purification was conducted by positive selection using a MACS Direct CD34 Progenitor Cell Isolation Kit (Miltenyi Biotech, Bergish‐Gladbach, Germany), and the purified cells were cryopreserved. Over 90% of the enriched cells were CD34+ as confirmed by flow cytometric analysis. The thawed cells were washed twice and viability was determined by trypan blue staining. Only samples containing more than 95% viable cells were used in further studies.
Hedgehog antagonists and chemical reagents. Anti‐Hh monoclonal antibody 5E1, which neutralizes the Shh and Ihh activity, was prepared from a hybridoma, which was purchased from the Iowa Hybridoma Bank, as previously described.( 17 ) Recombinant murine Hh‐interacting protein (Hip) was obtained from R & D Systems. Chemical hedgehog inhibitor cyclopamine or control compound tomatidine was purchased from Toronto Research Chemicals Inc. (North York, ON, Canada) or BIOMOL Research Laboratories, Inc. (Plymouth Meeting, PA, US), respectively. The structure and properties of tomatidine are similar to cyclopamine, but tomatidine does not inhibit Hh signaling. Cytarabine (Ara‐C; Nippon Shinyaku, Kyoto, Japan) was dissolved in sterile double‐distilled H2O before use.
Analysis of expression of Ihh and related genes by RT‐PCR. For RT‐PCR analysis, total RNA was prepared from cells using the QIAGEN RNeasy kit. Total RNA (1 µg) was reverse‐transcribed by SuperScript™II (Invitrogen, Tokyo, Japan) and amplified using the Advantage™ GC 2 Polymerase Mix (Clontech, Tokyo, Japan) with primers specific for Ihh (5′‐TGCGGGCCGGGTCGGGTG GTG‐3′ and 5′‐GCCGCCCGTCTTGGCTGC‐3′), Ptc (5′‐CTGTT GGCATAGGAGTGGAGTTCACC‐3′ and 5′‐CTGCTGGGCCTC GTAGTGCCGAAGC‐3′), Smo (5′‐CAGAACATCAAGTTCAACA GTTCAGGC‐3′ and 5′‐ATAGGTGAGGACCACAAACCAAACC ACACC‐3′) or human α‐actin (5′‐GCTCGTCGTCGACAACGG CTC‐3′ and 5′‐CAAACATGATCTGGGTCATCTTCTC‐3′) as previously reported.( 17 ) The primer pairs for GLI1 (5′‐CTCCCGAA GGACAGGTATGTAAC‐3′ and 5′‐CCCTACTCTTTAGGCACTA GAGTTG‐3′) and GLI2 (5′‐CGAGAAACCCTACATCTGCAAGA‐3′ and 5′‐GTGGACCGTTTTCACATGCTT‐3′) were synthesized according to a previous report.( 23 ) PCR amplification was carried out using 35 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 60 s. The PCR products were separated on a 2% or 4% (for GLI2) agarose gel.
Activity of hedgehog signaling in leukemic cell lines. The lhh reporter plasmid (TK‐6GBS‐Luc) expressed luciferase under the control of the thymidine kinase (TK) promoter and was a gift of Dr Jun Aruga (Developmental Neurobiology Laboratory, Brain Science Institute, Riken, Japan).( 24 ) The plasmid contained six tandem copies of the GLI‐target site, 5′‐GCGTGGACCACCCAAGACGAAATT‐3′,( 25 ) inserted upstream of the TK promoter. The parental luciferase reporter plasmid was used as a control (TK‐0GBS‐Luc). For analysis of Ihh‐N activity in leukemic cells, cells were exposed to 10 µg/mL 5E1 antibody or isotype control antibody for 12 h. Subsequently, cells were transfected with 200 ng of the luciferase reporters, TK‐0GBS‐Luc or TK‐6GBS‐Luc, together with 2.5 ng of phRL‐TK as an internal standard (renilla luciferase) using DMRIE‐C (Invitrogen, Carlsbad, CA, US). After transfected cells were incubated at 37°C and 5% CO2 for 12 h in the presence of 10 µg/mL 5E1 antibody or isotype control antibody, luciferase activities were measured according to the manufacturer's recommendation (Promega, Madison, WI, US) using a lumat LB 9507 luminometer (Berthold Technology, Bad Wildbad, Germany). Firefly luciferase activity was normalized to renilla luciferase activity. The relative fold‐activation was presented as the ratio of the normalized value to the activity observed in the cells transduced with the empty vector.
Immunofluorescent staining for assessment of cell morphology. Cells were cultured in complete medium in the presence of 10 µM tomatidine or cyclopamine. For nuclear staining, living cell staining solution SYTO16 (Invitrogen) was added to complete medium at a final concentration of 5 µM. After 5 min incubation at 37°C and 5% CO2, cells were examined using a Biozero BZ‐8000 laser scanning microscope (KEYENCE Laboratories, Osaka, Japan).
Drug cytotoxic assay. For drug cytotoxic assays, leukemic cells were washed once, resuspended in each adequate complete medium, and then 4 × 104 cells were added to each well of a 96‐well plate. To evaluate drug resistance against Ara‐C, 10−2–10−6 µM of Ara‐C or vehicle control was added to each well and incubated for 48 h. Alternatively, to assess the effect of Hh signaling inhibitors, 0–100 µM cyclopamine or control compound tomatidine, 0–100 µg/mL murine Hip, or 0–100 µg/mL anti‐Hh neutralizing antibody 5E1 was added to each well and incubated for 48 h. Additionally, the effect of 10 µM cyclopamine on drug resistance of leukemic cells against Ara‐C was also evaluated. The surviving cells were assessed by Annexin V‐FITC Apoptosis Detection Kit (Medical & Biological Laboratories, Tokyo, Japan) and Premix WST‐1 assay Cell Proliferation Assay System (Takara, Tokyo, Japan). The WST‐1 assay is based on mitochondrial conversion of WST‐1 to yellowish formazan, being indicative of the number of viable cells.( 26 ) The number of viable cells was evaluated by absorbance OD450 nm (Abs) using Model 680 microplate reader (Bio‐Rad Laboratories, Tokyo, Japan).
Flowcytometric analysis of intracellular Bcl‐2. Aliquots of leukemic cells were washed with 0.1% NaN3 1 × phosphate buffered saline (PBS). One million cells were fixed with BD Cytofix Fixation buffer containing 4% paraform aldehyde (BD Biosciences, Tokyo, Japan) and incubate for 20 min on ice. After washing with 0.1%NaN3 1 × PBS, cells were permeated by Perm Buffer I containing saponin (BD Biosciences) and were labeled with antihuman Bcl‐2 fluoroscein isothiocyanate (FITC) (DAKO, Tokyo, Japan) or an isotype control. The cells were analyzed by flow cytometry using a FACSCalibur. Cells were gated based on forward and side light scatter to exclude debris.
Statistical analysis. Each data set was first evaluated for normality of distribution by the Komolgorov‐Smirnov test to decide whether a non‐parametric rank‐based analysis or a parametric analysis should be used. Two groups were compared by either the Wilcoxon signed‐rank test or the Student's t‐test (two‐tailed test).
Results
Screening expression of Hh and its receptors in myeloid leukemic cells and BM CD34+ cells by RT‐PCR. We previously demonstrated that Ihh and receptor components were highly expressed in CB CD34+ cells.( 17 ) Hence, we first screened the expression of Ihh, Ptc, Smo, GLI1 and GLI2 in acute leukemic cells including CD34+ leukemic cell lines (Fig. 1). Ihh was expressed in most acute leukemic cell lines except for NB4. Interestingly, receptor components, including Ptc and Smo, were only detected in three out of four CD34+ cell lines (Kasumi‐1, Kasumi‐3 and TF‐1), which are known to be responsive to hematopoietic growth factors such as interleukin‐3, granulocyte‐macrophage colony‐stimulating factor (GM‐SCF) and erythropoietin.( 27 , 28 , 29 , 30 ) Regarding transcription factors, GLI1 was detected in most acute leukemic cell lines except for KG1, while GLI2 was detectable in all cell lines. It has been reported that GLI1 and GLI2 are the prime transcriptional effectors involved, and constitutive activation of at least one of them is of critical importance for cancer development.( 31 , 32 , 33 ) We further examined the expression of these Hh‐related molecules in primary CD34+ leukemic cells and normal BM CD34+ cells. Consequently, Ihh, receptor components such as Ptc and Smo, transcription factors such as GLI1 and GLI2 were expressed in primary CD34+ leukemic and normal BM CD34+ cells. These findings raise the possibility that Ihh signaling could transmit signals to cytokine‐responsive CD34+ leukemic cells and normal BM CD34+ cells.
Figure 1.
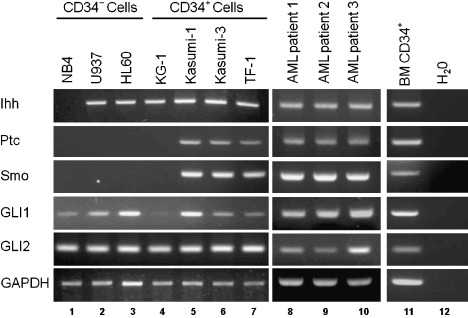
The mRNA expression of Ihh and receptor molecules. The expression of Indian hedgehog (Ihh), Patched (Ptc), Smoothened (Smo), glioma‐associated oncogene homolog (GLI)1 and GLI2 mRNA in leukemic cell lines and primary CD34+ leukemic cells was analyzed by reverse transcription – polymerase chain reaction. The expression of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as an internal standard. Lane 1, NB4; Lane 2, U937; Lane 3, HL60; Lane 4, KG1; Lane 5, Kasumi‐1; Lane 6, Kasumi‐3; Lane 7, TF‐1; Lane 8–10, primary CD34+ leukemic cells derived from three acute myeloid leukemia (AML) patients; Lane 11, bone marrow (BM) CD34+ cells; Lane 12, H2O.
Hh‐responsive luciferase assay in CD34+ cell lines in the presence or absence of cyclopamine. We next examined whether Hh signaling transmits through the transcription factor GLI using an activated GLI‐responsive luciferase assay. Leukemic cell lines U937, HL60, KG1, Kasumi‐1, Kasumi‐3 and TF‐1 were pretreated with 10 µg/mL antihedgehog neutralizing Ab 5E1 or isotype control antibody. Subsequently, GLI‐responsive reporter plasmid (TK‐6GBS‐Luc) or non‐responsive control plasmid (TK‐0GBS‐Luc) was transduced into these cells. Luciferase activity was measured 12 h after culturing in the presence of 10 µg/mL 5E1 antibody or isotype control antibody. As shown in Fig. 2, TK‐0GBS‐Luc‐transduced cells, Hh receptor negative cell lines U937, HL60 or KG1 did not exhibit any luciferase activity. Expectedly, TK‐6GBS‐Luc‐transduced Kasumi‐1, Kasumi‐3 and TF‐1 cells showed high luciferase activity, indicating that transcription factor GLI was activated in these cell lines. Furthermore, luciferase activity of these cells was significantly reduced in the presence of anti‐Hh neutralizing antibody 5E1. These results indicate that Ihh signaling transmits to these cytokine‐responsive CD34+ leukemic cell lines in an autocrine manner.
Figure 2.
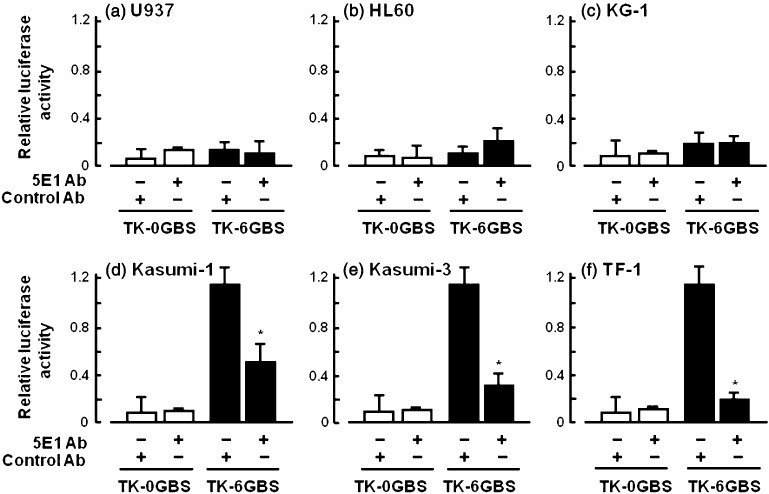
Endogenous hedgehog (Hh) signaling in leukemic cell lines was assessed by a Hh‐responsive luciferase assay. U937 (a), HL60 (b), KG1 (c), Kasumi‐1 (d), Kasumi‐3 (e) and TF‐1 (f) were treated with 10 µg/mL anti‐Hh antibody 5E1 or isotype control antibody for 12 h. Subsequently, cells were transfected with Hh‐responsive luciferase reporter plasmid (TK‐6GBS) or Hh‐non‐responsive control plasmid (TK‐0GBS). Transfected cells were further incubated in the presence of 10 µg/mL anti‐Hh antibody 5E1 or isotype control antibody for 12 h. Luciferase activity was normalized to the activity of an internal control (renilla luciferase). *P < 0.05, isotype control vs. 5E1 (Student's t‐test, two‐tailed).
The survival of CD34+ leukemic cell lines in the presence of hedgehog inhibitors and neutralized antibody. We previously demonstrated that Ihh contributes to the proliferation of CB CD34+ stem/progenitor cells.( 17 ) However, the roles of Hh signaling in CD34+ leukemic cell lines are uncertain. To gain insight into the effect of Hh signaling, CD34+ cell lines were exposed to the chemical inhibitor cyclopamine, endogenous Hh inhibitor HIP or anti‐Hh neutralizing antibody. Survival of CD34+ leukemic cells was analyzed by mitochondrial conversion of WST‐1 to yellowish formazan (Fig. 3). When cyclopamine was used, percent survival of Kasumi‐1, Kasumi‐3 and TF‐1 decreased in a dose‐dependent manner, although no changes were observed upon exposure of control compound tomatidine. In clear contrast, percent survival of KG‐1 was not affected by cyclopamine, probably because our KG‐1 does not express Smo, which is a target molecule of cyclopamine. These results suggest that the growth or survival of Kasumi‐1, Kasumi‐3 and TF‐1 cells depend upon the Hh signaling pathway. Subsequently, we investigated the effect of neutralization of Hh protein in supernatant by hedgehog‐interacting protein (HIP) and anti‐Ihh 5E1 antibody. As a result, percent survival of Kasumi‐1, Kasumi‐3 and TF‐1 was similarly reduced. These results suggest that Ihh released from these CD34+ leukemic cells transmits via receptor molecule Smo and that Ihh signaling plays an important role in the growth or survival of these leukemic cells.
Figure 3.

Survival of CD34+ leukemia cell lines in the presence of hedgehog (Hh) signaling inhibitors and neutralizing antibody 5E1. Survival of CD34+ leukemic cells was analyzed by mitochondrial conversion of WST‐1 to yellowish formazan. Each cell line was exposed to the chemical hedgehog signaling inhibitor cyclopamine (closed circle), control compound tomatidine (open circle) (a), Hh‐interacting protein (Hip) (b) or Hh‐neutralizing antibody 5E1 (c). The X‐axis indicates the dose of cyclopamine, tomatidine, Hip or 5E1 and the Y‐axis indicates percent survival. (i) Kasumi‐1 (LD50: cyclopamine, 33.3 µM) (ii) Kasumi‐3 (LD50: cyclopamine, 2.1 µM) (iii) TF‐1 (LD50: cyclopamine, 10.1 µM) and (iv) KG‐1 (LD50: cyclopamine, N.D.). (v) Kasumi‐1 (LD50: Hip, 8.3 µg/mL) (vi) Kasumi‐3 (LD50: Hip, 2.5 µg/mL) (vii) TF‐1 (LD50: Hip, 1.2 µg/mL) and (viii) KG‐1 (LD50: Hip, N.D.). (ix) Kasumi‐1 (LD50: 5E1, N.D.) (x) Kasumi‐3 (LD50: 5E1, 36.1 µg/mL) (xi) TF‐1 (LD50: 5E1, N.D.) and (xii) KG‐1 (LD50: 5E1, N.D.). Data represent three independent experiments, each done in triplicate. *P < 0.05, tomatidine versus cyclopamine (Student's t‐test, two‐tailed). † P < 0.05, vehicle vs. Hip or 5E1. N.D. = not determined.
Analysis of annexinV+/PI+ leukemic cells after cyclopamine treatment. We next observed morphological changes of CD34+ TF‐1 cells to assess the possibility of apoptosis after exposure to Hh inhibitor cyclopamine. Although no morphological or nuclear changes were observed after treatment with 10 µM tomatidine, 10 µM cyclopamine treatment drastically changed the morphology and nuclear fragmentation of TF‐1 (Fig. 4a). Hence, we subsequently conducted annexinV/PI assays for Hh receptor‐negative cells (U937, HL60 and KG‐1) and Hh receptor‐positive cells (Kasumi‐1, Kasumi‐3 and TF‐1) after exposure to 10 µM cyclopamine. Regarding Hh receptor‐negative cells, no apparent changes were observed after cyclopamine treatment (Fig. 4b). In clear contrast, annexinV+ cells of Kasumi‐1, Kasumi‐3 and TF‐1 increased after 12 h of cyclopamine treatment. The percentage of annexinV and PI+ cells further increased after 24 h. These results indicate that the hedgehog inhibitor cyclopamine blocked the autocrine loop of Ihh signaling in these CD34+ leukemic cells, thereby inducing apoptosis.
Figure 4.
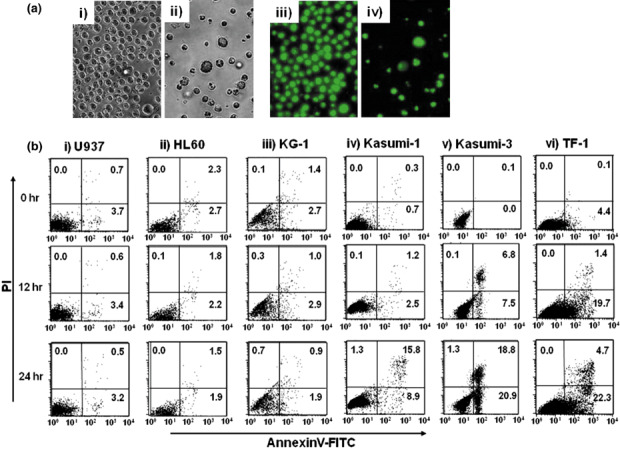
Morphological changes and annexin assay of hedgehog (Hh) receptor‐expressing CD34+ leukemic cell lines exposed to Hh inhibitors. (A) Morphological and nuclear findings of CD34+ cell line TF‐1 after cyclopamine treatment. Left side panels indicate morphological findings of cells and right side panels indicate specific nuclear staining by SYTO16 after 24‐h exposure to 10 µM tomatidine (i, iii) or 10 µM cyclopamine (ii, iv). (B) Leukemic cell lines were analyzed 0 h (upper panels), 12 h (middle panels) and 24 h (lower panels) after exposure to 10 µM cyclopamine. The X‐axis indicates annexinV‐fluorescein isothiocyanate. The Y‐axis indicates propidium iodide (PI). Data shown are from one representative experiment of three showing similar results.
Cytotoxicity of leukemic cells after exposure to Ara‐C in combination with cyclopamine. We finally examined whether a combinational effect between chemotherapeutic drugs and cyclopamine existed. Ara‐C was employed as a chemotherapeutic drug in this study because it is currently utilized in the chemotherapy of acute myeloid leukemia. CD34+ leukemic cells were exposed to 0–10−6 µM Ara‐C in combination with 10 µM cyclopamine. As shown in Fig. 5(a), no synergistic effect was observed in NB4, U937, HL60 or KG1 after exposure to Ara‐C and cyclopamine for 48 h. Kasumi‐1, Kasumi‐3 and TF‐1 are slightly resistant to Ara‐C. Consistent with the results of Fig. 3, treatment with 10 µM cyclopamine without Ara‐C (0 µM) reduced percent survival of these CD34+ cells. Surprisingly, in combination with cyclopamine, sensitivity to Ara‐C was dramatically enhanced in these cell lines. Moreover, similar effects were shown when we analyzed the sensitivity of primary CD34+ leukemic cells separated from three AML patients (Fig. 5b). We further investigated whether apoptosis of these leukemic cell lines was enhanced in combination with cyclopamine. Expectedly, no apparent difference of annexinV+ and PI+ cells were observed in Hh receptor‐negative cells (NB4, U937, HL60 and KG‐1) (Fig. 6a). In clear contrast, annexinV+/PI+ cells of Hh receptor positive cells (Kasumi‐1, Kasumi‐3 and TF‐1) in combination with cyclopamine increased as compared with the treatment of Ara‐C alone. Additionally, we assessed the expression of Bcl‐2 protein in leukemic cells 48 h after cyclopamine treatment because Bcl‐2 was proven to be a direct target of Hh signaling pathway.( 3 , 34 ) The Bcl‐2 expression was significantly reduced in Hh receptor‐positive cells, but not Hh receptor negative cells after cyclopamine treatment (Fig. 6b). These findings suggest the possibility that the Hh signaling inhibitor sensitized CD34+ leukemic cells against Ara‐C. In other words, cyclopamine may reduce drug resistance to chemotherapeutic drugs as demonstrated in solid tumor cells.( 35 , 36 , 37 )
Figure 5.
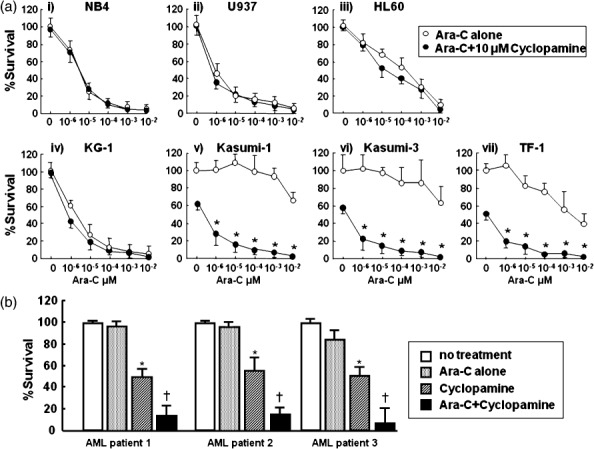
Cytotoxic assay of leukemic cells after exposure to Ara‐C in combination with 10 µM cyclopamine. (A) Survival of leukemic cells was analyzed by mitochondrial conversion of WST‐1 to yellowish formazan. Each cell line was exposed to Ara‐C. The X‐axis indicates dose of Ara‐C (µM) with (d) or without (s) 10 µM cyclopamine, and the Y‐axis indicates percent survival. (i) NB4 (ii) U937 (iii) HL60 (iv) KG1 (v) Kasumi‐1 (vi) Kasumi‐3 (vii) TF‐1. Data represent three independent experiments, each done in triplicate. *P < 0.01, Ara‐C alone vs. Ara‐C+ cyclopamine (Student's t‐test, two‐tailed). (B) Cytotoxic assay of primary leukemic cells. CD34+ leukemic cells were separated and exposed to 10−6 µM Ara‐C and/or 10 µM cyclopamine. Survival of leukemic cells was analyzed by WST‐1 assay. No treatment (open columns), Ara‐C alone (stippled columns), cyclopamine (diagonal columns) and Ara‐C+ cyclopamine (closed columns). *P < 0.05, no treatment vs. Ara‐C alone (Student's t‐test, two‐tailed). † P < 0.05, Ara‐C alone vs. Ara‐C and cyclopamine.
Figure 6.
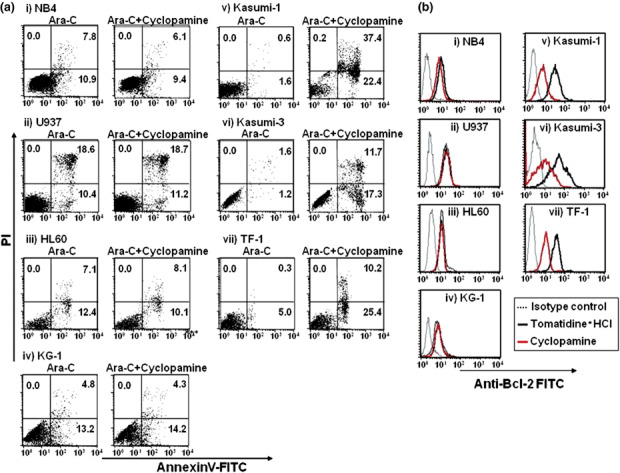
Annexin assay of leukemic cell lines 24 h after treatment of Ara‐C with or without cyclopamine and Bcl‐2 expression in leukemic cells. (A) Leukemic cell lines were exposed to 10−6 µM Ara‐C with or without 10 µM cyclopamine. After 24 h of treatment, cells were analyzed by annexinV/PI assay. (B) Expression of Bcl‐2 in leukemic cell lines. Flow cytometric analysis of Bcl‐2 in leukemic cell lines were conducted 48 h after exposure of 10 µM tomatidine or cyclopamine. Black thick line indicates the level of Bcl‐2 after tomatidine treatment (control). Red thick line indicates the level of Bcl‐2 after cyclopamine treatment. Thin dotted line indicates the result of isotype control.
Discussion
In the present study, we demonstrate that Hh receptor components Ptc and Smo are expressed in three CD34+ cell lines, while Hh‐transcription factor GLI1 or GLI2 is expressed in all acute leukemic cell lines that we screened. The Hh signaling pathway could transmit via GLI, as shown by the GLI‐reporter assay, in an autocrine manner in three cytokine‐responsive CD34+ cell lines. Furthermore, inhibition of Hh signaling induced apoptosis in these CD34+ cell lines and sensitized them against Ara‐C.
It has been demonstrated that Hh promotes up‐regulation of a drug transporter,( 38 ) Bcl‐2,( 3 , 34 ) and Bcl‐xL protein.( 39 ) Furthermore, the Hh inhibitor cyclopamine reduced the expression of phosphorylated human homolog of the v‐akt oncogene (Akt)/Protein kinase B and insulin‐like growth factor (IGF)‐II, which is associated with cell survival in several stressful circumstances.( 40 ) These findings support our results showing that inhibition of Hh signaling induced apoptosis in Hh‐responsive CD34+ cells. Among the cell survival factors described above, Bcl‐2 is relevant in response to Hh signaling since the expression of Bcl‐2 was proven to be up‐regulated via direct binding of Hh‐activated transcription factor GLI1 or GLI2 in solid cancer cells.( 3 , 34 ) Therefore, we investigated the expression of Bcl‐2 after cyclopamine treatment. Consequently, we found that cyclopamine treatment reduced expression levels of Bcl‐2 in Hh receptor‐positive CD34+ leukemic cells (Fig. 6b). It has been reported that Bcl‐2 expression could be correlated with resistance of cancer therapy such as radiation and paclitaxel.( 36 ) In agreement with these findings, cyclopamine treatment reduced drug resistance to Ara‐C in three CD34+ cells, Kasumi‐1, Kasumi‐3 and TF‐1. Collectively, inhibition of Hh signaling augments the therapeutic efficacy of Ara‐C in AML.
Toxicity of Hh signaling inhibitors should be our next concern since Hh signaling is involved in growth and healing in a variety of tissues. Regarding the hematopoietic system, the granulocytic/monocytic lineage was unaffected in terms of number of total cells, progenitor cells and morphology in the presence of 10 µM cyclopamine.( 18 ) In contrast, the number of erythroid colonies was reported to be significantly reduced.( 18 ) This observation may be explained by the expression level of Hh receptor components; in the erythroid lineage, expression levels were sustained (data not shown), whereas the expression of Hh receptor components in the granulocytic/monocytic lineage decreased during differentiation from hematopoietic stem cells in vitro.( 18 ) In the in vivo situation, bolus administration was limited by toxicity and rapid clearance in mice. In particular, even transient inhibition by Hh antagonist in young mice causes permanent defects in bone structure.( 41 ) Hence, Hh antagonist may not be applied with the infant patient. Thus, continuous infusion of cyclopamine was usually used for the adult mice model of medulloblastoma or pancreatic cancer,( 42 , 43 ) and in this procedure, the apparent toxicity of cyclopamine was not observed,( 42 , 43 ) Because Ara‐C is also administrated by continuous infusion in the treatment of AML, combination therapy between cyclopamine and Ara‐C would be feasible in a clinical setting.
Although inhibition of Hh signaling would be a useful strategy, cyclopamine or its derivatives are only applicable for leukemic cells that express Hh receptor components. In fact, in our screening, Hh receptor molecules are lost in several cell lines such as NB4, U937 and HL60. For these cells, blockade of the autocrine loop in Hh signaling would not be available. In the present study, we demonstrate that transcription factor GLI1 or GLI2 is expressed in all leukemic cell lines tested. Hence, GLI1 or GLI2 might be an alternative target for treatment of AML. One possible strategy is to use small interfering RNA (siRNA) specific for GLI1 or GLI2. Inhibition of GLI1 by siRNA is reported to suppress invasiveness and MMP‐9 expression in pancreatic cancer cells.( 44 ) However, efficient transfer of siRNA into hematopoietic cells is always problematic. In this regard, we have recently developed a vitamin A‐coupled liposome to deliver siRNA.( 45 ) This new retinol‐conjugated liposome could efficiently transfer siRNA into cells such as acute promyeloblastic leukemic cells and hematopoietic stem cells that are able to take up retinol (data not shown).( 46 , 47 , 48 , 49 ) This study is now ongoing.
In conclusion, Hh signaling could be involved in the survival and drug resistance of CD34+ leukemic cells. Inhibition of Hh signaling can be a therapeutic option to directly induce apoptosis and reduce drug resistance of AML.
Acknowledgments
The manuscript has been carefully reviewed by an experienced medical editor in NAI Inc. This work was supported in part by a grant from the Ministry of Health, Labour and Welfare of Japan to MK.
References
- 1. Denef N, Neubuser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 2000; 102: 521–31. [DOI] [PubMed] [Google Scholar]
- 2. Duman‐Scheel M, Weng L, Xin S, Du W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 2002; 417: 299–304. [DOI] [PubMed] [Google Scholar]
- 3. Bigelow RL, Chari NS, Unden AB et al . Transcriptional regulation of bcl‐2 mediated by the sonic hedgehog signaling pathway through gli‐1. J Biol Chem 2004; 279: 1197–205. [DOI] [PubMed] [Google Scholar]
- 4. Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci 2003; 6: 21–7. [DOI] [PubMed] [Google Scholar]
- 5. St‐Jacques B, Dassule HR, Karavanova I et al . Sonic hedgehog signaling is essential for hair development. Curr Biol 1998; 8: 1058–68. [DOI] [PubMed] [Google Scholar]
- 6. Spinella‐Jaegle S, Rawadi G, Kawai S et al . Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J Cell Sci 2001; 114: 2085–94. [DOI] [PubMed] [Google Scholar]
- 7. Dyer MA, Farrington SM, Mohn D, Munday JR, Baron MH. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development 2001; 128: 1717–30. [DOI] [PubMed] [Google Scholar]
- 8. Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small‐cell lung cancer. Nature 2003; 422: 313–7. [DOI] [PubMed] [Google Scholar]
- 9. Watkins DN, Berman DM, Baylin SB. Hedgehog signaling: progenitor phenotype in small‐cell lung cancer. Cell Cycle 2003; 2: 196–8. [PubMed] [Google Scholar]
- 10. Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. J Clin Oncol 2008; 26: 2806–12. [DOI] [PubMed] [Google Scholar]
- 11. Hegde GV, Munger CM, Emanuel K et al . Targeting of sonic hedgehog‐GLI signaling: a potential strategy to improve therapy for mantle cell lymphoma. Mol Cancer Ther 2008; 7: 1450–60. [DOI] [PubMed] [Google Scholar]
- 12. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG‐GLI1 signaling regulates human glioma growth, cancer stem cell self‐renewal, and tumorigenicity. Curr Biol 2007; 17: 165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peacock CD, Wang Q, Gesell GS et al . Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA 2007; 104: 4048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feldmann G, Dhara S, Fendrich V et al . Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res 2007; 67: 2187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berman DM, Karhadkar SS, Hallahan AR et al . Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002; 297: 1559–61. [DOI] [PubMed] [Google Scholar]
- 16. Bhardwaj G, Murdoch B, Wu D et al . Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol 2001; 2: 172–80. [DOI] [PubMed] [Google Scholar]
- 17. Kobune M, Ito Y, Kawano Y et al . Indian hedgehog gene transfer augments hematopoietic support of human stromal cells including NOD/SCID‐beta2m–/– repopulating cells. Blood 2004; 104: 1002–9. [DOI] [PubMed] [Google Scholar]
- 18. Detmer K, Walker AN, Jenkins TM, Steele TA, Dannawi H. Erythroid differentiation in vitro is blocked by cyclopamine, an inhibitor of hedgehog signaling. Blood Cells Mol Dis 2000; 26: 360–72. [DOI] [PubMed] [Google Scholar]
- 19. Kobune M, Kato J, Kawano Y et al . Adenoviral vector‐mediated transfer of the Indian hedgehog gene modulates lymphomyelopoiesis in vivo. Stem Cells 2008; 26: 534–42. [DOI] [PubMed] [Google Scholar]
- 20. Bai LY, Chiu CF, Lin CW et al . Differential expression of Sonic hedgehog and Gli1 in hematological malignancies. Leukemia 2008; 22: 226–8. [DOI] [PubMed] [Google Scholar]
- 21. Neering SJ, Bushnell T, Sozer S et al . Leukemia stem cells in a genetically defined murine model of blast‐crisis CML. Blood 2007; 110: 2578–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schepers H, Wierenga AT, van Gosliga D, Eggen BJ, Vellenga E, Schuringa JJ. Reintroduction of C/EBPalpha in leukemic CD34+ stem/progenitor cells impairs self‐renewal and partially restores myelopoiesis. Blood 2007; 110: 1317–25. [DOI] [PubMed] [Google Scholar]
- 23. Mori Y, Okumura T, Tsunoda S, Sakai Y, Shimada Y. Gli‐1 expression is associated with lymph node metastasis and tumor progression in esophageal squamous cell carcinoma. Oncology 2006; 70: 378–89. [DOI] [PubMed] [Google Scholar]
- 24. Mizugishi K, Aruga J, Nakata K, Mikoshiba K. Molecular properties of Zic proteins as transcriptional regulators and their relationship to GLI proteins. J Biol Chem 2001; 276: 2180–8. [DOI] [PubMed] [Google Scholar]
- 25. Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog‐induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem 1999; 274: 8143–52. [DOI] [PubMed] [Google Scholar]
- 26. Kobune M, Chiba H, Kato J et al . Wnt3/RhoA/ROCK signaling pathway is involved in adhesion‐mediated drug resistance of multiple myeloma in an autocrine mechanism. Mol Cancer Ther 2007; 6: 1774–84. [DOI] [PubMed] [Google Scholar]
- 27. Kozu T, Miyoshi H, Shimizu K et al . Junctions of the AML1/MTG8 (ETO) fusion are constant in t (8;21) acute myeloid leukemia detected by reverse transcription polymerase chain reaction. Blood 1993; 82: 1270–6. [PubMed] [Google Scholar]
- 28. Mochizuki N, Shimizu S, Nagasawa T et al . A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t (1;3) (p36;q21)‐positive leukemia cells. Blood 2000; 96: 3209–14. [PubMed] [Google Scholar]
- 29. Kitamura T, Tojo A, Kuwaki T et al . Identification and analysis of human erythropoietin receptors on a factor‐dependent cell line, TF‐1. Blood 1989; 73: 375–80. [PubMed] [Google Scholar]
- 30. Kitamura T, Tange T, Terasawa T et al . Establishment and characterization of a unique human cell line that proliferates dependently on GM‐CSF, IL‐3, or erythropoietin. J Cell Physiol 1989; 140: 323–34. [DOI] [PubMed] [Google Scholar]
- 31. Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997; 389: 876–81. [DOI] [PubMed] [Google Scholar]
- 32. Grachtchouk M, Mo R, Yu S et al . Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet 2000; 24: 216–7. [DOI] [PubMed] [Google Scholar]
- 33. Kimura H, Stephen D, Joyner A, Curran T. Gli1 is important for medulloblastoma formation in Ptc1+/– mice. Oncogene 2005; 24: 4026–36. [DOI] [PubMed] [Google Scholar]
- 34. Dierks C, Grbic J, Zirlik K et al . Essential role of stromally induced hedgehog signaling in B‐cell malignancies. Nat Med 2007; 13: 944–51. [DOI] [PubMed] [Google Scholar]
- 35. Lavie Y, Harel‐Orbital T, Gaffield W, Liscovitch M. Inhibitory effect of steroidal alkaloids on drug transport and multidrug resistance in human cancer cells. Anticancer Res 2001; 21: 1189–94. [PubMed] [Google Scholar]
- 36. Shafaee Z, Du Schmidt HW, Posner M, Weichselbaum R. Cyclopamine increases the cytotoxic effects of paclitaxel and radiation but not cisplatin and gemcitabine in Hedgehog expressing pancreatic cancer cells. Cancer Chemother Pharmacol 2006; 58: 765–70. [DOI] [PubMed] [Google Scholar]
- 37. Mimeault M, Johansson SL, Vankatraman G et al . Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Mol Cancer Ther 2007; 6: 967–78. [DOI] [PubMed] [Google Scholar]
- 38. Sims‐Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007; 26: 5674–9. [DOI] [PubMed] [Google Scholar]
- 39. Varas A, Hernandez‐Lopez C, Valencia J et al . Survival and function of human thymic dendritic cells are dependent on autocrine Hedgehog signaling. J Leukoc Biol 2008; 83: 1476–83. [DOI] [PubMed] [Google Scholar]
- 40. Levitt RJ, Zhao Y, Blouin MJ, Pollak M. The hedgehog pathway inhibitor cyclopamine increases levels of p27, and decreases both expression of IGF‐II and activation of Akt in PC‐3 prostate cancer cells. Cancer Lett 2007; 255: 300–6. [DOI] [PubMed] [Google Scholar]
- 41. Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 2008; 13: 249–60. [DOI] [PubMed] [Google Scholar]
- 42. Lipinski RJ, Hutson PR, Hannam PW et al . Dose‐ and route‐dependent teratogenicity, toxicity, and pharmacokinetic profiles of the hedgehog signaling antagonist cyclopamine in the mouse. Toxicol Sci 2008; 104: 189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Feldmann G, Habbe N, Dhara S et al . Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nagai S, Nakamura M, Yanai K et al . Gli1 contributes to the invasiveness of pancreatic cancer through matrix metalloproteinase‐9 activation. Cancer Sci 2008; 99: 1377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sato Y, Murase K, Kato J et al . Resolution of liver cirrhosis using vitamin A‐coupled liposomes to deliver siRNA against a collagen‐specific chaperone. Nat Biotechnol 2008; 26: 431–42. [DOI] [PubMed] [Google Scholar]
- 46. Cheung AM, Tam CK, Chow HC, Verfaillie CM, Liang R, Leung AY. All‐trans retinoic acid induces proliferation of an irradiated stem cell supporting stromal cell line AFT024. Exp Hematol 2007; 35: 56–63. [DOI] [PubMed] [Google Scholar]
- 47. Luo P, Wang A, Payne KJ et al . Intrinsic retinoic acid receptor alpha‐cyclin‐dependent kinase‐activating kinase signaling involves coordination of the restricted proliferation and granulocytic differentiation of human hematopoietic stem cells. Stem Cells 2007; 25: 2628–37. [DOI] [PubMed] [Google Scholar]
- 48. Chute JP, Muramoto GG, Whitesides J et al . Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA 2006; 103: 11707–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Evans T. Regulation of hematopoiesis by retinoid signaling. Exp Hematol 2005; 33: 1055–61. [DOI] [PubMed] [Google Scholar]