

Abstract
Genome‐wide DNA hypomethylation and concomitant site‐specific gene hypermethylation are among the most common molecular alterations in human neoplasia. Previous studies revealed that genetic reduction of the DNA methylation level results in opposing effects on tumor development, depending on the tumor cell type and on the different stages of the tumorigenesis. For instance, reduced levels of DNA methylation in mice strongly inhibited tumor development of the intestine, whereas they induced thymic lymphomas and liver tumors. In the present study, using DNA methyltrasferase 1 (Dnmt1) hypomorphic alleles to reduce genomic methylation, we examined the effects of DNA hypomethylation on a murine squamous carcinogenesis in the tongue and esophagus induced by 4‐nitroquinoline 1‐oxide. Genetic reduction of DNA methylation level led to the suppression of tumor formation in both tongue and esophagus. Histological analyses revealed that DNA hypomethylation preferentially inhibited the development of squamous cell carcinomas. The results suggest that genomic hypomethylation inhibits squamous carcinogenesis in the tongue and esophagus, and that pharmacological modification of epigenetic status might be useful for the prevention and treatment of cancers in the upper digestive tract. (Cancer Sci 2009; 100: 1186–1191)
Altered DNA methylation in the form of global hypomethylation and regional hypermethylation is one of the most consistent epigenetic changes in cancer.( 1 ) Global DNA hypomethylation, which is frequently observed at early stages of tumorigenesis in human cancer, promotes tumor development in mouse models and causes chromosomal instability in cultured fibroblasts.( 2 ) After the initial observation of DNA hypermethylation within the retinoblastoma tumor suppressor gene, a number of genes have been shown to be hypermethylated and transcriptionally silenced in tumors. Although the consequences of global hypomethylation and gene‐specific hypermethylation have been mechanistically connected to chromosome instability and transcriptional silencing, respectively,( 3 , 4 , 5 ) the causes of aberrant genomic methylation patterns are currently unknown.
DNA methylation is catalyzed by a family of three DNA methyltransferases: Dnmt1, Dnmt3a, and Dnmt3b.( 6 , 7 , 8 ) Although the three Dnmt partially cooperate to establish and maintain genomic methylation patterns, they also have distinctive functions. Dnmt1 has a preference for hemimethylated DNA, and indeed a hypomorphic allele of Dnmt1 has been shown to cause global DNA hypomethylation. Thus, Dnmt1 is considered to be the major maintenance methyltransferase. Using Dnmt1 hypomorphic alleles as a model for global DNA hypomethylation, previous studies have revealed that global DNA hypomethylation inhibits tumorigenesis in the intestine,( 5 ) whereas it induces or promotes T‐cell lymphomas,( 9 ) liver cancers,( 4 ) and fibrosarcomas.( 10 ) These results indicate that the forced reduction of genomic methylation levels leads to opposing effects on tumorigenesis depending on the cell type. Considering the fact that DNA hypomethylating agents have been used for cancer therapy in a subset of cancer,( 1 ) it is important to clarify the effect of global DNA hypomethylation on the risk for tumor development in various organs.
The potent carcinogenicity of 4‐nitroquinoline 1‐oxide (4NQO) depends on the formation of DNA adducts, in addition to the exertion of oxidative stress in target cells.( 11 , 12 ) 4NQO has been shown to produce squamous neoplasms preferentially in the tongue and a small number of tumors arise in the esophagus in rodents exposed to 4NQO.( 13 , 14 ) There is a histological sequence of epithelial changes from dysplasia through invasive squamous cell carcinoma (SCC) in this model.( 14 ) In the present study, we have investigated the effect of reduced DNA methylation levels on the squamous cell carcinogenesis induced by 4NQO and found that global DNA hypomethylation can suppress the tumorigenesis in both the tongue and esophagus.
Materials and Methods
Mice. All animal studies and breeding were carried out under the Regulations for Animal Experiments in Gifu University. Two mutant alleles of Dnmt1 were used: the null Dnmt1 c allele in the C57BL/6 background( 15 ) and the hypomorphic Dnmt1 chip allele in the 129Sv4 background.( 9 ) Dnmt1 c/+ mice were crossed with female Dnmt1 chip/chip mice (129Sv4) to generate all experimental mice in an isogenic F1 hybrid (C57 : 129) background. Previous study indicated that Dnmt1 chip/+ mice have the same levels of genomic methylation as Dnmt1 +/+ mice, whereas Dnmt1 chip/c mice have reduced DNA methylation contents at pricentromeric satellite repeats.( 4 ) Therefore, we analyzed 28 Dnmt1 chip/+ mice as a control cohort, and 28 Dnmt1 chip/c mice as a DNA hypomethylated cohort to quantify both the tongue and esophagus lesions at 25 weeks of age. Tumors were quantified along the surface of the tissue, and were further analyzed after dissection.
4NQO exposure. A total of 56 mice were used for the studies with 4NQO. We used both male and female mice in the present study. All mice were maintained under specific pathogen‐free conditions with isolated ventilation cages in an air‐conditioned room with a 12 : 12 L : D cycle. They were bred and maintained on a basal diet, CE‐2 (CLEA Japan, Tokyo, Japan) until termination of the study. Genotypes were identified by PCR analysis of tail DNA using allele‐specific primers. The mice were administrated with drinking water containing 4NQO at concentrations of 0.1 mg/mL (23 Dnmt1 chip/+ mice and 22 Dnmt1 chip/c mice) or no 4NQO (five Dnmt1 chip/+ mice and six Dnmt1 chip/c mice), starting at 5 weeks of age until the termination of the experiment. To examine tongue and esophagus lesions, 25‐week‐old Dnmt1 chip/c and age‐matched Dnmt1 chip/+ littermates were killed. As reported previously,( 9 ) we observed a decreased bodyweight in Dnmt1 chip/c mice in comparison with Dnmt1 chip/+ littermates.( 9 ) However, there was no detectable difference in consumption of the drinking water containing 4NQO (data not shown).
Preparation of tissue samples for tumor counting and histological analysis. All mice underwent a thorough postmortem examination at the time of death. The tongue and esophagus were removed and the esophagus was opened along the longitudinal axis. The number as well as the longest diameter of the tumors in the tongue and esophagus were measured using a dissecting microscope. Tumors >0.5 mm in diameter were mapped and counted. To eliminate interobserver error, all counts were done by a single observer blinded to the genotype of the mice. All of the cases were also counted by a second observer to confirm the results of the first observer. After tumor counting, the tongues were cut at the longitudinal center and were processed for the assessment of microscopic lesions. The incidences and multiplicities of microscopic lesions in the tongue were determined on histological sections at the longitudinal center of the tongue. In contrast to the tongue tumors, all esophageal tumors were processed for histological analysis. The esophagus was cut at the longitudinal axis and processed for the assessment of histological changes of squamous epithelium. The incidence and multiplicities of microscopic esophageal lesions were examined using histological sections of both macroscopically identified tumors and macroscopically normal‐looking mucosa. All of the excised tissues, including the neoplastic nodules in the esophagus, were fixed for 24 h in neutral‐buffered 10% formalin. The fixed samples were processed by standard methods, embedded in paraffin, sectioned at 5 µm, and stained with HE. The defining characteristics for dysplasia, papilloma, and SCC were adapted from previous reports in the literature.( 16 , 17 )
Immunostaining. The avidin–biotin–peroxidase complex (ABC) technique was used for immunohistochemical studies. Sections (5‐µm thick) were cut, deparaffinized, rehydrated in PBS, placed in 10 mmol/L citrate buffer (pH 6.0), and heated in a 750‐W microwave four times for 6 min. The endogenous peroxidase activity was blocked by incubation for 10 min in 0.3% H2O2. After washing three times with PBS, the sections were preincubated with normal blocking serum for 20 min at room temperature and then incubated with Ki‐67 (1:200; DAKO, Glostrup, Denmark) and CK19 (1:500; Abcam, Cambridge, MA, USA) overnight at 4°C. Subsequently, the sections were incubated with biotinylated secondary antibodies (Vectastain ABC kit; Vector Laboratories, Burlingame, CA, USA) for 30 min followed by incubation with avidin‐coupled peroxidase (Vector Laboratories) for 30 min. The sections were developed with 3,3′‐diaminobenzidine (DAB) using the DAKO Liquid DAB Substrate‐Chromogen System (DAKO) and then counterstained with hematoxylin. No specific staining was observed in the negative control slides prepared without primary antibody.
Results
Reduced DNA methylation levels suppressed the squamous tumorigenesis induced by 4NQO in both the tongue and esophagus. All mice with the exception of one Dnmt1 chip/c mouse survived at the end of the 20‐week 4NQO drinking, and there were no differences in the survival rate during 4NQO administrations between Dnmt1 chip/c mice and Dnmt1 chip/+ mice (n = 22 and 23 respectively). There were no detectable differences in pathological alterations in the liver, kidney, lung, or heart between Dnmt1 chip/+ and Dnmt1 chip/c mice with or without 4NQO administration. The mice without 4NQO exposure did not develop neoplasms in any organs examined, including the tongue and esophagus at the time of death.
Cohorts of Dnmt1 chip/+ or Dnmt1 chip/c genotypes were aged to 25 weeks and analyzed for macroscopic tumors in both the tongue and esophagus. Macroscopically, nodular and polypoid tongue tumors were observed in the dorsum and tip of the tongue in mice with both genotypes that were exposed to 4NQO (Fig. 1a–c). The incidences and multiplicities of macroscopic neoplasm in the tongue and esophagus are summarized in Table 1. In the present study, we did not observe any metastatic tumors. The incidence of macroscopic tongue tumors was 100 and 90.5% in Dnmt1 chip/+ and Dnmt1 chip/c mice respectively. The multiplicity of macroscopic tongue tumors was 4.91 ± 1.02 and 1.90 ± 0.81 in Dnmt1 chip/+ and Dnmt1 chip/c mice respectively. The multiplicity of macroscopic tongue tumors in Dnmt1 chip/c mice was significantly smaller than Dnmt1 chip/+ mice (Table 1; P < 0.05). In the present study, we did not observe significant differences in the development of tumors between male and female mice. The size of macroscopic tumors in the tongue was significantly smaller in Dnmt1 chip/c mice than Dnmt1 chip/+ mice (Table 1; P < 0.005). Next, we examined the development of microscopic lesions on the histological sections at the longitudinal center of the tongue. Histopathologically, dysplasia, papilloma, and SCC developed in the tongue of 4NQO‐drinking mice of both genotypes (Fig. 1d–f). Histological assessment of squamous lesions at the longitudinal center of the tongue is summarized in Table 2. Although the development of early lesions of the tongue (dysplasia and papilloma) was not significantly different between Dnmt1 chip/c and Dnmt1 chip/+ mice, both the incidence and multiplicity of SCC in Dnmt1 chip/c mice were significantly lower than those in the control Dnmt1 chip/+ mice (Table 2; P < 0.005 and P < 0.01 respectively).
Figure 1.
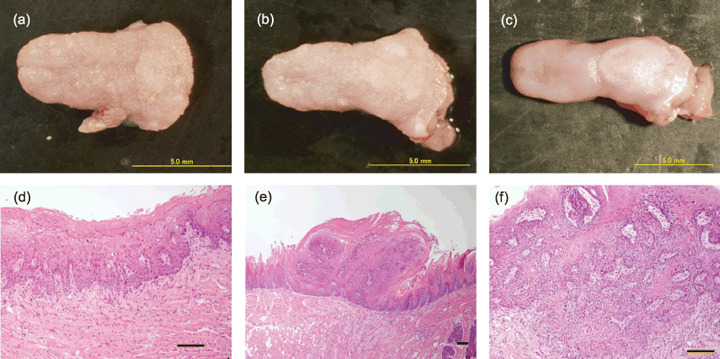
Representative macroscopic appearance of the tongue and histology of different stages of squamous lesions. (a–c) Macroscopic appearance of the tongue. (a) 4‐Nitroquinoline 1‐oxide (4NQO)‐exposed DNA methyltrasferase 1 (Dnmt1)chip/+, (b) 4NQO‐exposed Dnmt1 chip/c, and (c) the control Dnmt1 chip/+ mouse. Nodular and polypoid tumors are seen in the dorsum and tip of the tongue. (d–f) A histological sequence of epithelial changes from dysplasia through invasive squamous cell carcinoma. (d) Dysplasia, (e) papilloma, and (f) squamous cell carcinoma observed in the present study. Scale bars = 100 µm.
Table 1.
Incidences and multiplicities of macroscopic tumors of the tongue and esophagus
| Allele | Tongue | Esophagus | |
|---|---|---|---|
| Incidence (%) | chip/+ | 100.0 | 78.3 |
| chip/c | 90.5 | 42.9* | |
| Multiplicity (mean ± SD) | chip/+ | 4.91 ± 1.02 | 1.30 ± 1.04 |
| chip/c | 1.90 ± 0.81* | 0.62 ± 0.81* | |
| Diameter (mm) (mean ± SD) | chip/+ | 2.07 ± 0.57 | 1.82 ± 1.04 |
| chip/c | 1.30 ± 0.67** | 1.38 ± 0.68 |
Significantly different from chip/+ (P < 0.05).
Significantly different from chip/+ (P < 0.005).
chip/+, control mice; chip/c, hypomethylated mice.
Table 2.
Incidences and multiplicities of microscopic tumors of the tongue and esophagus
| Allele | Tongue † | Esophagus | |||||
|---|---|---|---|---|---|---|---|
| Dysplasia | Papilloma | SCC | Dysplasia | Papilloma | SCC | ||
| Incidence | chip/+ | 91.3 | 17.4 | 60.9 | 100.0 | 13.0 | 82.6 |
| (%) | chip/c | 81.0 | 4.8 | 19.0* | 71.4** | 19.0 | 42.9** |
| Multiplicity | chip/+ | 1.61 ± 0.99 | 0.17 ± 0.39 | 0.91 ± 0.85 | 2.26 ± 1.66 | 0.14 ± 0.34 | 1.30 ± 1.22 |
| (Mean ± SD) | chip/c | 1.52 ± 1.21 | 0.05 ± 0.22 | 0.19 ± 0.40** | 1.48 ± 1.47 | 0.19 ± 0.40 | 0.48 ± 0.60*** |
Significantly different from chip/+ (P < 0.005).
Significantly different from chip/+ (P < 0.01).
Significantly different from chip/+ (P < 0.05).
Incidences and multiplicities of microscopic tumors in the tongue were determined on the histological sections at the longitudinal center of the tongue.
chip/+, control mice; chip/c, hypomethylated mice.
The incidence of macroscopic esophageal tumors was 78.3 and 42.9% with a multiplicity of 1.30 ± 1.04 and 0.62 ± 0.81 in Dnmt1 chip/+ and Dnmt1 chip/c mice respectively (Table 1). Both the incidence and multiplicity of the macroscopic esophageal tumors were significantly smaller in Dnmt1 chip/c mice than Dnmt1 chip/+ mice (P < 0.05). However, there was no significant difference in tumor size between two cohorts, with size of 1.82 ± 1.04 and 1.38 ± 0.68 mm in Dnmt1 chip/+ and Dnmt1 chip/c mice respectively. Histological analyses revealed that both Dnmt1 chip/+ and Dnmt1 chip/c mice with 4NQO administration developed dysplasia, squamous papilloma, and SCC in the esophagus (Table 2). Consistent with the results in the tongue, the incidence and multiplicity of microscopic SCC in the esophagus were significantly smaller in Dnmt1 chip/c mice than in Dnmt chip/+ mice (Table 2; P < 0.01 and P < 0.05 respectively), whereas those of papillomas were not different between the two cohorts.
DNA hypomethylation suppresses increased cell proliferation activity in the squamous epithelium exposed to 4NQO. In order to clarify the inhibitory mechanisms of the reduced DNA methylation levels on squamous tumorigenesis, we assessed the cell proliferation activity in non‐tumoral squamous epithelium in the tongue by immunostaining for Ki‐67, a marker for proliferating cells. 4NQO administration resulted in an increased ratio of Ki‐67‐positive cells in non‐tumoral squamous epithelium of both Dnmt1 chip/+ and Dnmt1 chip/c mice in comparison with non‐treated mice (Fig. 2a). The results suggest that 4NQO induces abnormal cell proliferation even in the non‐tumoral epithelium, which may support the concept of ‘field cancerization’.( 18 , 19 ) Importantly, the Ki‐67‐positive ratio was significantly lower in the tongue of Dnmt1 chip/c mice in comparison with Dnmt1 chip/+ mice, indicating that reduced levels of genomic methylation suppress the cell proliferation activity that is activated by 4NQO administration (Fig. 2a). Additionally, the Ki‐67‐positive cell ratio in non‐tumoral epithelium of the esophagus of 4NQO‐exposed Dnmt1 chip/c mice was significantly lower than that of 4NQO‐exposed Dnmt1 chip/+ mice (Fig. 2b).
Figure 2.
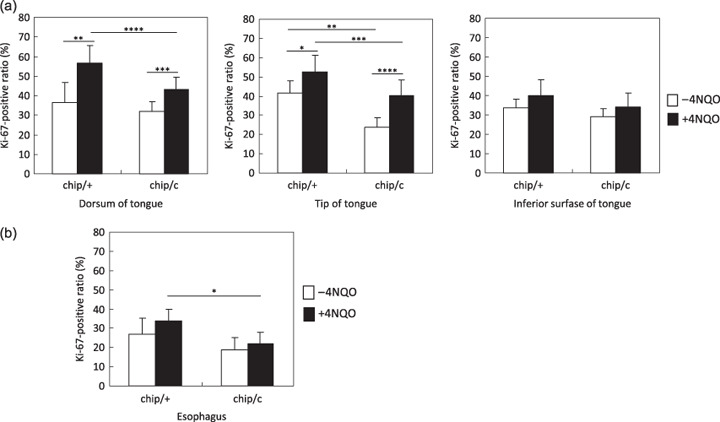
Percentage of Ki‐67‐positive cells in non‐tumoral squamous epithelium of both DNA methyltrasferase 1 (Dnmt1)chip/+ and Dnmt1 chip/c mice. (a) The Ki‐67‐positive ratio at the non‐tumoral squamous epithelium of the tongue. 4‐Nitroquinoline 1‐oxide (4NQO) administration resulted in an increased ratio of Ki‐67‐positive cells in squamous epithelium at the dorsum and tip of the tongue where direct exposure of 4NQO is expected. In contrast, the ratio at the inferior surface where 4NQO exposure is expected to be less than other sites did not change. The Ki‐67‐positive ratio is significantly lower in Dnmt1 chip/c mice in comparison with Dnmt1 chip/+ mice, suggesting that DNA hypomethylation suppresses cell proliferative activity. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001. (b) The Ki‐67‐positive ratio at the non‐tumoral squamous epithelium of the esophagus. The Ki‐67‐positive ratio in Dnmt1 chip/c mice exposed to 4NQO is significantly lower than that in Dnmt1 chip/+ mice exposed to 4NQO. *P < 0.001.
Expansion of CK19‐positive progenitor cells by 4NQO was blocked in DNA hypomethylated mice. In order to examine the differentiation status of squamous epithelium, we carried out immunostaining for CK19, a marker of undifferentiated cells at the basal–suprabasal cell layer of squamous epithelium in the tongue.( 20 , 21 ) Previous study revealed that the SCC contain an increased number of CK19‐expressing cells in comparison with normal squamous epithelium,( 21 ) suggesting that expansion of undifferentiated cells is involved in the tumorigenesis. Interestingly, non‐tumoral mucosa exposed to 4NQO revealed a thickening of the CK19‐positive layer in comparison with non‐exposed tongue (Fig. 3a,b,d). An increased ratio of CK19‐expressing layer to total epithelial thickness was also observed in 4NQO‐exposed tongue (Fig. 3e). The results suggest that 4NQO exposure increases the number of CK19‐expressing progenitor cells and inhibits the cellular differentiation of squamous epithelium. Importantly, the expansion of squamous progenitors by the administration of 4NQO was blocked in DNA hypomethylated mice (Fig. 3c–e), thus indicating that DNA methylation plays a role in the suppression of cellular differentiation in the tongue.
Figure 3.
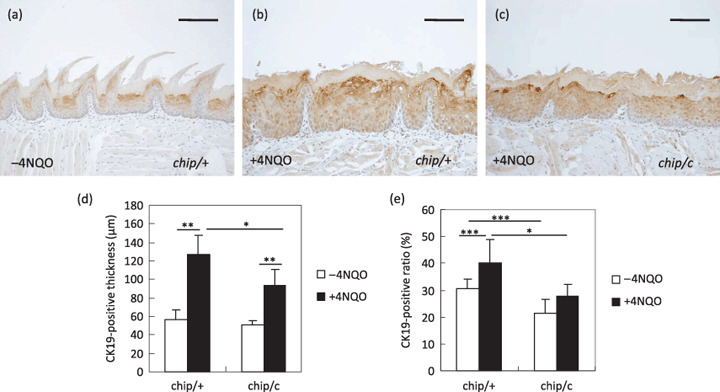
Expansion of the CK19‐positive cell layer in 4‐nitroquinoline 1‐oxide (4NQO)‐exposed tongue epithelium and its suppression by genomic hypomethylation. (a–c) CK19 immunohistological staining of tongue squamous epithelium. (a) 4NQO non‐exposed DNA methyltrasferase 1 (Dnmt1) chip/+ mice. (b) 4NQO exposed Dnmt1 chip/+ mice. (c) 4NQO exposed Dnmt1chip/c mice. Note that 4NQO exposure resulted in increased thickness of the CK19‐positive layer, whereas such an effect was less prominent in DNA hypomethylated mice. Scale bars = 100 µm. (d) Thickness of CK19‐positive cells (µm). (e) The CK19‐positive ratio at the dorsum of the tongue. *P < 0.001, **P < 0.005, ***P < 0.05.
Discussion
Here we have shown that genetic reduction of DNA methylation levels suppresses squamous tumorigenesis in both tongue and esophagus. The findings that genomic hypomethylation significantly inhibited SCC development in both tongue and esophagus suggest that DNA methylation plays a role in the progression stage of the squamous carcinogenesis in the upper digestive tract.
Expansion of stem and progenitor cells is a driving force in tumorigenesis.( 22 ) Consistent with the notion, many tumors express genes that are expressed in the undifferentiated cells of the target organs.( 22 ) In the present study, we showed that 4NQO exposure increases the number of CK19‐expressing progenitor cells( 20 , 21 ) and that global DNA hypomethylation suppresses such expansion of progenitor cells in the tongue. Given the fact that squamous progenitors proliferate actively, these findings suggest that the suppression of cellular differentiation, which is associated with DNA methylation, causes the expansion of proliferating progenitor cells, eventually leading to tumorigenesis in the tongue. The hypothesis is also consistent with our findings that the Ki‐67‐positive cell ratio is suppressed in the tongue of DNA hypomethylated mice. In the present study, suppression of the Ki‐67‐positive ratio by DNA hypomethylation was also observed in the esophagus. Recently, mouse esophageal stem cells have been identified using a label‐retaining method. It would be of interest to examine the effect of the genomic DNA hypomethylation on the number of stem and progenitor cells in the esophageal tumors.
It is also noteworthy that a previous study indicated that expansion of undifferentiated cells is directly associated with invasive growth of epithelial cells.( 22 ) Our findings that DNA hypomethylation promotes cellular differentiation and suppresses the development of invasive tumors may provide an additional link between the maintenance of undifferentiated states and the invasive growth of cancer cells. The observation that abnormal DNA methylation is associated with the maintenance of undifferentiated states is also consistent with previous findings, in which targets of abnormal DNA methylation in cancer cells are shown to be frequently overlapping with targets of the polycomb complex that is important to suppress cellular differentiation by silencing differentiation‐associated genes in embryonic stem (ES) cells.( 23 , 24 ) It is possible that the genes that are involved in cellular differentiation of the squamous epithelium are the targets of site‐specific DNA hypermethylation in 4NQO‐induced carcinogenesis.
Target genes of the abnormal DNA methylation in squamous lesions of this mouse model are not yet clear. Although it has been reported that the promoter regions of p16Ink4a and p15Ink4b are methylated in 4NQO‐induced rat tongue cancers,( 25 ) we failed to detect DNA hypermethylation at p16Ink4a by methylation‐specific PCR in the present study (data not shown). Understanding the epigenetic controls in the cellular differentiation of normal squamous epithelium might uncover the mechanisms underlying DNA methylation‐associated tumorigenesis in the tongue and esophagus. Further analyses are required to identify genes that connect the abnormal genomic hypermethylation and altered differentiating potential involved in cancer cells.
Acknowledgment
We thank Kyoko Takahashi, Ayako Suga, and Yoshitaka Kinjyo for the technical assistance and animal care. This study was supported by grants from the Ministry of Health, Labour, and Welfare of Japan, and grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 2. Chen RZ, Pettersson U, Beard C, Jackson‐Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature 1998; 395: 89–93. [DOI] [PubMed] [Google Scholar]
- 3. Toyota M, Ahuja N, Ohe‐Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamada Y, Jackson‐Grusby L, Linhart H et al . Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc Natl Acad Sci USA 2005; 102: 13 580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laird PW, Jackson‐Grusby L, Fazeli A et al . Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995; 81: 197–205. [DOI] [PubMed] [Google Scholar]
- 6. Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69: 915–26. [DOI] [PubMed] [Google Scholar]
- 7. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99: 247–57. [DOI] [PubMed] [Google Scholar]
- 8. Linhart HG, Lin H, Yamada Y et al . Dnmt3b promotes tumorigenesis in vivo by gene‐specific de novo methylation and transcriptional silencing. Genes Dev 2007; 21: 3110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gaudet F, Hodgson JG, Eden A et al . Induction of tumors in mice by genomic hypomethylation. Science 2003; 300: 489–92. [DOI] [PubMed] [Google Scholar]
- 10. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300: 455. [DOI] [PubMed] [Google Scholar]
- 11. Nakahara W, Fukuoka F, Sugimura T. Carcinogenic action of 4‐nitroquinoline‐N‐oxide. Gan 1957; 48: 129–37. [PubMed] [Google Scholar]
- 12. Ikenaga M, Ishii Y, Tada M, Kakunaga T, Takebe H. Excision‐repair of 4‐nitroquinolin‐1‐oxide damage responsible for killing, mutation, and cancer. Basic Life Sci 1975; 5B: 763–71. [DOI] [PubMed] [Google Scholar]
- 13. Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen‐treated mice. Clin Cancer Res 2004; 10: 301–13. [DOI] [PubMed] [Google Scholar]
- 14. Tanaka T, Makita H, Ohnishi M et al . Chemoprevention of 4‐nitroquinoline 1‐oxide‐induced oral carcinogenesis in rats by flavonoids diosmin and hesperidin, each alone and in combination. Cancer Res 1997; 57: 246–52. [PubMed] [Google Scholar]
- 15. Lei H, Oh SP, Okano M et al . De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 1996; 122: 3195–205. [DOI] [PubMed] [Google Scholar]
- 16. Gunji A, Uemura A, Tsutsumi M et al . Parp‐1 deficiency does not increase the frequency of tumors in the oral cavity and esophagus of ICR/129Sv mice by 4‐nitroquinoline 1‐oxide, a carcinogen producing bulky adducts. Cancer Lett 2006; 241: 87–92. [DOI] [PubMed] [Google Scholar]
- 17. Ide F, Oda H, Nakatsuru Y et al . Xeroderma pigmentosum group A gene action as a protection factor against 4‐nitroquinoline 1‐oxide‐induced tongue carcinogenesis. Carcinogenesis 2001; 22: 567–72. [DOI] [PubMed] [Google Scholar]
- 18. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953; 6: 963–8. [DOI] [PubMed] [Google Scholar]
- 19. Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol 2007; 40: 142–50. [DOI] [PubMed] [Google Scholar]
- 20. Lindberg K, Rheinwald JG. Suprabasal 40 kd keratin (K19) expression as an immunohistologic marker of premalignancy in oral epithelium. Am J Pathol 1989; 134: 89–98. [PMC free article] [PubMed] [Google Scholar]
- 21. Chen S, Takahara M, Kido M et al . Increased expression of an epidermal stem cell marker, cytokeratin 19, in cutaneous squamous cell carcinoma. Br J Dermatol 2008; 159: 952–5. [DOI] [PubMed] [Google Scholar]
- 22. Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct‐4 blocks progenitor‐cell differentiation and causes dysplasia in epithelial tissues. Cell 2005; 121: 465–77. [DOI] [PubMed] [Google Scholar]
- 23. Widschwendter M, Fiegl H, Egle D et al . Epigenetic stem cell signature in cancer. Nat Genet 2007; 39: 157–8. [DOI] [PubMed] [Google Scholar]
- 24. Ohm JE, McGarvey KM, Yu X et al . A stem cell‐like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39: 237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ogawa K, Tanuma J, Hirano M et al . Selective loss of resistant alleles at p15INK4B and p16INK4A genes in chemically‐induced rat tongue cancers. Oral Oncol 2006; 42: 710–17. [DOI] [PubMed] [Google Scholar]