

Abstract
Heparan sulfate proteoglycan syndecan‐1 (CD138) is well known to be associated with cell proliferation, adhesion and migration in various types of malignancies. In the present study, we focused on the role of syndecan‐1 in human prostate cancer. Immunohistochemical analysis revealed either no or rare expression of syndecan‐1 in normal secretory glands and prostate cancer cells at hormone naïve status, whereas the expression was significantly increased in viable cancer cells following neo‐adjuvant hormonal therapy. Syndecan‐1 expression was much higher in the androgen independent prostate cancer cell lines DU145 and PC3, rather than the androgen‐dependent LNCaP, but the level in LNCaP was up‐regulated in response to long‐term culture under androgen deprivation. Silencing of syndecan‐1 by siRNA transfection reduced endogenous production of reactive oxygen species through down‐regulating NADPH oxidase 2 and induced apoptosis in DU145 and PC3 cells. Consistently, NADPH oxidase 2 knockdown induced apoptosis to a similar extent. Subcutaneous inoculation of PC3 cells in nude mice demonstrated the reduction of tumor size by localized injection of syndecan‐1 siRNA in the presence of atelocollagen. Moreover, the mouse model and chorioallantoic membrane assay demonstrated significant inhibition of vascular endothelial growth factor and tumor angiogenesis by silencing of syndecan‐1. In conclusion, syndecan‐1 might participate in the process of androgen‐dependent to ‐independent conversion, and be a new target molecule for hormone resistant prostate cancer therapy. (Cancer Sci 2009; 100: 1248–1254)
Syndecan‐1 (CD138), a member of four mammalian heparin sulfate proteoglycans, is highly expressed in many types of normal epithelial cells and malignant counterparts where it plays an important role in cell growth, adhesion, migration, epithelial morphogenesis, and angiogenesis.( 1 ) Syndecan‐1 is overexpressed in normal and malignant plasma cells, contributing to plasma cell myeloma development:( 2 , 3 ) heparin sulfate–epidermal growth factor ligands were shown to be concentrated by syndecan‐1 at the cell membrane, resulting in activation of ErbB‐mediated cell survival signals.( 4 ) Derksen et al.( 5 ) reported that syndecan‐1 functions as a co‐receptor for hepatocyte growth factor (HGF) that promotes HGF/Met signaling and myeloma cell proliferation. In addition, previous investigations have identified that syndecan‐1 can be a therapeutic target for plasma cell myeloma: heparin or its nonanticoagulant derivatives compete with heparin sulfate and prevent myeloma cells from binding growth factors.( 6 ) Of note, Yang et al.( 7 ) showed that treatment with an inhibitor of human heparanase, an enzyme that synergizes with syndecan‐1 in promoting myeloma progression, or knockdown of syndecan‐1 expression by RNAi, diminished myeloma tumor development in vivo. Together, these data suggest syndecan‐1 could relate to malignant potential, particularly in plasma cell myeloma; however, the biological function is yet to be completely understood in other types of malignancies, including prostate cancer.
Reactive oxygen species (ROS) are implicated in both stimulation and inhibition of cellular proliferation, apoptosis, and senescence.( 8 , 9 ) ROS trigger genetic programs associated with transformation, resulting in alteration of genes by manipulating the cell cycle and signal transduction.( 10 ) Similarly, generation of oxygen radicals is increased in Ras‐transformed fibroblasts( 11 ) and antioxidants can block DNA synthesis, suggesting involvement of ROS in mitogenic signaling in the course of neoplastic transformation. NADPH oxidase (NOX) is one of the major sources for cellular ROS.( 12 , 13 ) NOX enzymes are the structural homologs of phagocytic NOX (gp91phox–NOX2) and consist of both single (NOX‐1‐5), and dual oxidases (DuOX1 and 2).( 14 ) Emerging evidence continues to accumulate that implicates low levels of ROS generated by NOX enzymes as mediators in inflammation, apoptosis, cell growth, and angiogenesis in various types of human cancers; moreover, these might be attractive targets for therapeutic intervention in cancer development.
In the present study, the expression of syndecan‐1 is found to be increased with the progression to hormone resistance, and contributes to cell survival through regulating NOX‐mediated ROS generation in androgen‐independent prostate cancer cells. Our results clearly showed blocking syndecan‐1 signaling or gene silencing offers the promise of a new therapeutic strategy for prostate cancer.
Materials and Methods
Cell culture, plasmids, and chemicals. Human prostate cancer cell lines PC‐3, DU145, and LNCaP were purchased from American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI supplemented with 10% fetal bovine serum. An androgen‐independent LNCaP cell line was established following long‐term subculturing of in DMEM and 5% charcoal‐stripped fetal bovine serum.( 15 ) Anti‐poly (ADP‐ribose) polymerase (PARP) antibody was purchased from Cell Signaling (MA, USA), antiactin and antivascular endothelial growth factor (VEGF) antibodies were purchased from Santa Cruz Biotech (CA, USA), anti‐NOX2 antibody was from Abcam (Cambridge, UK), and antisyndecan‐1 (CD138) and anti‐CD31 antibodies were purchased from Dako (Tokyo, Japan).
Preparation of cell lysates and Western blot analysis. Cell lysates were resolved in SDS‐polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) that were then blocked in 5% skim milk at room temperature for 1 h. Membranes were incubated with the indicated primary antibody for 1 h, and then incubated with horseradish peroxidase‐conjugated antimouse or rabbit IgG (Amersham Pharmacia Biotech, Osaka, Japan). Peroxidase activity was detected on X‐ray film using an enhanced chemiluminescence detection system.
Reverse transcription PCR. Using the One‐step RT‐PCR kit (Qiagen, Germantown, MD, USA), we extracted total RNA using Trizol reagent and subjected it to reverse transcription PCR (RT‐PCR). PCR conditions were 95°C for 30 s, 55–60°C for 30 s, and 72°C for 1 min through a total of 30 cycles. The PCR primer sequences for syndecan‐1 were: 5′‐GGCTGTAGTCCTGCCAGAAG‐3′ (sense) and 5′‐GTTGAGGCCTGATGAGTGGT‐3′ (antisense). The primers for NOX2 were 5′‐GGGCTGTTCAATGCTTGTGGCT‐3′ (sense), and 5′‐ACATCTTTCTCCTCATCATGGTGC‐3′ (antisense). For glyceraldehyde‐3‐phosphate dehydrogenase (G3PDH), the primers used were 5′‐ACCACAGTCCATGCCATCAC‐3′ (sense) and 5′‐TCCACCACCCTGTTGCTGTA‐3′ (antisense).
Measurement of H2O2 production. We assessed the production of intracellular H2O2 with 5,6‐chloromethyl‐2′,7′‐dichlorodihydrofluorescein diacetate (CM–H2DCF–DA, Wako, Osaka, Japan) using a method modified from Bae et al.( 16 ) Briefly, cells were plated on chambered slides and transfected with either control RNA or with syndecan‐1 or NOX2 small interfering RNA (siRNA) and incubated for 60 h, or were treated with 1500 U/mL of catalase for 30 min. Then, we incubated them with a 5 µM CM–H2DCF–DA solution for 20 min at 37°C. Excess CM–H2DCF–DA was washed and H2O2 formation in the cells was visualized using a Leica CTR6000 photomicroscope and analyzed by measuring the fluorescence intensity of 50–100 randomly selected cells.( 17 )
siRNA transfection of syndecan‐1 or NOX2. For our transfection analyses, 106 cells from each prostate cancer cell line were seeded in 6‐cm plates and transfected with either 100 nM of control RNA (Santa Cruz, CA, USA) or with the siRNA of syndecan‐1 or NOX2. Transfections were carried out using the Lipofectamine system (Invitrogen, Tokyo, Japan) in accordance with the manufacturer's protocol. The syndecan‐1 siRNA duplexes, generated with 3′‐dTdT overhangs and prepared by Qiagen, were chosen against the DNA target sequences as: 5′‐CACCATTCTGACTCGGTTTCT‐3′. NOX2 siRNA (gp91phox siRNA) was purchased from Santa Cruz.
Terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling assay. Formalin‐fixed and paraffin‐embedded 5‐µm‐thick sections of all tumor samples were used to identify apoptotic cells by terminal uridine deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining using a tumor TACS in situ apoptosis detection kit (R & D Systems, Mineapolis, MN, USA). The apoptotic index (per microscopic field of 400× magnification) was calculated as the number of apoptotic cells × 100 per total number of cells.( 18 )
Cell proliferation assay. Cells were stimulated with various reagents for a given period, after which MTS reagent (3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfonyl)‐2H‐tetrazolium, inner salt; Promega, Tokyo, Japan) was added. After a 3‐h incubation, optical absorbance at 490 nm was measured using a microplate reader. Cell viability was expressed as mean percentages with standard deviations of absorbance before and after treatment with various reagents. All experiments were performed in triplicate.
Apoptosis detection assay. After transfection with siRNA (syndecan‐1 or NOX2), cells were collected and stained by propidium iodide (PI) and FITC conjugated Annexin V (AV) according to the manufacturer's protocol (TACS Annexin V‐FITC kit, R & D Systems). Then, cells undergoing apoptosis were quantified by measurement of those that bound AV but negative for PI. All experiments were performed at least three times in duplicate.
Tissue samples and immunohistochemistry. Eighteen patients with primary prostate carcinomas receiving no therapies, and 38 patients with neo‐adjuvant androgen deprivation therapies at radical prostatectomy, were examined in this study. All fixation and tissue processing procedures are described in previous reports.( 19 , 20 ) Slides were reviewed for the Gleason score for prostate carcinomas. Informed consent was obtained from all patients before the collection of specimens as appropriate. The study was approved by the ethics committee of Nara Medical University. Sections were incubated with the indicated primary antibodies for 16 h at 4°C and the reactions were visualized using a Histofine kit (Nichirei, Tokyo, Japan), using diaminobenzidine as the chromogen, with hematoxylin counterstaining. Percentages of cells positive for syndecan‐1 were expressed per greater than 1000 cells examined.
Chorioallantoic membrane assay. PC3 cells (106) from each cell line transfected with pEGFP (Clontech, Mountain View, CA, USA) were seeded on the chorioallantoic membranes (CAMs) of 11‐day‐old chick embryos. After a 24‐h cultivation, we added control RNA or 10 µM of syndecan‐1 siRNA combined with atelocollagen (Atelogene, Koken Co., Tokyo, Japan); according to the manufacturer's protocol. Five days post‐implantation, CAM samples were collected and fixed. We assessed angiogenesis as the number of visible blood vessel branch points within a defined area of the sample.( 21 ) At least three CAMs were used for each experiment.
Tumor xenograft study. To establish PC3 cells in mice, PC3 cells were grown in culture, then detached by trypsinization, washed, and resuspended in serum‐free RPMI. Six‐week‐old athymic nu/nu male mice (Oriental Bioservice, Kyoto, Japan) were injected s.c. with 5 × 106 PC3 cells to initiate tumor growth. After 7 days, we subcutaneously injected to tumor foci either control RNA (n = 8) or 10 µM of the CD138 siRNA + atelocollagen mixture (n = 10) as previously described (Koken Co.) Body weight and food consumption were recorded twice weekly throughout the study. After xenografts started growing, their sizes were measured twice weekly. Tumor volume was calculated by the formula 0.5326 × L1 × (L2)2, where L1 is the long axis and L2 is the short axis of the tumor.( 22 ) Fourteen days afterwards, tumors were excised, weighed, and one part fixed in buffered formalin with the remaining portion stored at –80°C until further analysis. Tumor microvessel density was quantified by counting CD31‐positive cells and the total number of cells at 10 randomly selected fields at 400× magnification.( 23 )
Statistical analysis. Data were statistically analyzed using either the Student's t‐test or non‐parametric Kruskal–Wallis test.( 19 , 20 ) Results were considered significant when P < 0.05.
Results
Overexpression of syndecan‐1 in androgen‐independent prostate cancer cells. Expression of syndecan‐1 was immunohistochemically examined using human radical prostatectomy samples either receiving or not receiving neo‐adjuvant hormonal therapy (NAT). In normal glands and prostate cancer cells at hormone naïve status, syndecan‐1 was either not or seldom expressed, with the exception of basal cells. In contrast, it was statistically highly expressed in viable prostate cancer cells following NAT (Fig. 1a,b). From a Western blot shown in Fig. 1(c), syndecan‐1 was observed as a single band, indicating the antibodies selected were of high specificity, and the expression pattern was consistent with the immunohistochemical analysis. Even though viable cancer cells after androgen withdrawal can not always exhibit hormone independence, cell growth or proliferation in response to androgen in these cells is considered to be less sensitive. Therefore, these data mean that syndecan‐1 expression is closely dependent on hormone sensitivity in prostate cancer cells.
Figure 1.

Immune profile of syndecan‐1 in human prostate cancer. (a) Immunohistochemical and (b) statistical analyses of syndecan‐1 in normal glands, and prostate cancer cells with and without neo‐adjuvant androgen withdrawal therapy (NAT). (c) Western blot analysis confirmed antibody specificity (four cases for each). The percentage of immuno‐positive cells was calculated per 1000 cells per high‐power field. A positive correlation between the percentages of syndecan‐1 immuno‐positive cells and tumor grade is shown. Each value is the mean ± SE.
Syndecan‐1 contributes to cell survival in androgen‐independent prostate cancer cells. We examined the expression profile of syndecan‐1 in human androgen‐dependent and ‐independent prostate cancer cell lines LNCaP, PC3, and DU145. Both mRNA and protein expression levels were much higher in PC3 and DU145 than LNCaP. Interestingly, syndecan‐1 in LNCaP was up‐regulated following long‐term culture using 5% charcoal–dextran‐treated fetal bovine serum by which LNCaP cells could continue to proliferate even in androgen‐free conditions; moreover, sensitivity to androgen, by regulation of prostate specific antigen by dihydrotestosterone, was strongly canceled.( 15 ) Syndecan‐1 gene silencing induced apoptosis as determined by AV staining and PARP cleavage. Percentages of apoptotic cells after syndecan‐1 siRNA transfection increased up to 30%, whereas cell growth rate was decreased by 20–40% compared to cases with control RNA transfection (Fig. 2d), suggesting apoptosis induction is mainly involved in cytotoxicity mediated by syndecan‐1 knockdown. Altogether, syndecan‐1 is closely associated with androgen dependency, which could be up‐regulated in the process of androgen‐dependent to ‐independent conversion, and contributes to cell survival in androgen‐independent prostate cancer cells.
Figure 2.

Effect of syndecan‐1 knockdown on cell survival in prostate cancer cells. (a) mRNA and protein expression of syndecan‐1 in LNCaP, DU145, and PC3 cells were examined by RT‐PCR and Western blot analysis. (b) After long‐term culture of LNCaP cells under 5% charcoal‐stripped fetal bovine serum, two clones were selected and syndecan‐1 expression was examined and compared to parental LNCaP. (c) PC3 and DU145 cells were transfected by control RNA or syndecan‐1 siRNA (100 nM). After 48 h‐cultivation, AV and PI double staining and PARP cleavage were analyzed by flow cytometry and Western blot, respectively. (d) Percentages of apoptotic cells, AV(+)/PI(–), were measured by flow cytometry (left panel). Cell growth rate in 48 h‐cultivation after control RNA or syndecan‐1 siRNA transfection was examined (right panel). Each value is the mean ± SE.
Syndecan‐1 is essential for ROS generation through NOX2. Recently, ROS generation through NOX is required for aggressive phenotypes of prostate cancer; therefore, we examined whether syndecan‐1 affects NOX–ROS signals. As shown in Figure 3(a,b), syndecan‐1 silencing resulted in strong reduction of NOX2 mRNA–protein expression, and the endogenous level of ROS was decreased in PC3 and DU145 cells. Similarly to syndecan‐1 knockdown, NOX2 silencing decreased endogenous ROS levels and induced apoptosis to the same extent (Fig. 3c,d). This result indicates that NOX2 and its dependent generation of ROS are an important downstream signal of syndecan‐1, which is essential for prostate cancer cell survival.
Figure 3.

Endogenous generation of hydrogen peroxide was suppressed by syndecan‐1 siRNA transfection in prostate cancer cells. (a) DU145 and PC3 cells were transfected with syndecan‐1 siRNA or control RNA and were incubated for 48 h. H2O2 formation was visualized after exposure to 5 µM CM–H2DCF–DA by fluorescence microscopy. (b) After 48 h of cultivation following control RNA or syndecan‐1 siRNA, expression of NOX2 was examined by RT‐PCR and Western blot analysis. (c) After transfection with 100 nM of syndecan‐1 siRNA, DU145, and PC3 cells were exposed to CM–H2DCF–DA, cellular H2O2 formation was analyzed, and expression of NOX2 was examined by Western blot analysis. Each value indicates percentage of fluorescence intensity to the case without treatment. (d) After siRNA transfection, cells were stained by AV and PI, then percentages of AV(+)/PI(–) cells (apoptotic cells) were measured by flow cytometry. Each value is the mean ± SE.
Knockdown of syndecan‐1 reduced tumor size and angiogenesis in vivo. Finally, we examined the role of syndecan‐1 in tumor progression in vivo. The CAM assay clearly showed knockdown of syndecan‐1 or NOX2 strongly reduced tumor angiogenesis to the same extent in both grafts of PC3 and DU145 cells (Fig. 4a). VEGF expression was consistently, reduced by the silencing of these genes (Fig. 4b). Figure 4(c) illustrates the subcutaneous inoculation of PC3 cells in nude mice. Animals receiving a localized injection of syndecan‐1 siRNA mixed with atelocollagen showed syndecan‐1 down‐regulation, producing approximately two‐fold decreases in tumor size and vessel density (4, 5). Knockdown effects by single injection of syndecan‐1 siRNA with atelocollagen on subcutaneously inoculated prostate cancer cells could be sustained for approximately 5–7 days (K. Shimada et al. unpubl. data, 2008); therefore, we sacrificed mice and collected tumor samples at 7 days after siRNA treatment.
Figure 4.
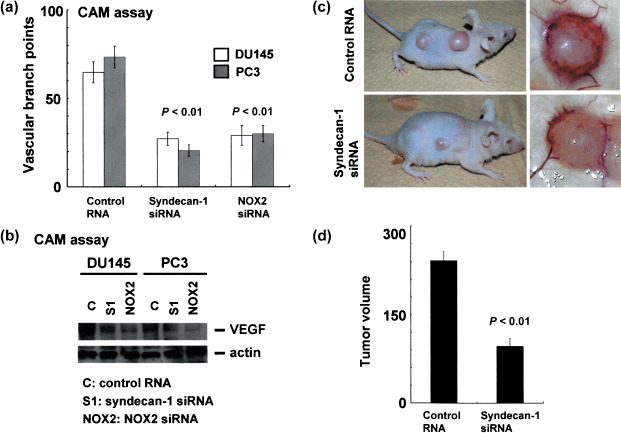
Silencing syndecan‐1 reduced tumor growth and angiogenesis in vivo. DU145 and PC3 cells were seeded on top of the CAMs of 11‐day‐old chicks and atelocollagen‐control RNA or siRNA of syndecan‐1 mixture was added, and incubated for 3 days. (a) Vascular branch points were measured. Each value is the mean ± SE of at least three experiments. (b) CD138 and NOX2 expression in cancer grafts were examined by RT‐PCR and Western blot analysis. (c) Upper and lower panels are macroscopic view of cancer graft (PC3 cells) subcutaneously inoculated after injection of either control RNA, or syndecan‐1 siRNA in the presence of atelocollagen, respectively. (d) Tumor volume was measured 14 days after implantation of PC3 cells and is shown as the ratio to the value at day 0.
Figure 5.

Subcutaneous injection of syndecan‐1 siRNA suppressed VEGF expression and induced apoptosis in vivo in xenograft models. (a) Immunohistochemical staining for syndecan‐1, VEGF, and TUNEL. A representative image has been shown for each transfection group. Magnification 400×. (b) CD31‐positive cells, and (c) TUNEL‐positive cells were calculated by number of positive cells × 100/total number of cells counted under 400× magnification in 10 randomly selected areas in each tumor sample. Columns represent the mean of two samples from individual mice in each group; each value is the mean ± SE. (d) Prostate cancer samples were resected and lyzed, and expression of syndecan‐1, NOX2, and VEGF was examined by Western blot analyses. Expression of actin was used as control.
TUNEL assay showed a significant increase in the number of cells undergoing apoptosis by syndecan‐1 knockdown (control RNA versus syndecan‐1 siRNA, 1.2 ± 0.26%versus 9.9 ± 1.1%) (Fig. 5a,c). Immunohistochemical analysis and Western blot analysis further confirmed that syndecan‐1 expression as well as NOX2 and VEGF were strongly decreased. In contrast, the number of TUNEL positive cells increased in vivo by this single injection of syndecan‐1 siRNA versus control RNA, using actin expression as a positive internal control (Fig. 5a,d). This model also proves the efficacy of atelocollagen to successfully deliver syndecan‐1 siRNA to subcutaneously implanted prostate cancer.
Discussion
We demonstrate here for the first time that syndecan‐1 (CD138) plays important roles in the survival of androgen‐independent prostate cancer cells through NOX‐dependent ROS generation. In vitro experiments using androgen dependent and independent prostate cancer cell lines clearly suggested that syndecan‐1 could be up‐regulated by acquisition of androgen independency, partly supported by immunohistochemical analysis showing that syndecan‐1 was overexpressed in viable prostate cancer cells following neo‐adjuvant androgen withdrawal therapy. Thus, the function of syndecan‐1 is closely associated with hormone sensitivity in human prostate cancer. Syndecan‐1 with its downstream pathway is considered to be one of important survival signals for androgen‐independent prostate cancer cells as well as Ras and mitogen protein kinases. It is well known that NOX2–ROS signals can be executed by various factors other than syndecan‐1; however, the present research clearly shows that NOX2–ROS signals are ineffective under suppression of syndecan‐1. Therefore, syndecan‐1 might be the most important upstream molecule that regulates NOX2‐dependent ROS generation, at least in androgen‐independent prostate cancer cells. Stimulation by testosterone or dihydrotestosterone showed no significant effects on syndecan‐1 expression in LNCaP cells treated with androgen deprivation for a long time (K. Shimada et al. unpubl. data, 2008). Therefore, once syndecan‐1 is overexpressed following androgen withdrawal, the expression could not be modified by androgen re‐introduction. Recently, Kumar et al.( 24 ) demonstrated inhibition of ROS generation by NOX inhibitors increased cell death and suppressed invasion capacity in prostate cancer cells. Thus, syndecan‐1, upstream of the NOX–ROS signal, is required for prostate cancer progression. Kumar et al.'s( 24 ) report showed that a pan‐inhibitor of NOX suppressed matrix metalloproteinase (MMP) 9, but MMPs were not significantly modified even though ROS was similarly suppressed in our study. It is speculated that there are NOX–ROS signal‐dependent and ‐independent mechanisms by which NOX inhibitor (diphenyliodonium) exhibits antitumor action, or other types of NOX than NOX2 may regulate MMPs in prostate cancer cells. Androgen and estrogen are well known to mediate oxidative stress as well as nitrosative stress in epithelial cells, resulting in prostatic carcinogenesis;( 25 ) however, syndecan‐1 was seldom or minimally expressed in normal secretory cells and malignant counterparts at hormone naïve status. Therefore, syndecan‐1 is considered to be involved in the development of prostate cancer but not in early phase of carcinogenesis. One more important effect by syndecan‐1 is regulation of VEGF and angiogenesis. This finding is consistent with several lines of evidence: ROS generation through NOX4 contributes to induction of angiogenesis through up‐regulation of VEGF and hypoxia inducible factor‐1α in ovarian cancer cells.( 26 ) It has been demonstrated that oncogenic Ras‐induced up‐regulation of VEGF and angiogenesis is mediated by NOX1.( 27 ) Moreover, Abid et al.( 28 ) found NOX activity is required for VEGF receptor‐mediated cell signaling; for example, phosphoinositide 3 kinase‐Akt‐forkhead pathways, and NOX modulates the effects of VEGF on endothelial cell phenotype. These reports clearly demonstrate that VEGF induction requires oxidative stresses mainly through NOX, and upstream of syndecan‐1 of NOX–ROS–VEGF signals in various types of tumor cells. We can not explain in detail how syndecan‐1 affects downstream NOX signals, but syndecan‐1 plays a major biological role as a co‐receptor for heparin‐binding growth factors and chemokines in epithelial cells,( 29 , 30 ) which may be associated with activation of NOX‐dependent pathways.
In the mouse model, we found that a single injection of syndecan‐1 siRNA, in the presence of atelocollagen, strongly down‐regulated syndecan‐1, NOX2, and VEGF protein expression, resulting in apoptosis induction and tumor volume reduction. This result was supported by the CAM assay demonstrating syndecan‐1 silencing exhibits suppression of tumor angiogenesis. Since apoptosis induction determined by TUNEL assay was not as prominent as seen in vitro experiments, the anti‐angiogenesis effect might play an important role in reduction of tumor volume by syndecan‐1 gene silencing in human prostate caner. A specific NOX inhibitor for research use, such as diphenyliodonium, has been developed; however, it can not be used as a clinically available drug, probably due to adverse reactions. Several health supplements including multivitamins can achieve neutralization of ROS, but the antitumor effect may be against any real expectation. In our study, treatment alone with ROS neutralizing dugs, for example N‐acetyl‐l‐cysteine, failed to suppress tumor growth and angiogenesis in vitro and in vivo (K. Shimada et al. unpubl. data, 2009). Instead of using such drugs, syndecan‐1 targeting therapy using siRNA plus atelocollagen and treatment with bacterial heparinase III, an enzyme that degrades heparan sulfate chains, or with an inhibitor of human heparanase, an enzyme that synergizes with syndecan‐1, could be more useful therapeutic tools.
In summary, syndecan‐1 was up‐regulated accompanied by hormonal resistance and contributed to the progression of androgen‐independent prostate cancer cells by regulating NOX2‐mediated ROS generation and VEGF‐dependent tumor angiogenesis. We can not deny the possibilities that syndecan‐1 could function by mechanisms other than NOX2, but activation of NOX2 is a mainstay of syndecan‐1‐mediated cell survival signals and may be closely associated with other pathways. Silencing syndecan‐1 by siRNA transfection suppressed the malignant potential in vitro and in vivo; moreover, siRNA combined with atelocollagen may be good delivery system to achieve successful down‐regulation of the gene and provide a new strategy for the treatment of advanced prostate cancer (Fig. 6).
Figure 6.

The role of syndecan‐1 in prostate cancer development. Syndecan‐1 is either not or seldom expressed in androgen‐dependent prostate cancer (PCa) cells, but was strongly up‐regulated under convergence of androgen‐dependent to ‐independent status, and much more highly expressed in androgen‐independent prostate cancer. In addition, syndecan‐1 contributes to cell survival and proliferation signals through maintaining NOX2‐mediated ROS generation in hormone resistant prostate cancer cells. Syndecan‐1‐mediated NOX2–ROS signals could be a new therapeutic target for advanced prostate cancer.
This research was supported in part by a Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (19390104).
References
- 1. Bernfield M, Gotte M, Park PW et al . Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 1999; 68: 729–77. [DOI] [PubMed] [Google Scholar]
- 2. Dhodapkar MV, Abe E, Theus A et al . Syndecan‐1 is a multifunctional regulator of myeloma pathobiology: control of tumor cell survival, growth, and bone cell differentiation. Blood 1998; 91: 2679–88. [PubMed] [Google Scholar]
- 3. Wijdenes J, Vooijs WC, Clement C et al . A plasmocyte selective monoclonal antibody (B‐B4) recognizes syndecan‐1. Br J Haematol 1996; 94: 318–23. [DOI] [PubMed] [Google Scholar]
- 4. Mahtouk K, Cremer FW, Reme T et al . Heparan sulphate proteoglycans are essential for the myeloma cell growth activity of EGF‐family ligands in multiple myeloma. Oncogene 2006; 25: 7180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Derksen PW, Keehnen RM, Evers LM, Van Oers MH, Spaargaren M, Pals ST. Cell surface proteoglycan syndecan‐1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood 2002; 99: 1405–10. [DOI] [PubMed] [Google Scholar]
- 6. Kragh M, Loechel F. Non‐anti‐coagulant heparins: a promising approach for prevention of tumor metastasis (review). Int J Oncol 2005; 27: 1159–67. [PubMed] [Google Scholar]
- 7. Yang Y, MacLeod V, Dai Y et al . The syndecan‐1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood 2007; 110: 2041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet‐derived growth factor signal transduction. Science 1995; 270: 296–9. [DOI] [PubMed] [Google Scholar]
- 9. Shibanuma M, Arata S, Murata M, Nose K. Activation of DNA synthesis and expression of the JE gene by catalase in mouse osteoblastic cells: possible involvement of hydrogen peroxide in negative growth regulation. Exp Cell Res 1995; 218: 132–6. [DOI] [PubMed] [Google Scholar]
- 10. Doudican NA, Song B, Shadel GS, Doetsch PW. Oxidative DNA damage causes mitochondrial genomic instability in Saccharomyces cerevisiae. Mol Cell Biol 2005; 25: 5196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Irani K, Xia Y, Zweier JL et al . Mitogenic signaling mediated by oxidants in Ras‐transformed fibroblasts. Science 1997; 275: 1649–52. [DOI] [PubMed] [Google Scholar]
- 12. Babior BM. The respiratory burst oxidase. Curr Opin Hematol 1995; 2: 55–60. [DOI] [PubMed] [Google Scholar]
- 13. Segal AW, Shatwell KP. The NADPH oxidase of phagocytic leukocytes. Ann N Y Acad Sci 1997; 832: 215–22. [DOI] [PubMed] [Google Scholar]
- 14. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004; 4: 181–9. [DOI] [PubMed] [Google Scholar]
- 15. Shimada K, Nakamura M, Ishida E et al . Prostate cancer antigen‐1 contributes to cell survival and invasion though discoidin receptor 1 in human prostate cancer. Cancer Sci 2008; 99: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bae YS, Kang SW, Seo MS et al . Epidermal growth factor (EGF)‐induced generation of hydrogen peroxide. Role in EGF receptor‐mediated tyrosine phosphorylation. J Biol Chem 1997; 272: 217–21. [PubMed] [Google Scholar]
- 17. Tsujikawa K, Koike K, Kitae K et al . Expression and sub‐cellular localization of human ABH family molecules. J Cell Mol Med 2007; 11: 1105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Singh RP, Tyagi A, Sharma G, Mohan S, Agarwal R. Oral silibinin inhibits in vivo human bladder tumor xenograft growth involving down‐regulation of survivin. Clin Cancer Res 2008; 14: 300–8. [DOI] [PubMed] [Google Scholar]
- 19. Shimada K, Matsuyoshi S, Nakamura M, Ishida E, Konishi N. Phosphorylation status of Fas‐associated death domain‐containing protein (FADD) is associated with prostate cancer progression. J Pathol 2005; 206: 423–32. [DOI] [PubMed] [Google Scholar]
- 20. Konishi N, Nakamura M, Kishi M, Nishimine M, Ishida E, Shimada K. Heterogeneous methylation and deletion patterns of the INK4a/ARF locus within prostate carcinomas. Am J Pathol 2002; 160: 1207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shimada K, Nakamura M, Ishida E et al . c‐Jun NH2 terminal kinase activation and decreased expression of mitogen‐activated protein kinase phosphatase‐1 play important roles in invasion and angiogenesis of urothelial carcinomas. Am J Pathol 2007; 171: 1003–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singh RP, Mallikarjuna GU, Sharma G et al . Oral silibinin inhibits lung tumor growth in athymic nude mice and forms a novel chemocombination with doxorubicin targeting nuclear factor kappaB‐mediated inducible chemoresistance. Clin Cancer Res 2004; 10: 8641–7. [DOI] [PubMed] [Google Scholar]
- 23. Singh RP, Deep G, Chittezhath M et al . Effect of silibinin on the growth and progression of primary lung tumors in mice. J Natl Cancer Inst 2006; 98: 846–55. [DOI] [PubMed] [Google Scholar]
- 24. Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 2008; 68: 1777–85. [DOI] [PubMed] [Google Scholar]
- 25. Azzali G. Tumor cell transendothelial passage in the absorbing lymphatic vessel of transgenic adenocarcinoma mouse prostate. Am J Pathol 2007; 170: 334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res 2007; 67: 10 823–30. [DOI] [PubMed] [Google Scholar]
- 27. Komatsu D, Kato M, Nakayama J, Miyagawa S, Kamata T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras‐induced vascular endothelial growth factor expression. Oncogene 2008; 27: 4724–32. [DOI] [PubMed] [Google Scholar]
- 28. Abid MR, Spokes KC, Shih SC, Aird WC. NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J Biol Chem 2007; 282: 35 373–85. [DOI] [PubMed] [Google Scholar]
- 29. Rapraeger AC. Syndecan‐regulated receptor signaling. J Cell Biol 2000; 149: 995–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Couchman JR. Syndecans. proteoglycan regulators of cell‐surface microdomains? Nature Rev 2003; 4: 926–37. [DOI] [PubMed] [Google Scholar]