

Abstract
Transfer of genetic information during mitosis is accurately conducted by proper condensation and segregation of chromosomes, for which condensins play a central role. Both condensin I and II have common structural maintenance of chromosomes subunits, named hCAP‐C and hCAP‐E. Pyothorax‐associated lymphoma (PAL) is a non‐Hodgkin's lymphoma developing in the pleural cavity of patients with long‐standing pyothorax. Mutations of hCAP‐C and hCAP‐E were investigated in 24 leukemia–lymphoma cell lines including eight PAL cell lines, and their influences in chromosome morphology were evaluated. Heterozygous point mutations within hCAP‐C were found in two PAL cell lines and corresponding tumor samples (OPL‐3 and OPL‐7). Deletion of exon 24 within hCAP‐E and a point mutation at the donor splice site of intron 24 were detected in OPL‐5 and original tumor samples. OPL‐5 showed an extensive reduction in expression of not only hCAP‐E but also hCAP‐C proteins. OPL‐5 occasionally showed the chromosome bridge in anaphase and telophase, indicating that segregation is not accurate. OPL‐7 showed reduced hCAP‐C protein expression, abnormality in chromosome length and width, and abnormal aggregates of hCAP‐C protein. These findings indicated that condensin gene alteration might play a role in genome instability, which accelerates the accumulation of other gene alterations in PAL. (Cancer Sci 2007; 98: 1041–1047)
Abbreviations
- DAPI
4’,6’‐diamidino‐2‐phenylindole dihydrochloride
- gDNA
genomic DNA
- PAL
pyothorax‐associated lymphoma
- PBS
phosphate‐buffered saline
- RT‐PCR
reverse transcription–polymerase chain reaction
- siRNA
, small interfering RNA
- SMC
structural maintenance of chromosomes
In a mitotic process, two copies of genomic DNA (gDNA) must be properly segregated into each daughter cell after a replication.( 1 , 2 , 3 ) Mitotic cells have to manage with two mechanical problems: the size of gDNA is enormous with regard to the mitotic apparatus; and sister chromatids as well as different chromosomes are topologically entangled. Therefore chromosome segregation needs any tangles to loosen and chromosomes to be highly compacted. Cells that are defective for either chromosome compaction or resolution fail to faithfully segregate replicated chromosomes and become aneuploid, which induces genome instability and subsequent malignant transformation.( 4 ) The compaction and resolution processes are likely to be coupled.
Both topoisomerase II and other condensation factors are components of the mitotic chromosomes. Among chromosomal components, condensins are multisubunit protein complexes that play a central role in mitotic chromosome assembly and segregation.( 1 ) The core of the complexes contains hCAP‐C and hCAP‐E, which are SMC subunits. hCAP‐C is located on chromosome 3q26, hCAP‐E is located on 9q31. There are two different condensin complexes in vertebrate cells, condensins I and II. Both complexes contain hCAP‐C and hCAP‐E, together with other non‐SMC subunits. Condensin I contains CAP‐D2, CAP‐G and CAP‐H,( 2 , 3 ) and condensin II contains CAP‐D3, CAP‐G2 and CAP‐H2.( 5 ) Condensin II participates in an early stage of chromosome condensation within the prophase nucleus. Condensin I gains access to chromosomes only after the breakdown of the nuclear envelope, and collaborates with condensin II to assemble metaphase chromosomes with fully resolved sister chromatids.( 5 ) Recent studies have shown that the condensins play a crucial role not only for chromosome segregation during mitosis and meiosis, but also chromosome‐wide gene regulation.( 4 , 6 , 7 , 8 )
Malignant lymphoma frequently develops in the pleural cavity of patients with over a 20‐year history of pyothorax.( 9 ) The term PAL has been proposed by Dr Aozasa for this type of tumor.( 10 ) This tumor is now being listed as a distinct clinicopathological entity in the recent World Health Organization Classification of Tumors.( 11 ) Most PAL are diffuse large lymphomas of B‐cell type, but unusual types of PAL, such as tumor with biphenotypic character of B and T cells, T‐cell rich B‐cell lymphoma, and Ki‐1 positive anaplastic large cell lymphoma of T‐cell phenotype, were also reported.( 9 ) PAL cell lines in the previous reports showed interesting features, which mirror the unique etiology of PAL. al Saati et al.( 12 ) reported a PAL‐derived cell line with dual B‐cell and T‐cell phenotype. Daibata et al.( 13 ) reported one cell line of PAL that expresses both CD2 and CD20, but did not express representative B‐cell markers or T‐cell markers CD1, CD3, CD5, CD7, and CD9. We previously established six B‐cell lines from the biopsy specimens of six PAL patients that had highly complex karyotypes,( 14 , 15 ) suggesting an increase in genome instability.
In the present study, mutations of hCAP‐C and hCAP‐E in 24 leukemia–lymphoma cell lines including eight PAL cell lines were analyzed. Three of the eight PAL cell lines have either hCAP‐C or hCAP‐E heterozygous mutations, indicating that condensin mutations were relatively frequent in PAL.
Materials and Methods
Cell lines. A total of 24 leukemia–lymphoma cell lines derived from PALs (OPL‐1, OPL‐2, OPL‐3, OPL‐4, OPL‐5, OPL‐7, Deglis and Pal‐1),( 12 , 13 , 14 , 15 ) Hodgkin's lymphomas (HDLM‐2, HD‐My‐Z, KM‐H2, L‐428, L‐540 and L‐1236), Burkitt's lymphomas (Daudi, Raji, Ramos), NK/T‐cell lymphomas (MTA, NK‐92), acute lymphoblastic leukemia (NALM‐6, HPB‐ALL, Jurkat, P30/Ohkubo), acute myeloid leukemia (P31/Fujioka),( 16 ) and HeLa were used in this study. Two lymphoblastoid cell lines (IB‐4 and LCL‐T) were used as controls.( 17 ) PAL cell lines, OPL‐1, ‐2, ‐3, ‐4, ‐5, and ‐7 were established in our laboratory from patients with PAL who had a history of between 39 and 66 years of chronic pyothorax resulting from artificial pneumothorax for the treatment of lung tuberculosis or tuberculous pleuritis.( 14 , 15 ) Pal‐1, Deglis, and IB‐4 were kind gifts from Drs M. Daibata, T. al Saati, and E. Kieff, respectively. Daudi, Raji, Ramos, Jurkat, MTA, P30/Ohkubo, P31/Fujioka, and HeLa were obtained from Japanese Collection of Research Bioresources (Tokyo, Japan). HDLM‐2, HD‐My‐Z, KM‐H2, L‐428, L‐540, L‐1236, NALM‐6, HPB‐ALL, and NK‐92 were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). All cell lines were incubated in RPMI‐1640 medium (Sigma, St Louis, MO) supplemented with 10% heat‐inactivated fetal bovine serum and antibiotics at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Patients. Tumor samples obtained by biopsy and peripheral blood samples from three patients with PAL admitted to hospitals in the area of Osaka, Japan, were used to extract genomic DNA. Informed consent was obtained in accordance with the Declaration of Helsinki. The three cell lines OPL‐3, ‐5, and ‐7 were established from these three patients.
Isolation of total RNA, RT‐PCR and detection of mutations. Total RNA was extracted from the cell lines using TRIzol reagent (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Five micrograms of total RNA was reverse transcribed by random hexamer priming using the Superscript first strand synthesis system (Invitrogen). Semi‐nested RT‐PCR was carried out to amplify the hCAP‐C and hCAP‐E transcripts using two sets of primers spanning the whole open reading frame (Table 1). After electrophoresis on 1.0% agarose gel, DNA fragments were excised and purified using the Wizard SV gel extraction kit (Promega, Madison, WI). The purified DNA was used as the template and directly sequenced with appropriate primers. When the potential mutations were detected, corresponding mutations were confirmed using gDNA from cell lines as well as tumor biopsy samples from which the cell lines were derived. The PCR products were cloned into the plasmid pCR2.1‐TOPO (Invitrogen), then 8–10 clones were sequenced to confirm the mutations and their frequency.
Table 1.
Oligonucleotide primers used for amplification of hCAP‐C and hCAP‐E genes
| First primer sequence | Second primer sequence | PCR products (bp) † | ||
|---|---|---|---|---|
| cDNA primers | ||||
| Cap‐C | F | 5′‐GGAGGTGGGTACTACAC‐3′ | 5′‐CACAACCGTCTCCAGCCTT‐3′ | 2063 (nt. 76–2138) |
| R | 5′‐CCCATACAGCCATCTTATC‐3′ | 5′‐CCCATACAGCCATCTTATC‐3′ | ||
| F | 5′‐GACGTGGCTATATCATCCTG‐3′ | 5′‐GACGTGGCTATATCATCCTG‐3′ | 2032 (nt. 1994–4025) | |
| R | 5′‐CAACTCATAATCCTTTACAAATAG‐3′ | 5′‐GAAAAAAATCAGTAATACACTG‐3′ | ||
| Cap‐E | F | 5′‐GCAGCGAACTGGTTTGTG‐3 | 5′‐CCTGTTTGATTCCTGTCAGAG‐3′ | 1928 (nt. 167–2094) |
| R | 5′‐GTTGTTCCAAAGACAAACTCC‐3′ | 5′‐GTTGTTCCAAAGACAAACTCC‐3′ | ||
| F | 5′‐CTGAAACGTCGATACACTATAATTCCACTC‐3′ | 5′‐CTGAAACGTCGATACACTATAATTCCACTC‐3′ | 1931 (nt. 1925–3855) | |
| R | 5′‐CAAATTAGTTATCTCAAGTCCT‐3′ | 5′‐GTTTACATTTAAAAAAACAGGTC‐3′ | ||
| Genomic primers | ||||
| Cap‐C exon 9 | F | 5′‐GGTAACATAAAGATGTATGCTC‐3′ | – | 185 |
| exon 9 | R | 5′‐AGCCCTTGTGTTTCCTG‐3′ | – | |
| Cap‐C exon13 | F | 5′‐CTCCTGAAAATACTCCTCG‐3′ | – | 187 |
| exon13 | R | 5′‐GACTGTTCTATGATTTGTCC‐3′ | – | |
| Cap‐E exon 24 | F | 5′‐GTTTATTTCTGACCAAATTCCAG‐3′ | – | 241 |
| exon 24 | R | 5′‐AGAGTATAACTCCACTGATGA‐3′ | – | |
Western blotting. Western blotting was carried out as described previously.( 18 ) Briefly, whole cells were lyzed in 1 × sample buffer, separated with 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis for hCAP‐C and hCAP‐E protein, then blotted to polyvinylidene difluoride membrane using a wet‐blotting apparatus. Anti‐hCAP‐C antibody (BL551) was purchased from Bethyl (Montgomery, TX) and anti‐hCAP‐E antibody was kind gift from Professor T. Hirano (Cold Spring Harbor Laboratory, New York, NY, USA). The epitope of hCAP‐C locates on exon 23, and hCAP‐E on the exon 27 encoding sequence. Anti‐actin antibody (Sigma) was used as the control. Signals were visualized with ECL Plus chemiluminescent reagents (GE Healthcare Bio‐Sciences, Piscataway, NJ).
Chromosome length and width measurement. Mitotic cells were collected after 2 h incubation with 4 µL/mL colcemid (Sigma). HeLa cell lines at mitotic phase were collected by tapping the plate for 1 min. The cells were incubated with 75 mM KCl in 37°C for 20 min, fixed in 3:1 (v/v) HPLC‐grade absolute methanol/glacial acetic acid, and mixed well gently before spreading onto slides.( 19 ) Chromosomes staining were carried out in 4% Giemsa solution for 10 min. Cells were observed using an Axioplan‐2 microscope (Carl Zeiss, Oberkochen, Germany), and chromosome lengths (the telomere‐to‐centromere‐to‐telomere length) and widths in cells at prometaphase were measured using IP Laboratory Spectrum v.3.1 (Signal Analytics, Vienna, VA). We analyzed chromosomes that had just entered prometaphase, in which sister chromatids were almost attached, because longitudinal chromosome compaction in metaphase proceeds during the colcemid treatment. Experiments were carried out in triplicate and 150–210 chromosomes were counted per cell line.
RNA interference silencing. The siRNA duplexes mixture specific to hCAP‐C and hCAP‐E were purchased from B‐Bridge International (Sunnyvale, CA). The sequences of the sense strands are as follows: hCAP‐C, 5′‐GGGUAAAGAUGGUGGAAAATT‐3′, 5′‐AAAAGAAAGUCGAGAGAAATT‐3′, 5′‐GCGAAAAAGAUGACCGAAATT‐3′; hCAP‐E, 5′‐CUUAAAUGGUAGTGGGAAATT‐3′, 5′‐GCACAGAAAACUAUAGAAAUU‐3′, 5′‐GGAAAUACCUGGAAAGAAAUU‐3′. RNA interference was carried out with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. Briefly, HeLa cells were grown at 50% starting confluence without antibiotics in a 6‐cm dish culture and transfected with 50 nM RNA duplexes using 4 µL of Lipofectamine RNAiMAX. Control cells were transfected with a mixture containing no siRNA. The cells were processed for further analysis for 24 h.
Transfection of hCAP‐C and hCAP‐E mutant genes. The constructs of hCAP‐C and hCAP‐E were kindly provided by Professor T. Hirano (Cold Spring Harbor Laboratory). Point mutations in C1410T and G2294A, and deletion of exon 24 were introduced into hCAP‐C and hCAP‐E, respectively. The constructs were cloned to p3XFLAG‐CMV‐7.1 expression vector (Sigma), which was designed to express 5′‐FLAG tagged proteins. These constructs were transfected into the HeLa cells with FuGENE 6 Transfection Reagent (Roche, Basel, Switzerland) according to the manufacturer's instructions and cultured at 37°C for 48 h. After replacing the medium with PBS, cells were collected by tapping the dish for 3 min, incubated with 0.5 × PBS hypotonic solution in room temperature for 30 min, then subjected to the further analysis.
Flow cytometry. Flow cytometry analysis was carried out using FACS (Becton Dickinson, Franklin Lakes, NJ). In brief, 1 × 106 cells were fixed with 2.0% formaldehyde in PBS for 5 min, and permeabilized with 0.5% Triton X‐100 in PBS. The cells were then incubated with anti‐hCAP‐E and anti‐hCAP‐C rabbit antibodies, followed with Alexa Fluor 488 goat antirabbit immunoglobulin G (Invitrogen). Data were collected and analyzed with the Cell Quest DNA quantization program (Becton Dickinson).
Immunofluorescence microscopy. Slides for condensin immunofluorescence staining were prepared by cytospin of each cell in Autosmear CF‐12D (Sakura, Nagano, Japan) with 500 g for 5 min. PAL cells were fixed with 2.0% formaldehyde in PBS for 5 min. PAL cells were then permeabilized with 0.5% Triton X‐100 in PBS for observation. HeLa cells were fixed with methanol–aceton solution, 1:1 concentration, for 1 min. After serum blocking, anti‐hCAP‐E, ‐hCAP‐G, ‐hCAP‐H2, and ‐hCAP‐G2 antibodies (kind gifts from Dr T. Hirano), anti‐hCAP‐C antibody (Bethyl), and Cy3‐conjugated anti‐FLAG antibody (Sigma) were applied and incubated for 1 h at room temperature. Slides were then treated with Alexa Fluor 488 goat anti‐rabbit immunoglobulin G (Invitrogen) for 1 h at 37°C. After thorough washing, DAPI solution (Wako, Osaka, Japan) were applied and slides were covered with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). LSM 510 META confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany) was used for analysis.
Statistical analysis. Comparisons were made using Student's t‐test (unpaired). P‐values less than 0.05 were taken as statistically significant.
Results
hCAP‐C and hCAP‐E mutations. Mutational status of hCAP‐C and hCAP‐E in 24 leukemia–lymphoma cell lines were examined. Missense mutations of the hCAP‐C gene were detected in two PAL cell lines (Fig. 1a). Mutations involved codon 728 (GCA → ACA; Ala → Thr) in OPL‐3 and codon 433 (GCC → GTC; Ala → Val) in OPL‐7 (GenBank accession number AB019987). The identical heterozygous point mutations within hCAP‐C in OPL‐3 and OPL‐7 were detected using gDNA from each cell line as well as from original tumor samples, but not from peripheral blood samples as template (data not shown).
Figure 1.

hCAP‐C and hCAP‐E gene alterations detected in pyothorax‐associated lymphoma cell lines. (a) Two cell lines had heterozygous hCAP‐C mutations, and one had heterozygous hCAP‐E mutations. (b) A transition of the invariable G ⇒ A at position +1 of the donor splice site of intron 24 was found in OPL‐5, which resulted in aberrant splicing in exon 24 of hCAP‐E.
A 117 bp deletion corresponding to exon 24 within hCAP‐E was detected in OPL‐5 (Fig. 1b). A heterozygous transition of the invariable G → A at position +1 at the donor splice site of intron 24 within hCAP‐E in OPL‐5 was detected using gDNA from each cell line as well as from original tumor samples, but not from peripheral blood samples as template (data not shown). These findings indicated that the mutations were somatic ones occurring in each PAL patient.
Mutations were not found in the two lymphoblastoid cell lines (IB‐4, LCL‐T), or cell lines from six Hodgkin lymphomas (HDLM‐2, HD‐My‐Z, KM‐H2, L‐428, L‐540, L‐1236), three Burkitt's lymphomas (Daudi, Raji, Ramos), two NK/T cell lymphomas (MTA, NK‐92), four acute lymphoblastic leukemias (NALM‐6, HPB‐ALL, Jurkat, P30/Ohkubo), or one acute myeloid leukemia (P31/Fujioka).
Western blotting. hCAP‐C and hCAP‐E protein expression was examined by Western blotting (Fig. 2). Of the two cell lines harboring aberrant hCAP‐C transcripts, OPL‐7 showed approximately 60% reduced expression of hCAP‐C protein, whereas OPL‐3 showed comparable expression with control IB4. OPL‐4 and OPL‐5 showed a decreased hCAP‐C expression (approximately 30% and >90% reductions) although no mutations were detected in the coding sequence within hCAP‐C. OPL‐5 harboring heterozygous 117 bp deletions within hCAP‐E transcripts showed more than 90% reduction in hCAP‐E protein expression, and no aberrant proteins were detected. The karyotypes and aberrations in hCAP‐C/‐E expressions and mutations are summarized in Table 2.
Figure 2.
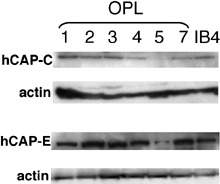
hCAP‐C and hCAP‐E protein expression in pyothorax‐associated lymphoma cell lines. Reduction of hCAP‐C was observed in OPL‐4, OPL‐5, and OPL‐7. Reduction of hCAP‐E was observed in OPL‐5. Anti‐actin labeling shows equivalence of loading of cell lysates.
Table 2.
Summary of karyotypes and aberrations in hCAP‐C and hCAP‐E expressions and mutations in pyothorax‐associated lymphoma (PAL) cell lines
| Name | Karyotypes | Aberrations in hCAP‐C and hCAP‐E expressions and mutations | Ref. |
|---|---|---|---|
| OPL‐1 | 47–48, X, add(1)(p11), der(2)add(2)(p23)add(2)(q35), add(4)(q21), der(4)(p16)add(4)(q35), + add(5)(p13), add(5)(q13), del(6)(q21), der(8)add(8)(p11)add(8)(q24), add(11)(q23), add12(q24), –13, add(13)(p11), add(14)(q24), add(15)(p11), der(15)t(10; 15)(q11;q22), –16, del(17)(q24), –18, add(20)(p11), del (20)(q11), der(21)t(13; 21)(q12;p11) | † | |
| OPL‐2 | 46–207, del(1)(q21) × 1–2, add(2)(p11.1), add(7)(q32 × 2, der(11)t(11; I4)(p15; q11) × 2, add (15)(p13), der(l9)t(1; 19)(q21; p13.3) X 2. Band 14q32.33 | ( 15 ) | |
| OPL‐3 | 43–49, XY, add (3)(q25), del (7)(q32), add (8)(p11), der (8), t(1;8)(q24;q24), del (12)(p11), add (13)(p11), add (17)(p11), add (18)(q21), –21, der (22), t (1; 22)(q12;p11), del(1)(q21q25), +0–3 mar | hCAP‐C; G2294A (Ala → Thr) | ( 14 ) |
| OPL‐4 | 54–55, X, + add (1)(q11), der (1), add (1)(p36), add (1)(q21), + add (2)(p25), der (2), add (2)(p13), add (2)(q33), der (4), t (4; 9)(p16;q11), + add (5)(q11), der (5) del (5)(p15) add (5)(q31), i (5)(q10), –6, add (6)(p21), +7, –9, del (9)(p21), add (10)(p11), add (11)(q23), add (11)(q23), add (12)(q24) × 2, add (13)(q32), +14, –15, add (15)(p11), –16, add (16)(q22), –17, +20, mar1, mar2, mar3, mar4, mar5, +2–4mar | reduced hCAP‐C protein expression | ( 14) |
| OPL‐5 | 40–42, XX, –8, add(9)(p23), –10, der(12)del(p?)add(12)q(24), add(13)(p11), –15, add(15)(q22), –18, add(18)(q21), + add(19)(p13), add(19)(p13), –21, + mar1, +0–1 mar | hCAP‐E; del exon 24 reduced hCAP‐C and hCAP‐E protein expression | ( 14 ) |
| OPL‐7 | 47, X, add(X)(q11), t(1; 14)(q21;q22), t(2; 13)(p10;p10), inv(5)(p15q33),−6, −7,add(8)(p21), add(9)(q34), der(10)t(6; 10)(p11;p15), add(11)(p15), der(12)del(12)(p?)add(12)(q22), –13, add(19)(p11), add(19)(p13), der(19)t(7; 19)(q11;p13), +mar1, +mar2 × 2 | hCAP‐C; C1410T (Ala fi Val) reduced hCAP‐C protein expression | ( 14 ) |
| Deglis | 46, XY, −2, + der(2)(?:: 2p23 +2q37::?), −4, + der(4)t(4;?)(q34;?), i(6p), del(6)(p22), + del(6)(p22), −8, + der(8)t(8;?) (q24;?), t(13; lS)(pll; pl l), t(21; 22)(pll; pl l), + mar | ( 12 ) | |
| Pal‐1 | 48–49. X, t(1; 14)(p34;q32), der(3)t(3;7)(p24:q21)ins(3;?)8p24;?), der(8)t(8; 14)(p11;q11)del(8)(q24), –14, +20, –22, mar1, mar2, mar3 | (13) |
Takakuwa T et al., unpublished data, 2003.
Morphology and distribution of condensin protein during cell division in PAL cell lines. The effect of hCAP‐C and hCAP‐E mutations on the morphology and distribution of condensin protein during cell division was analyzed on the DAPI stained preparations using confocal laser scanning microscopy. Chromosome bridges were occasionally observed at the anaphase and telophase in OPL‐5 (Fig. 3a). No apparent changes were observed in other cell lines. Distribution of condensin proteins was examined with immunostaining. In IB‐4 and OPL‐2, hCAP‐C and hCAP‐E were localized both in the nucleus and cytoplasm of cells during interphase. When the cells entered mitosis (arrowhead), hCAP‐E was localized in a chromosomal pattern (Fig. 3b), while hCAP‐C predominantly localized in the cytoplasm. Both hCAP‐E and hCAP‐C showed dot‐like ‘aggregate’ during mitosis (arrowhead) in OPL‐3 and more conspicuously located mainly on the chromosomes and occasionally in the cytoplasm in OPL‐7. Similar dot‐like ‘aggregate’ during mitosis was observed, although high detection sensitivity was required for analyzing OPL‐5. The distribution pattern of hCAP‐C and hCAP‐E at the interphase was not different among PAL and control lines (data not shown). Distribution of other condensin subunits, hCAP‐G, hCAP‐G2 and hCAP‐H2, was not apparently different among PAL and control lines (data not shown).
Figure 3.
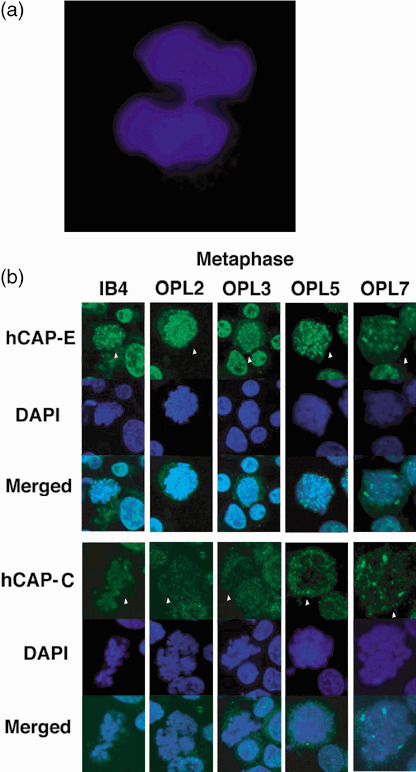
(a) Chromosome bridges in anaphase and telophase observed by 4’,6’‐diamidino‐2‐phenylindole dihydrochloride staining. Chromosome bridges in anaphase and telophase were occasionally observed in OPL‐5. (b) hCAP‐C and hCAP‐E localization in mitotic cells. In IB‐4 and OPL‐2 cell lines, immunofluorescent staining for hCAP‐E condensin subunit showed a chromosomal pattern of distribution, whereas hCAP‐C showed predominantly in the cytoplasm. Dot‐like ‘aggregates’ of hCAP‐E and hCAP‐C in the chromosomes and occasionally in the cytoplasm were observed in OPL‐3, OPL‐5, and OPL‐7 during mitosis (arrowhead).
Morphology of chromosomes in PAL cells. The chromosome morphology was then compared between the cell lines (Fig. 4a,b). The mean chromosome length was significantly shorter in OPL‐4 (mean ± SD; 3.00 ± 0.55 µm), OPL‐5 (3.25 ± 0.18 µm) and OPL‐7 (3.21 ± 0.24 µm) than in IB‐4 (4.47 ± 0.50 µm) and LCL‐T (4.47 ± 0.56 µm). The width of chromosomes was significantly broader in OPL‐3 (mean ± SD; 0.93 ± 0.09 µm), OPL‐4 (1.05 ± 0.08 µm), OPL‐5 (1.10 ± 0.12 µm), and OPL‐7 (0.97 ± 0.09 µm) than in IB‐4 (0.71 ± 0.14 µm) and LCL‐T (0.70 ± 0.14 µm).
Figure 4.
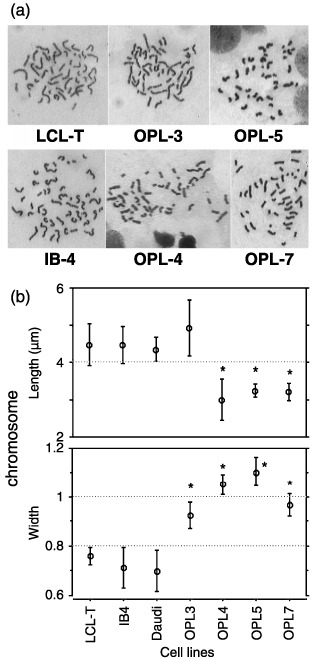
Comparison of chromosomal morphology among pyothorax‐associated lymphoma (PAL) cell lines. (a) Representative chromosome spreads from PAL cell lines are shown. (b) Chromosome lengths and widths in prometaphase from PAL cell lines were measured as described in ‘Materials and Methods’. The mean chromosome length was significantly shorter in OPL‐4 (mean ± SD; 3.00 ± 0.55 µm), OPL‐5 (3.25 ± 0.18 µm) and OPL‐7 (3.21 ± 0.24 µm) than in IB‐4 (4.47 ± 0.50 µm) and LCL‐T (4.47 ± 0.56 µm). The mean chromosome width was significantly broader in OPL‐3 (mean ± SD; 0.93 ± 0.09 µm), OPL‐4 (1.05 ± 0.08 µm), OPL‐5 (1.10 ± 0.12 µm), and OPL‐7 (0.97 ± 0.09 µm) than in IB‐4 (0.71 ± 0.14 µm) and LCL‐T (0.70 ± 0.14 µm).
Morphology of chromosomes in HeLa cells with hCAP‐C and hCAP‐E depletion. To analyze the effect of depletion of condensin subunits on chromosomal morphology, chromosome spreads were prepared from both HeLa cells transfected with and without hCAP‐C siRNA duplex. In the HeLa cells with approximately 70% of hCAP‐C and 55% of hCAP‐E depletion, the shape of chromosomes was ‘blurred’ and not compact enough to control cells (Fig. 5a,b). The shape around the telomere regions was ambiguous. The chromosomes in hCAP‐C and hCAP‐E depleted cells were significantly shorter in length and broader in width than that in control lines. The mean length ± SD for hCAP‐C depleted cells, hCAP‐E depleted cells and control HeLa cells was 3.87 ± 1.79 µm, 4.52 ± 2.25 µm and 6.7 ± 3.25 µm, respectively, and the mean width ± SD was 2.02 ± 0.46 µm, 1.89 ± 0.41 µm and 1.43 ± 0.22 µm, respectively. Immunofluorescence staining was prepared from HeLa cells transfected with hCAP‐C/‐E siRNA duplex and control (no treatment of anti‐hCAP‐C/‐E antibody in HeLa) after hypotonic treatment (Fig. 5c). The signal from the control series, both for hCAP‐C and hCAP‐E, was very strong and stained along with the chromosomes. HeLa cells transfected with siRNA duplex specific to hCAP‐C/‐E gave significantly weaker dot‐like signals compared to the control. Chromosomes tended to be more dispersed, wider, and curly in shape, as shown by DAPI staining.
Figure 5.
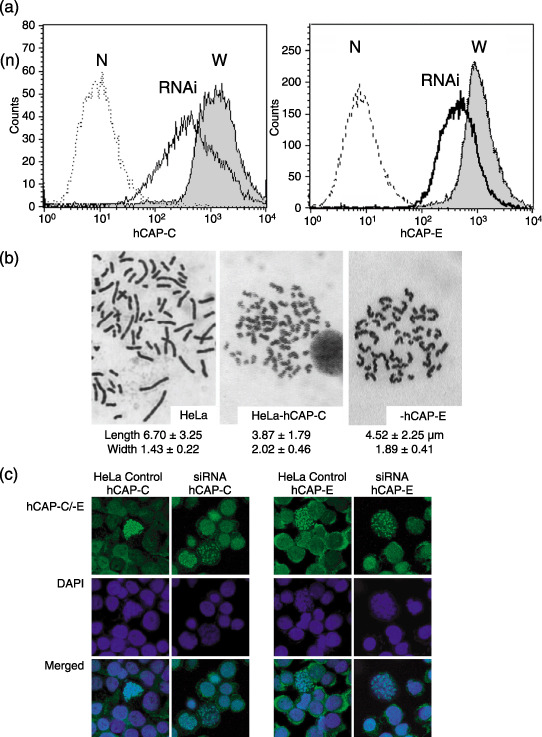
Depletion of hCAP‐C and hCAP‐E in the HeLa cell line. (a) Flow cytometry revealed that transfection of HeLa cells with small interfering (si)RNA duplex specific to hCAP‐C and hCAP‐E results in reduction of condensin to 30% and 45%, respectively, as compared to control HeLa cells. N, no treatment of anti‐hCAP‐C and anti‐hCAP‐E antibody in HeLa cells as negative control; RNAi, HeLa cells transfected with siRNA duplex specific to hCAP‐C and hCAP‐E; W, HeLa cells transfected without siRNA. (b) HeLa cells without depletion of hCAP‐C showed proper condensation during mitosis, resulting in a “compact” chromosome, 6.70 ± 3.25 µm in length and 1.43 ± 0.22 µm in width. Depletion of hCAP‐C and hCAP‐E using siRNA duplex results in impaired condensation as revealed by shortening and broadening of chromosomes, 3.87 ± 1.79 µm in length and 2.02 ± 0.46 µm in width for hCAP‐C, and 4.52 ± 2.25 µm in length and 1.89 ± 0.41 µm in width for hCAP‐E. (c) Immunofluorescent staining prepared from HeLa cells transfected with hCAP‐C/‐E siRNA duplex and control (no treatment of anti‐hCAP‐C/‐E antibody in HeLa) after hypotonic treatment. The signal from the control series, both for hCAP‐C and hCAP‐E, was very strong and stained along with chromosomes. HeLa cells transfected with siRNA duplex specific to hCAP‐C/‐E gave significantly weaker and dot‐like signals compared to the control. Chromosomes tended to be more dispersed, wide, and curly in shape as shown by 4’,6’‐diamidino‐2‐phenylindole dihydrochloride staining.
Morphology of chromosomes in HeLa cells with FLAG‐tagged hCAP‐C and hCAP‐E mutations. To examine whether the observed alterations in PAL cell lines result in the change in chromosome morphology, these alterations were introduced into HeLa cell lines. Anti‐FLAG staining showed that the introduced protein located mainly in cytoplasm at interphase, whereas some parts of the protein were moved to the chromosomes at mitosis (Fig. 6). The same intensity of signals at interphase was observed in all series of hCAP‐C and hCAP‐E staining. Cells transfected with wild‐type hCAP‐C and hCAP‐E showed stronger signals compared to cells with mutated hCAP‐C and hCAP‐E. The hCAP‐C and hCAP‐E signals were observed along with chromosomes in cells with wild‐type hCAP‐C and hCAP‐E. In contrast, the signals were dot‐like and arranged in curly shapes on the chromosomes in cells with mutated hCAP‐C and hCAP‐E. DAPI staining revealed that chromosomes became dispersed and broad in cells with mutated hCAP‐C and hCAP‐E. Weaker signals were observed in cells with mutated hCAP‐E compared to those in wild‐type hCAP‐E. These chromosomal morphologies were similar to those in cells with depletion of hCAP‐C and hCAP‐E, as well as other condensin components.( 5 , 20 , 21 , 22 )
Figure 6.
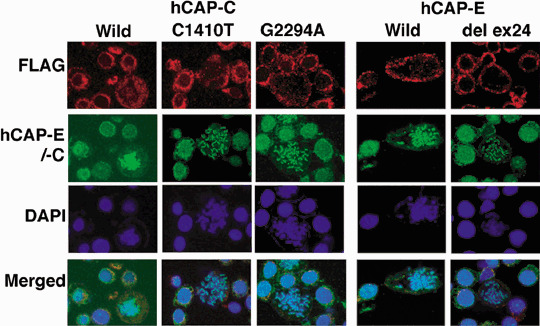
Comparison of chromosomal morphology among HeLa cells in which FLAG‐tagged hCAP‐C and hCAP‐E were introduced. hCAP‐C and hCAP‐E staining was carried out on FLAG‐tagged hCAP‐C and hCAP‐E transfected cells, respectively. The same intensity of signals at interphase was observed in all series of hCAP‐C and hCAP‐E staining. Cells transfected with wild‐type hCAP‐C and hCAP‐E showed stronger signals compared to cells with mutated hCAP‐C and hCAP‐E. The hCAP‐C and hCAP‐E signals were observed along with chromosomes in cells with wild‐type hCAP‐C and hCAP‐E. In contrast, the signals were dot‐like and arranged in curly shapes on chromosomes in cells with mutated hCAP‐C and hCAP‐E.
Discussion
Chromosome condensation and segregation play an essential role in ensuring the fidelity of genetic information transfer during mitosis. Among the chromosomal components, condensins are multisubunit protein complexes that play a central role in assembly and segregation of mitotic chromosomes.( 1 , 2 , 3 , 5 ) Impairment of the genes encoding condensins might affect the structural integrity of mitotic chromosomes, such as conformational change of DNA and proper binding of a number of essential non‐histone proteins to mitotic chromosomes.( 23 ) Cells that are defective for either chromosome condensation or resolution fail to faithfully segregate their replicated chromosomes, become aneuploid, then induce genome instability.( 5 ) In the present study, hCAP‐C and hCAP‐E gene mutations were found in two and one PAL cell line, respectively. The identical mutations were detected in the corresponding tumor samples from which the cell lines were derived but not in the peripheral blood leukocytes, indicating that the mutations were somatic mutations occurring in each PAL patient. No mutations were detected in 16 non‐PAL lymphoma–leukemia cell lines. PAL cell lines have complex karyotypes with no common abnormalities, suggesting that the chromosome abnormality occurred randomly. Most hemato‐lymphoid malignancies have relatively constant patterns of chromosomal abnormality, such as t(8; 14)(q24;q32) in Burkitt's lymphoma and t(9; 22)(q34;q11) in acute lymphocytic leukemia and chronic myeloid leukemia. Condensin alterations might be prone to induce random, rather than specific, chromosome abnormalities as observed in PAL cell lines.( 24 ) Further study will be necessary to elucidate whether mutations of hCAP‐C and hCAP‐E are characteristic in PAL, a lymphoma developing in a specific circumstance of chronic inflammation.
In the present study, depletion of hCAP‐C and hCAP‐E in HeLa cells brought about the change in chromosome morphology and its measurement as well, that is, shorter in length and broader in width. The method used in the present study to prepare chromosome spreads includes incubation with hypotonic [0.075 M] KCl solution.( 19 ) Because the impairment of condensin subunits leads to weakness against hypotonic shock, the morphological changes observed in the present study, such as ‘blurred’ and ‘amorphous’ chromosome structures, were in agreement with those reported in the condensin subunits‐depleted cells.( 5 , 20 , 21 , 22 ) Hudson et al. reported chromosomes become wider after depletion of hCAP‐C.( 23 )
The similar change in chromosome morphology was observed in the present cell lines with hCAP‐C and hCAP‐E mutations. The severity of the morphological changes might correlate with hCAP‐C and hCAP‐E expression levels. OPL‐5 with aberrant skipping of exon 24 in hCAP‐E showed an extensive reduction in expression of not only hCAP‐E but also hCAP‐C proteins. OPL‐5 occasionally showed the chromosome bridge in anaphase and telophase, which was described previously in the cells with condensing subunits depletion.( 22 , 25 , 26 ) This phenomenon indicates that segregation is not accurate, and resultant genome instability in OPL‐5. Of the two cell lines (OPL‐3, OPL‐7) with point mutations in hCAP‐C, OPL‐7 showed reduced hCAP‐C protein expression, abnormality in chromosome length and width, and abnormal aggregates of hCAP‐C protein. OPL‐3 showed no obvious reduction in protein expression, abnormality in chromosome width but not in length, and occasional aggregates of the proteins. In OPL‐4 with no mutations within hCAP‐C, reduced hCAP‐C protein expression and similar chromosome morphology as found in the hCAP‐C depleted cells were also present. Other mechanisms, such as mutations and epigenetic change in promoter regions, might be involved in the reduction of the protein expression and abnormality in chromosome morphology.
The present study showed that identical hCAP‐C and hCAP‐E mutations are present in the tumor samples of PAL and the cell lines derived from PAL. hCAP‐C and hCAP‐E mutations were accompanied with reduced protein expression and functional abnormalities in condensins, such as change in chromosome morphology and inaccurate segregation during mitosis. Previous in vitro studies have shown the contribution of condensins to chromosome stability.( 22 , 24 , 25 ) Impairment in condensins might induce genome instability and gene alterations, which eventually lead to accumulation of other gene alterations in PAL.
Acknowledgments
We thank Drs M. Daibata, T. al Saati and E Kieff for providing Pal‐1, Deglis, and IB‐4 cell lines, respectively. We thank Professor T. Hirano for providing anti‐hCAP‐G, ‐hCAP‐G2, ‐hCAP‐H2, ‐hCAP‐C, and ‐hCAP‐E antibodies as well as hCAP‐C and hCAP‐E constructs. Supported by grants from the Ministry of Education, Science, Culture, and Sports, Japan (14031213, 14770073, 15026209, 15406013, 15590340, 16390105 and 18014015).
References
- 1. Hirano T. At the heart of the chromosome: SMC proteins in action. Nat Rev Mol Cell Biol 2006; 7: 311–22. [DOI] [PubMed] [Google Scholar]
- 2. Swedlow JR, Hirano T. The making of the mitotic chromosome: modern insights into classical questions. Mol Cell 2003; 11: 557–69. [DOI] [PubMed] [Google Scholar]
- 3. Hagstrom KA, Meyer BJ. Condensin and cohesin: more than chromosome compactor and glue. Nat Rev Genet 2003; 4: 520–34. [DOI] [PubMed] [Google Scholar]
- 4. Legagneux V, Cubizolles F, Watrin E. Multiple roles of condensins: a complex story. Biol Cell 2004; 96: 201–13. [DOI] [PubMed] [Google Scholar]
- 5. Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 2003; 115: 109–21. [DOI] [PubMed] [Google Scholar]
- 6. Lehmann AR. The role of SMC proteins in the responses to DNA damage. DNA Repair (Amst) 2005; 4: 309–14. [DOI] [PubMed] [Google Scholar]
- 7. Heale JT, Ball AJ, Schmiesing JA et al . Condensin I interacts with the PARP‐1‐XRCC1 complex and functions in DNA single‐strand break repair. Mol Cell 2006; 21: 837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen ES, Sutani T, Yanagida M. Cti1/C1D interacts with condensin SMC hinge and supports the DNA repair function of condensin. Proc Natl Acad Sci USA 2004; 101: 8078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakatsuka S, Yao M, Hoshida Y, Yamamoto S, Iuchi K, Aozasa K. Pyothorax‐associated lymphoma: a review of 106 cases. J Clin Oncol 2002; 20: 4255–60. [DOI] [PubMed] [Google Scholar]
- 10. Iuchi K, Aozasa K, Yamamoto S et al . Non‐Hodgkin's lymphoma of the pleural cavity developing from long‐standing pyothorax. Summary of clinical and pathological findings in thirty‐seven cases. Jpn J Clin Oncol 1989; 19: 249–57. [PubMed] [Google Scholar]
- 11. Banks PM, Harris NL, Warnke RA, Gaulard PH. Lymphomas. In: Travis WD, Brambilla E, Mueller‐Hermelink HK, Harris CC, eds. World Health Organization Classification of Tumors: Pathology and Genetics of Tumors of the Lung, Pleura, Thymus and Heart. Lyon: IARC Press, 2004: 137–40. [Google Scholar]
- 12. Al Saati T, Delecluze HJ, Chittal S et al . A novel human lymphoma cell line (Deglis) with dual B/T phenotype and gene rearrangements and containing Epstein‐Barr virus genomes. Blood 1992; 80: 209–16. [PubMed] [Google Scholar]
- 13. Daibata M, Taguchi T, Nemoto Y et al . Epstein‐Barr virus (EBV)‐positive pyothorax‐associated lymphoma (PAL): chromosomal integration of EBV in a novel CD2‐positive PAL B‐cell line. Br J Haematol 2002; 117: 546–57. [DOI] [PubMed] [Google Scholar]
- 14. Takakuwa T, Luo WJ, Ham MF, Mizuki M, Iuchi K, Aozasa K. Establishment and characterization of unique cell lines derived from pyothorax‐associated lymphoma which develops in long‐standing pyothorax and is strongly associated with Epstein‐Barr virus infection. Cancer Sci 2003; 94: 858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kanno H, Yasunaga Y, Ohsawa M et al . Expression of Epstein‐Barr virus latent infection genes and oncogenes in lymphoma cell lines derived from pyothorax‐associated lymphoma. Int J Cancer 1996; 67: 86–94. [DOI] [PubMed] [Google Scholar]
- 16. Ham MF, Takakuwa T, Luo WJ, Liu A, Horii A, Aozasa K. Impairment of double‐strand breaks repair and aberrant splicing of ATM and MRE11 in leukemia‐lymphoma cell lines with microsatellite instability. Cancer Sci 2006; 97: 226–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. King W, Thomas PA, Raab TN, Hawke M, Kieff E. Epstein‐Barr virus RNA. V. Viral RNA in a restringently infected, growth‐transformed cell line. J Virol 1980; 36: 506–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu A, Takakuwa T, Fujita S et al . Alterations of DNA damage‐response genes ATM and ATR in pyothorax‐associated lymphoma. Lab Invest 2005; 85: 436–46. [DOI] [PubMed] [Google Scholar]
- 19. Bangs CD, Donlon TA. Cytogenetics. Metaphase chromosome preparation from cultured peripheral blood cells. In: Haines JL, Korf BR, Morton CC, Seidman CE, Seidman JG, Smith DR, eds. Current Protocols in Human Genetics. New York: John Wiley & Sons, 2005: 4.1.1–4.1.19. [DOI] [PubMed] [Google Scholar]
- 20. Hirota T, Gerlich D, Koch B, Ellenberg J, Peters JM. Distinct functions of condensin I and II in mitotic chromosome assembly. J Cell Sci 2004; 117: 6435–45. [DOI] [PubMed] [Google Scholar]
- 21. Ono T, Fang Y, Spector DL, Hirano T. Spatial and temporal regulation of condensins I and II in mitotic chromosome assembly in human cells. Mol Biol Cell 2004; 15: 3296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oliveira RA, Coelho PA, Sunkel CE. The condensin I subunit Barren/CAP‐H is essential for the structural integrity of centromeric heterochromatin during mitosis. Mol Cell Biol 2005; 25: 8971–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hudson DF, Vagnarelli P, Gassmann R, Earnshaw WC. Condensin is required for nonhistone protein assembly and structural integrity of vertebrate mitotic chromosomes. Dev Cell 2003; 5: 323–36. [DOI] [PubMed] [Google Scholar]
- 24. Harris NL, Jaffe ES, Diebold J et al . The World Health Organization classification of neoplastic diseases of the haematopoietic and lymphoid tissues: report of the Clinical Advisory Committee Meeting, Airlie House, Virginia, November 1997. Histopathology 2000; 36: 69–86. [DOI] [PubMed] [Google Scholar]
- 25. Gerlich D, Hirota T, Koch B, Peters JM, Ellenberg J. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr Biol 2006; 16: 333–44. [DOI] [PubMed] [Google Scholar]
- 26. Watrin E, Legagneux V. Contribution of hCAP‐D2, a non‐SMC subunit of condensin I, to chromosome and chromosomal protein dynamics during mitosis. Mol Cell Biol 2005; 25: 740–50. [DOI] [PMC free article] [PubMed] [Google Scholar]