

Abstract
The antitumor effects of tumor necrosis factor (TNF‐α) were evaluated following transfection of TNF‐α plasmid DNA into solid mouse tumors using the nanobubbles (NBs) and ultrasound (US) gene delivery system. Murine breast carcinoma (EMT6) cells expressing luciferase (1 × 106 cells) were injected intradermally into the flanks of 6–7‐week‐old male SCID mice on day 0. Ten microliters of TNF‐α (5 μg/μL) or TNF‐α mock plasmid DNA (5 μg/μL) with/without NBs (15 μL) and saline was injected intratumorally in a total volume of 30 μL, and tumors were exposed to US (frequency, 1 MHz; intensity, 3.0 W/cm2; duty cycle, 20%; number of pulses, 200; and exposure time, 60 s) on days 2, 4, 7, and 9. Changes in tumor size were measured with an in vivo bioluminescent imaging system and a mechanical caliper. Changes in tumor vessel area were quantified using contrast‐enhanced US imaging with Sonazoid and a high frequency US imaging system (40 MHz) and immunohistochemistry (CD31). At the mRNA level, expression of TNF‐α, caspase‐3, and p53 were quantified using real‐time quantitative RT‐PCR. At the protein level, expression of caspase‐3 and p53 were confirmed by immunohistochemistry. We show that repeated TNF‐α gene delivery using NBs and US can lead to the local production of TNF‐α. This results in antitumor effects, including activation of p53‐dependent apoptosis, decrease in tumor vessel density, and suppression of tumor size. In this study, we showed the effectiveness of using NBs and US for TNF‐α gene delivery into tumor cells. (Cancer Sci 2011; 102: 2082–2089)
Tumor necrosis factor (TNF)‐α is a cytokine that mediates a broad range of biological actions and it possesses potent antitumor activity.( 1 , 2 ) Tumor necrosis factor‐α induces the further production of TNF‐α and the antitumor activity is mediated by direct cytotoxicity on tumor cells and an increase in vascular permeability.( 3 , 4 , 5 , 6 ) This induces apoptosis through a signaling pathway that causes caspase activation, hemorrhagic necrosis, and consequently tumor regression. Administration of TNF‐α can mediate the regression of many tumors and it has, therefore, attracted much attention as a potential antitumor agent. However, the dose‐limiting toxicity of this agent has been quite apparent in human trials.( 4 , 5 ) Severe side‐effects, such as systemic inflammatory response syndrome and multiple organ failure, are often observed in cancer patients receiving TNF‐α treatment.( 7 , 8 ) To avoid such toxicity, TNF‐α is given to patients through a locoregional drug delivery system, for example, to an isolated limb perfusion or an isolated hepatic perfusion.( 9 , 10 ) These procedures require surgery, so they are used for the treatment of locally advanced solid tumors, such as limb‐threatening soft tissue sarcomas and primary or metastatic unresectable liver tumors.( 9 )
By induction of a high local concentration of TNF‐α in a tumor through an autocrine and paracrine mechanism of TNF‐α production from transfected cells, gene therapy offers the potential to advance the treatment of cancer.( 11 , 12 ) Recently, many gene therapy strategies have been explored, including use of plasmid or recombinant viral vectors carrying therapeutic genes.( 1 ) In clinical trials, TNF‐α, expressed by an adenovirus, showed antitumor action and synergism with radiation to produce cytotoxic effects.( 13 ) However, administration of recombinant cytokines has proven to be toxic.( 13 , 14 ) Because of high transfection efficiency, regional bolus cytokine delivery has led to systemic toxicity.( 15 , 16 ) Such barriers to the use of cytokines could be overcome by the use of plasmids encoding cytokines and by controlling expression of these cytokine genes over extended time periods. To achieve the full potential of gene therapy, additional improvements in the delivery of therapeutic genes to tumor cells are required.
Gene delivery using nanobubbles (NBs) and ultrasound (US) is a non‐viral vector method, similar to the use of microbubbles, that has certain advantages, including easy operation, low toxicity, low immunogenicity, low invasiveness, high tissue selectivity, and repeated applicability.( 17 ) This system enhances cell permeabilization through the impulsive pressure generated by either collapsing bubbles or the cavitation bubbles created by the collapse. Subsequently, exogenous molecules enter nearby cells.( 18 , 19 , 20 ) Thus, it allows the non‐invasive delivery of therapeutic compounds into specific target cells. Notably, the use of NBs improves transfection efficiency because transfection efficiency is increased by increasing the number of bubbles that physically interact with cells.( 18 , 19 , 21 ) Nanobubbles are smaller than microbubbles; therefore, the area induced by impulsive pressure from collapsing NBs can be limited and direct damage to non‐targeted cells caused by the impulsive pressure is reduced. Increasing the concentration of NBs per unit volume can increase adherence to the cell membrane, resulting in an increase in transfection efficiency.( 21 ) Thus, NBs are applicable for gene delivery through the enhanced permeability and retention effect.( 22 ) In addition, accumulation of NBs in tumors can be confirmed using US imaging because NBs can be used as US contrast agents, hence US‐triggered transfection is possible.( 18 , 22 )
Our ultimate goal is to develop a gene delivery system using NBs and US that can spatially and temporally control gene delivery. In this study, as the first step, we have evaluated the antitumor effects of TNF‐α following transfection of TNF‐α plasmid DNA into solid mouse tumors using the NBs and US gene delivery system.
Materials and Methods
Animal tumor models. This study was approved by the Animal Care Committee of Tohoku University (Sendai, Japan). Murine breast carcinoma (EMT6) cells were obtained from ATCC (Manassas, VA, USA). EMT6 cells stably expressing the luciferase gene (EMT6‐luc) were prepared by transfection of EMT6 cells with pEGFPLuc (BD Biosciences, Franklin Lakes, NJ, USA) using Lipofectin Transfection Reagent (Invitrogen, Carlsbad, CA, USA).( 23 ) EMT6 and EMT6‐luc were cultured as previously described.( 23 ) EMT6 cells either with or without luciferase expression (1 × 106 cells) in 100 μL saline were injected intradermally into the right and left flanks of 6–7‐week‐old male CB17/Icr‐Prkcscid/CrlCrlj (SCID) mice (Charles River Japan, Kanagawa, Japan). The day of inoculation was defined as day 0. The mouse’s back was depilated using a commercial electric shaver and cream on day‐1.
Plasmid vectors. The pGL3‐Control Vector plasmid, which includes SV40 promoter and encodes the luciferase reporter (pGL3, 5256 bp; Promega, Madison, WI, USA), pORF9‐mTNFα containing the mouse TNFα gene (pTNFα, 3746 bp; Invitrogen, San Diego, CA, USA), and pOFR9‐mcs, the mock of pORF9‐mTNFα, (pTNFα‐MOCK, 3023 bp; Promega) were used in this study. Plasmid DNA was purified by an endotoxin‐free Qiagen Mega plasmid purification kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol.
Preparation of NBs. The NBs used in this study were acoustic liposomes (ALs).( 24 ) The ALs (lipid concentration, 1 mg/mL) were composed of 1,2‐distearoyl‐sn‐glycero‐phosphatidylcholine (DSPC; NOF, Tokyo, Japan) and 1,2‐distearoyl‐sn‐glycero‐3‐phosphatidyl‐ethanolamine‐methoxy‐polyethyleneglycol [DSPE‐PEG (2k)‐OMe; 94:6 (m/m); NOF] containing C3F8 gas and were prepared according to a previously described method.( 18 , 22 ) The particle size and the zeta potential of ALs were 199 ± 84.4 nm and −2.1 ± 0.9 mV, respectively.( 22 )
Plasmid transfection. Ten microliters of pGL3 (1 μg/μL), pTNFα (5 μg/μL), or pTNFα‐MOCK (5 μg/μL) with/without NBs (15 μL) and saline were injected intratumorally in a total volume of 30 μL, and tumors were exposed to US on days 2, 4, 7, and 9 (two times/week for 2 weeks). Ultrasound (intensity, 3.0 W/cm2; duty cycle, 20%; number of pulses, 200; and exposure time, 60 s) was generated by a 1 MHz submersible US probe with a diameter of 30 mm (Honda Electronics, Toyohashi, Japan). Mice in the control group were given intratumoral injections of 30 μL saline on days 2, 4, 7, and 9. Each trial was independent but the mean values in the control group were used for evaluation of treatment efficiency of single or repeated NBs and US transfection of TNF‐α gene and antitumor effect of NBs and US transfection of the TNF‐α gene because the condition in the control group is the same. The tumors were positioned 10 cm from the transducer surface in tap water at 37°C. The detailed experimental procedures are described in our previous study.( 23 , 25 )
Monitoring gene expression and measurement of tumor volume. To evaluate transfection efficiency in vivo, pGL3 (expressing luciferase) was transfected to EMT6 cells (which do not express luciferase). To evaluate treatment efficiency of TNF‐α delivery, TNF‐α plasmid, which does not express luciferase, was transfected to EMT6‐luc cells that do express luciferase. Levels of gene expression and tumor volume were quantified on days 2, 4, 7, 9, and 11. In vivo bioluminescent imaging was carried out using an IVIS Lumina (Xenogen, Alameda, CA, USA) and the luciferase activity was quantified as previously described.( 23 , 25 ) Tumor size was measured using a mechanical caliper and tumor volume was estimated according to the formula: 0.5 × (short axis)2 × (long axis) [mm3].( 26 )
Vessel imaging and quantification of tumor vessel area using contrast‐enhanced US imaging. Vessel imaging of tumors was carried out using contrast‐enhanced US imaging. Tumors were imaged using a high‐frequency US imaging system (Vevo770; VisualSonics, Toronto, Canada) with a 40 MHz transducer (RMV‐704; VisualSonics) on day 1 (before treatment), day 5 (after two treatments), and on day 10 (after four treatments). Sonazoid (Daiichi Sankyo, Tokyo, Japan) was used as a US contrast agent and prepared according to the manufacturer’s instructions. Before and 3 min after i.v. tail vein bolus injection of Sonazoid in a total volume of 100 μL, consecutive B‐mode images with a slice thickness of 100 μm were captured throughout the whole tumor for 3D imaging. The difference in video intensities between pre‐injection and post‐injection image frames was indicated in green overlay on the B‐mode anatomical images using the accompanying software (VisualSonics). The difference indicated in green area is considered as the extracted vessel image. Vessel densities in tumor, where the tumor boundaries were manually traced in sequential, parallel US scans according to the anatomical and acoustic characteristics, were calculated by the analysis software (VisualSonics). Intratumoral vessel area was calculated by multiplying the percentage of vessel area by the tumor volume. Throughout the imaging session, mice were kept anesthetized with 2% isoflurane in oxygen at 1 L/min on a heated stage according to the manufacturer’s protocol. Respiratory gating was used to synchronize data acquisition with the mouse respiratory cycle to reduce motion artifact during image analysis.
Blood biochemistry tests. To evaluate the acute toxicity of this transfection method, biochemical serum tests were used. Liver and kidney injuries were evaluated by measuring blood urea nitrogen (BUN), aspartate aminotransferase (AST), and alanine aminotransferase (ALT). On day 11, each mouse in the control and treatment groups was subjected to blood sampling from the caudal vena cava under general anesthesia. Thirty minutes after sampling, blood samples were centrifuged (1200g, 5 min) to obtain serum to measure BUN, AST, and ALT levels (Oriental Yeast, Tokyo, Japan).
Tumor tissue sampling. On day 5 or after blood sampling on day 11, solid tumors were removed and cut perpendicularly against the tumor surface into two parts to coincident each cross‐section with the B‐mode image; half was used for mRNA quantification and the other half was used for histological analysis. The samples for mRNA quantification were frozen in liquid nitrogen and stored at −80°C until use. The samples for histological analysis were mounted in OCT compound (Sakura Fineteck Japan, Tokyo, Japan), taking care not to slant the sample and frozen with liquid nitrogen and stored at −80°C until use.
Real‐time quantitative PCR. For mRNA quantification, frozen tissue was ground using a mortar and pestle and total RNA was isolated with an RNeasy Mini Kit (Qiagen) and treated with the RNase‐Free DNase Set (Qiagen) according to the manufacturer’s protocol. Reverse transcription was carried out using the RNA PCR Kit (AMV; Takara Bio, Shiga, Japan) according to the manufacturer’s protocol. For quantitative real‐time PCR (qPCR), duplicate reactions of 300 ng cDNA were amplified with gene‐specific primers using Brilliant II Fast SYBR Green (Stratagene, Santa Clara, CA, USA) and run in an MX3000P (Stratagene) according to the manufacturer’s protocol. In the optimization experiment for qPCR, transferin receptor (TFRC) and β‐actin were chosen for housekeeping genes. There was difference in cycle threshold (CT) values with <0.5 cycles between the control and the treatment group for TFRC, but there was with more than 2.6 for β‐actin (data not shown). Expression of the genes of interest (TNF‐α, caspase3, and p53) was normalized against a housekeeping gene (TFRC) and fold change was determined relative to the control. The PCR primers used were as follows (forward, reverse, accession number): murine (m)‐TFRC, 5′‐TCCGCTCGTGGAGACTACTT‐3′, 5′‐ACATAGGGCGACAGGAAGTG‐3′, NM_008084; m‐TNF‐α, 5′‐ATGGCCTCCCTCTCATCAGT‐3′, 5′‐CACTTGGTGGTTTGCTACGA‐3′, NM_013693; m‐caspase3, 5′‐AATGGGCCTGTTGAACTGAA‐3′, 5′‐CATTGAGACAGACAGTGGGA‐3′, NM_009810; m‐p53, 5′‐GAAGACAGGCAGACTTTTCG‐3′, 5′‐TAAGGATAGGTCGGCGGTTC‐3′, NM_011640.
Histological analysis. With day 5 samples, duplicate serial sections cut at 8 μm were prepared for anti‐CD31 + DAPI and H&E staining. With day 11 samples, duplicate serial sections cut at 8 μm were prepared for anti‐cleaved‐caspase‐3 + anti‐p53 + DAPI and H&E staining. For immunohistological analysis, expression of CD31, cleaved caspase‐3, and p53 were analyzed in tumor samples. Frozen sections were fixed in 4% paraformaldehyde for 15 min at room temperature and washed with PBS. The primary antibodies: purified rat anti‐mouse CD31 (MED13.3) (BD Pharmingen, San Diego, CA, USA) at 1:100 dilution; cleaved caspase‐3 (Asp175) (5A1E) rabbit mAb (Cell Signaling Technology, Danvers, MA, USA) at 1:200; and p53 (1C12) mouse mAb (Cell Signaling Technology) at 1:1000 were diluted in PBS with 3% BSA and 0.1% Triton X (CD31) or with 5% BSA and 0.3% Triton X (caspase3 and p53). The antibodies were applied overnight at 4°C. After washing with PBS, slides were incubated at 4°C with the secondary antibodies: goat anti‐rat Alexa 555 (CD31), goat anti‐rabbit Alexa Fluor 555 (cleaved caspase‐3), or goat anti‐mouse Alexa 488 (p53) (all Invitrogen, Carlsbad, CA, USA) diluted in PBS with DAPI (100 ng/mL) for 40 min in a moisture chamber. After washing with PBS, sections were mounted using Vectorshield mounting medium (Vector, Burlingame, CA, USA). Cytotoxic area was analyzed following H&E staining carried out according to standard procedures. All histological images were captured using a fluorescence microscope (BX51; Olympus, Tokyo, Japan) at ×100 magnification. For quantification of vessel density, the number of pixels that represents positive staining by CD31 was quantified using Photoshop CS3 (Adobe Systems, San Jose, CA, USA). For quantification of vessel density in the peripheral area of tumors, two randomly selected images (control, n = 7 [four mice] and treatment, n = 6 [four mice]) avoiding cytotoxic areas were captured and the percentage of vessel area from each sample was obtained. For quantification of vessel density and cytotoxic area over the entire tumor area, the images acquired with ×100 magnification were merged using Photoshop CS3 (Adobe).
Statistical analysis. Data are expressed as the mean ± SEM where indicated. Statistical differences were analyzed using the Kruskal–Wallis test and the Mann–Whitney U‐test. Differences of P < 0.05 were considered significant.
Results
Comparison of transfection efficiency between single and repeated NBs and US transfection. The method with NBs and US is able to deliver exogenous molecules repeatedly. First, we investigated the transfection efficiency between single and repeated transfection by NBs and US. Figure 1(A) shows time‐dependent gene expression of a luciferase reporter gene (pGL3) transfected into EMT6 cells using NBs and US. With a single transfection, gene expression was maximal the day after transfection and decreased gradually to the background level by day 11. In contrast, repeated transfection resulted in persistent gene expression. After the second transfection, the luciferase gene expression in the repeated transfection group was increased compared to that in the single transfection group. After the third transfection, the gene expression reached the maximum and became persistent by the following transfection. There was no significant difference in tumor growth between single and repeated transfection groups (Fig. 1B); the gene expression levels did not depend on the tumor size.
Figure 1.
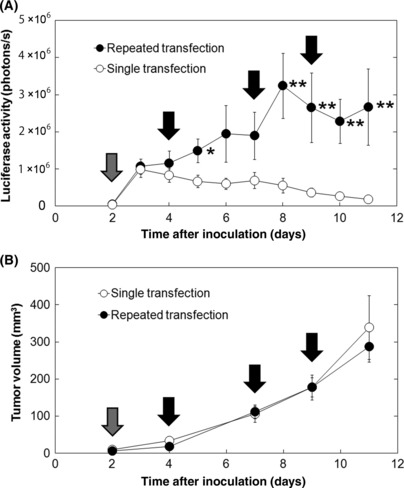
Evaluation of transfection efficiency of single or repeated transfection of the reporter gene by nanobubbles and ultrasound. (A) Time‐dependent changes in gene expression level. (B) Changes in tumor volume. Luciferase reporter plasmid DNA (pGL3 control vector) was transfected on day 2 in the single transfection group and on days 2, 4, 7, and 9 in the repeated transfection group. Gray arrows indicate the days on which tumors both in the single and repeated transfection groups were transfected (day 2). Black arrows indicate the days on which tumors in the repeated transfection group were transfected (days 4, 7, and 9). n = 12 (six mice) in each group; mean ± SEM. *P < 0.05; **P < 0.01.
Comparison of treatment efficiency between single and repeated NBs and US transfection of the TNF‐α gene. Next, treatment efficiency of repeated NBs and US transfection of the TNF‐α gene was evaluated by transfecting pTNF‐α into EMT6 cells expressing luciferase. Luciferase activity, which represents tumor size,( 23 ) was decreased after the first treatment (both single and repeated transfection groups) compared with that of the control group (Fig. 2A). However, after the second measurement, there was no significant difference in the luciferase activity between the single transfection group and the control group, whereas the treatment effect was sustained in the repeated transfection group. Figure 2(A,B) shows significant tumor regression by repeated transfection of the TNF‐α gene. The TNF‐α protein level in tumor on day 11 both in the control and treatment group was not detected with ELISA (data not shown). At the mRNA level, TNF‐α expression in tumors on day 11 was 2.4 times higher in the single transfection group (P < 0.05) and 6.9 times higher in the repeated transfection group (P < 0.01) compared to that in the control group (Fig. 2C).
Figure 2.
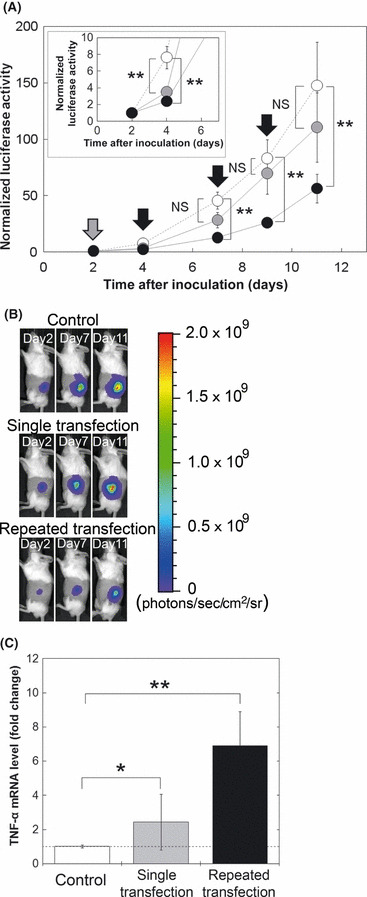
Evaluation of treatment efficiency of single or repeated transfection of the tumor necrosis factor‐α (TNF‐α) gene by nanobubbles and ultrasound. (A) Normalized luciferase activity. Control (white circles), n = 64 (32 mice); single transfection (gray circles), n = 16 (eight mice); repeated transfection (black circles), n = 42 (21 mice). Data are normalized against data on day 2. Gray arrows indicate the days on which tumors both in the single and repeated transfection groups were transfected (day 2). Black arrows indicate the days on which tumors in the repeated transfection group were transfected (days 4, 7, and 9). (B) Representative images of luciferase bioluminescence in each group. (C) Fold change of TNF‐α mRNA levels in tumor quantified by quantitative PCR, normalized against expression of the TFRC housekeeping gene. Samples taken on day 10 were used for quantitative analysis (n = 6, three mice in each group). NS, not significant.
Effectiveness of NBs and US transfection of the TNF‐α gene. Effectiveness of repeated NBs and US transfection of TNF‐α was evaluated by transfecting pTNF‐α or pTNF‐α‐MOCK into EMT6 cells expressing luciferase (Fig. 3). Tumor volume (Fig. 3A) and luciferase activity (Fig. 3B) decreased significantly only in the TNF‐α + NB + US group compared to that in the control group from day 7 and 4, respectively. There was a difference in the number of days needed to show a statistical difference in tumor size between the measurements of luciferase activity, measured by the bioluminescent imaging system, and tumor volume, calculated from mechanical caliper measurements. This is because of the difference in measurement accuracy. Luciferase activity is measured from the expression of each single cell and the level of expression represents tumor size.( 27 ) However, mechanical calipers measure overlying skin and surrounding non‐tumor tissue in addition to the tumor itself, leading to inaccuracy of tumor volume, especially in small tumors. There were no significant differences between the tumor volume (Fig. 3A) and luciferase activities (Fig. 3B) in the control, mock + NB + US, or TNF‐α groups.
Figure 3.
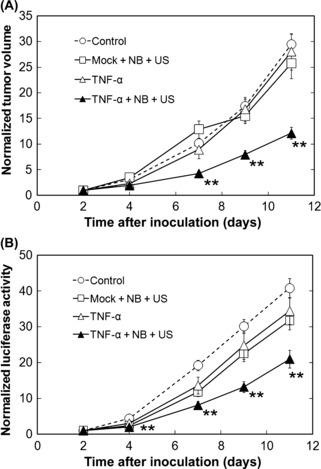
Antitumor effect of nanobubbles (NBs) and ultrasound (US) transfection of the tumor necrosis factor‐α (TNF‐α) gene. (A) Normalized tumor volume. (B) Normalized luciferase activity. Plasmid DNA (Mock or TNF‐α) or saline was injected on days 2, 4, 7, and 9. pTNF‐α or pTNF‐α‐MOCK was transfected with US and NB, and saline was injected in the control group, on days 2, 4, 7, and 9. Control (white circles), n = 64 (32 mice); mock + NB + US (white squares), n = 24 (12 mice); TNF‐α (white triangles), n = 18 (nine mice); TNF‐α + NB + US (black circles), n = 42 (21 mice). Data are normalized against data on day 2. Mean ± SEM. **P < 0.01.
Acute toxicity by NBs and US. To determine whether there were any toxic responses with this method, bloods samples and changes in body weight during the study were analyzed and compared with those of control animals (Table 1). Mice in the control group were injected with saline in their solid tumors, whereas mice in the treatment group were transfected with pTNF‐α in their solid tumors using NBs and US. There were no significant differences in BUN, AST, or ALT levels between groups, indicating that there was no acute toxicity to liver or kidney resulting from this method. In addition, there was no significant difference in the body weight increase between the groups.
Table 1.
Evaluation of acute toxicity
| Control | Treatment | Statistical significance | |
|---|---|---|---|
| BUN (mg/dL) | 25.9 ± 2.50 | 27.6 ± 4.02 | NS |
| AST (IU/L) | 46.8 ± 7.04 | 51.7 ± 7.69 | NS |
| ALT (IU/L) | 23.2 ± 2.48 | 26.5 ± 6.06 | NS |
| Body weight change (g) | +2.26 ± 0.99 | +2.54 ± 1.25 | NS |
Blood sample was obtained on day 11. n = 6 (six mice) in each group. Values are represented as mean ± SEM. NS, not significant; BUN, blood urea nitrogen; AST, aspartate aminotransferase; ALT, alanine aminotransferase.
Change in tumor vessel density. To evaluate these antitumor effects, vessel area in tumors was analyzed using contrast‐enhanced US imaging and histological analysis. Mice in the control group were injected with saline in their solid tumors, whereas mice in the treatment group were transfected with pTNF‐α in their solid tumors using NBs and US. The same volume of saline into the tumor was injected in the control group as the treatment group. In this study, CD31 antibody was used to identify endothelial cells in tumors. Significant reductions in normalized tumor vessel area from contrast‐enhanced US images in the treatment group were observed on days 5 and 10 (Fig. 4A). Tumor vessel density was significantly decreased by the TNF‐α treatment on day 5 but not on day 10 (Fig. 4B). Day 5 tumor sections stained for CD31 showed a significant decrease in vessel area in the treatment group, consistent with the contrast‐enhanced US imaging results (Fig. 4C). Cytotoxic areas in tumors were increased by treatment on day 5 but there was no significant difference between cytotoxic area in tumor in the control group and that in the treatment group on day 10 (Fig. 4D).
Figure 4.
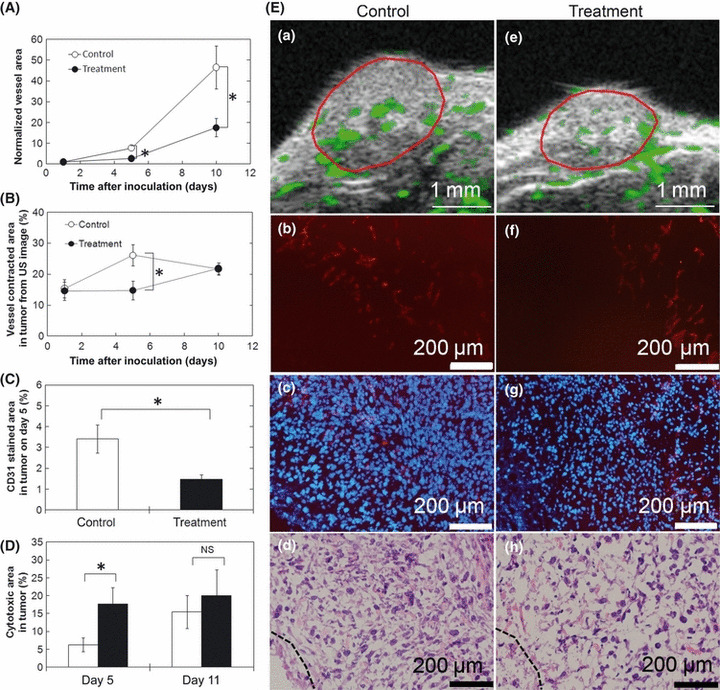
Evaluation of vessel density in tumors. (A) Normalized vessel extracted area in tumors by ultrasound (US) imaging system. Images were acquired on days 1, 5, and 10. n = 6 (six mice) in each group. Vessel area was normalized with that on day 1. (B) Vessel extracted densities in tumors by US imaging system. Images were acquired on days 1, 5, and 10. n = 6 (six mice) in each group. (C) CD31 stained area in tumors. (D) Cytotoxic area in tumors from H&E stained images on day 5 and day 10. (E) Vessel extraction images by the US imaging system (Ea,e, extracted vessels are indicated in green and tumor area are enclosed in red lines); fluorescence images stained with anti‐CD31 (Eb,f, red); merged images of (Eb) and (Ef) with DAPI nuclear staining (Ec,g, CD31 in red and DAPI in blue); and H&E stained images (Ed,h, dashed lines indicate boundaries to obvious cytotoxic area). Plasmid DNA (tumor necrosis factor‐α) was transfected with US and nanobubbles in the treatment group and saline was injected in the control group on days 2, 4, 7, and 9. Tumor samples on day 5 were used for immunohistological analysis (for anti‐CD31 staining; C, Eb–h). Tumor samples on days 5 and 10 were used for H&E staining. Control, n = 7 (four mice); treatment, n = 6 (four mice). Eb (the same section as Ec) and Ed, and Ef (the same section as Eg) and Eh are serial sections. Mean ± SEM. NS, not significant. *P < 0.05.
Apoptosis and tumor suppression factors. To evaluate the cytotoxic activity of TNF‐α in NBs and US transfected tumors, levels of caspase‐3 and p53 expression were evaluated by immunohistological analysis and by qPCR (Fig. 5). Mice in the control group were injected with saline in their solid tumors, whereas mice in the treatment group were transfected with pTNF‐α in their solid tumors using NBs and US. The same volume of saline into the tumor was injected in the control group as the treatment group. Colocalization between p53 and DAPI was checked with sections stained for those in higher magnification (×600) and p53‐positive cells were detected only in the treatment group but not the control group (data not shown). Figure 5(Aa,b) shows merge images of cleaved caspase‐3 (Fig. 5c,d, red), p53 images (Fig. 5e,f, green), and DAPI (blue) showing the localization of expressing cleaved caspase‐3 and p53 at the center of the tumor. Cleaved caspase‐3 was detected in either treated or untreated tumors (Fig. 5Ac,d). Quantitative PCR showed that caspase‐3 mRNA levels in the treatment group were two times higher compared to that in the control group. These results indicate that apoptosis was induced in tumors in the control group and activated by the treatment. At the same time, in the treatment group, apparent increases in p53 expression were detected, both at the protein and mRNA levels (Fig. 5Ae,f,C).
Figure 5.
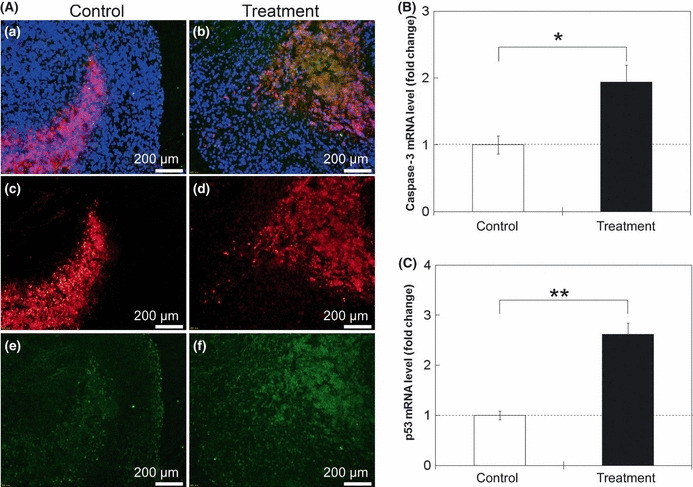
Evaluation of apoptosis and tumor suppressor factors in tumor in response to treatment with tumor necrosis factor‐α (TNF‐α). (A) Fluorescence images. Merged images (Aa,b) of cleaved caspase‐3 (Ac,d, red), p53 images (Ae,f, green), and DAPI (blue). (B) Fold change of caspase‐3 mRNA levels in tumors by quantitative PCR, normalized to the expression of the TFRC housekeeping gene, determined relative to controls. n = 6 (three mice) in each group. (C) Fold change of p53 mRNA levels in tumors by quantitative PCR, normalized to the expression of the TFRC housekeeping gene determined relative to controls. Day 11 tumor samples were used. n = 6 (three mice) in each group. Plasmid DNA (TNF‐α) was transfected with ultrasound and nanobubbles in the treatment group and saline was injected in the control group on days 2, 4, 7, and 9. Mean ± SEM. *P < 0.05; **P < 0.01.
Discussion
Tumor necrosis factor‐α is a cytokine that can initiate tumor cell apoptosis by disrupting vasculature and can exert cytotoxic effects on a wide range of tumor cells. It shows promise for the treatment of cancer and its efficacy has been widely studied. However, the clinical use of TNF‐α has been limited because of severe systemic toxicity. Here we show that repeated TNF‐α gene delivery using NBs and US could lead to the local production of TNF‐α within a tumor that permitted antitumor effects, including activation of p53‐dependent apoptosis, decrease in tumor vessel density, and suppression of tumor size, but that reduced the acute toxic effects.
Sonoporation using NBs and US has been reported to result in effective gene transfection of tumors; however, the transgene expression level is transient and decreases with time.( 23 , 28 ) Peak expression following DNA delivery was dependent on tissues, for example, gene expression reaches its peak at the next day in solid tumor( 23 ) and periodontal tissue( 17 ) and at the fourth day in skeletal muscle( 19 ) after transfection with NBs and US. Using NBs and US, 2.04 ± 1.48 cells per mm2 were transfected when β‐galactosidase encoding plasmid DNA (7.2 kb) was transfected into mouse skeletal muscle.( 19 ) In Figure 1, persistent transgene expression by repeated NBs and US transfection was confirmed by the delivery of a reporter gene. Although gene expression was transient by this method, repeated transfection by NBs and US could maintain the level of gene expression. In fact, Figure 2(A) shows significant tumor regression by repeated transfection of the TNF‐α gene. In Figure 2(C), at the mRNA level, gene expression of TNF‐α was induced even with a single transfection and was sustained in the tumor; however, the expression level was not enough to confirm a treatment effect. Repeated transfection of the TNF‐α gene is needed to induce significant tumor regression. The TNF‐α protein levels in tumor on day 11 after TNF‐α treatment was lower than the detection limit of the ELISA (15 pg/mL). Regulation of protein synthesis at the translational level was depressed on day 11 because the secretion of TNF‐α protein is suppressed within 48 h of TNF‐α gene transfection in tumor in vivo ( 15 , 29 ) where the last treatment was on day 9. The low levels of secretion of TNF‐α protein was sufficient to affect the tumor growth and to reduce the acute toxic effects.
One of the antitumor activities of TNF‐α is mediated by cytotoxicity to endothelial cells.( 3 , 4 , 5 , 6 ) To evaluate these antitumor effects, vessel area in tumors was analyzed using contrast‐enhanced US imaging and histological analysis (Fig. 4). Significant reductions in normalized tumor vessel area were observed in the treatment group, measured with contrast‐enhanced US imaging (Fig. 4A). This result indicates that the TNF‐α treatment induced anti‐angiogenic effects. On day 5, a significant reduction in tumor vessel density were observed in the treatment group, measured with both contrast‐enhanced US imaging (Fig. 4B) and sections stained for CD31 (Fig. 4C). Cytotoxic area confirmed with H&E staining was increased by the treatment on day 5 (Fig. 4D). The results on day 5 indicate the TNF‐α treatment induces anti‐angionic effects resulting cell death in tumor. However, on day 10, there was no difference between vessel density in the control group and that in the treatment group (Fig. 4B). In the control group, intrinsically the cell death area was developed and increased as tumor grew due to lack of nutrients at the tumor center.( 30 ) In the TNF‐α treatment group, the cytotoxic area was expanded. Therefore, there was no significant difference between the cytotoxic area in the control group and that in the treatment group on day 10 (Fig. 4D).
Direct cytotoxicity on tumor cells through apoptosis, another antitumor activity of TNF‐α, was also evaluated.( 6 ) In Figure 5, to evaluate the cytotoxic activity of TNF‐α in NBs and US transfected tumors, levels of caspase‐3 and p53 gene expression were evaluated by immunohistological analysis and by qPCR. Apoptosis induced by TNF‐α is mediated through caspase activation and caspase‐3 acts as initiator and executor at the end of the cascade in the apoptotic process.( 31 ) Expression in cleaved caspase‐3 is increased by the absence of oxygen.( 32 ) Because cell death in the center of solid tumor was detected both in the control and treatment groups (Fig. 4D), expression of cleaved caspase‐3 was confirmed both at mRNA and protein levels, although the expression of cleaved caspase‐3 was increased by the TNF‐α treatment. A nuclear transcription factor, p53 regulates genotoxic stress to block cell cycle progression and induces apoptosis in TNF‐α transfected cells.( 33 , 34 ) p53 is involved in tumor susceptibility to the cytotoxic action of TNF‐α and has a role of triggering apoptosis in response to TNF‐α.( 33 ) Figure 5 shows increase in expression of p53 at mRNA and protein levels by the TNF‐α treatment. In this study, TNF‐α treatment resulted in the accumulation of p53 and in the induction of apoptosis at the center of the tumor where pTNF‐α was injected and transfected. Although the cellular mediator of TNF‐α‐promoted p53 accumulation and genotoxic stress is unknown, p53 was required to promote TNF‐α‐induced cell death and to help achieve the significant therapeutic effect.
The NBs used in this study were ALs, composed of pegylated liposomes encapsulating C3F8 gas and liquid.( 22 ) Acoustic liposome numbers can be increased at the tumor site by active accumulation using binding antibodies or ligands against tumor cells attached to PEG on the AL surface and by passive accumulation through the enhanced permeability and retention effect because of the size distribution.( 22 ) Moreover, oligonucleotides can be encapsulated with ALs.( 35 ) Acoustic liposomes provide enhanced echogenicity as echo contrast agents both in vitro and in vivo when they concentrate and the scattering area becomes close to that of microbubbles.( 18 , 22 ) Using the properties of NBs effectively, plasmid DNA encoding TNF‐α, encapsulated in NBs, can be transfected to tumor cells in a spatial and temporal manner as triggered by US exposure while accumulation at the tumor site is confirmed by a US imaging device. This opens new possibilities for TNF‐α cancer gene therapy using the NBs and US gene delivery system.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
S. Horie received a Grant‐in‐Aid for Japan Society for the Promotion Science (JSPS) Fellows (21‐7271) and a Grant‐in‐Aid for Young Scientists (Start‐up) (19800002). Y. Watanabe received a Grant‐in‐Aid for JSPS Fellows (21‐7073). M. Shiro acknowledges a Grant‐in‐Aid for Scientific Research (B) (22390378) and a Grant‐in‐Aid for Challenging Exploratory Research (22659363). T. Kodama acknowledges a Grant‐in‐Aid for Scientific Research (B) (23300183) and a Grant‐in‐Aid for Challenging Exploratory Research (221650124).
References
- 1. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer 2009; 9: 361–71. [DOI] [PubMed] [Google Scholar]
- 2. Meng Y, Mauceri HJ, Khodarev NN et al. Ad.Egr‐TNF and local ionizing radiation suppress metastases by interferon‐beta‐dependent activation of antigen‐specific CD8+ T cells. Mol Ther 2010; 18: 912–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Horssen R, Ten Hagen TL, Eggermont AM. TNF‐alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist 2006; 11: 397–408. [DOI] [PubMed] [Google Scholar]
- 4. Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin 2008; 29: 1275–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burton ER, Libutti SK. Targeting TNF‐alpha for cancer therapy. J Biol 2009; 8: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hao S, Su L, Guo X, Moyana T, Xiang J. A novel approach to tumor suppression using microencapsulated engineered J558/TNF‐alpha cells. Exp Oncol 2005; 27: 56–60. [PubMed] [Google Scholar]
- 7. Li CY, Huang Q, Kung HF. Cytokine and immuno‐gene therapy for solid tumors. Cell Mol Immunol 2005; 2: 81–91. [PubMed] [Google Scholar]
- 8. Rügg C, Dormond O, Oguey D, Lejeune FJ. Tumor necrosis factor: clinical use and mechanisms of action. Drug Resist Updat 2000; 3: 271–6. [Google Scholar]
- 9. Cai W, Kerner ZJ, Hong H, Sun J. Targeted cancer therapy with tumor necrosis factor‐alpha. Biochem Insights 2008; 1: 5–21. [PMC free article] [PubMed] [Google Scholar]
- 10. Lejeune FJ, Ruegg C, Lienard D. Clinical applications of TNF‐alpha in cancer. Curr Opin Immunol 1998; 10: 573–80. [DOI] [PubMed] [Google Scholar]
- 11. Sato Y, Niitsu Y. Gene therapy of cancer using TNF gene. Tanpakushitsu Kakusan Koso 1995; 40: 2694–9. [PubMed] [Google Scholar]
- 12. Wu S, Boyer CM, Whitaker RS et al. Tumor necrosis factor alpha as an autocrine and paracrine growth factor for ovarian cancer: monokine induction of tumor cell proliferation and tumor necrosis factor alpha expression. Cancer Res 1993; 53: 1939–44. [PubMed] [Google Scholar]
- 13. Drutskaya MS, Efimov GA, Kruglov AA, Kuprash DV, Nedospasov SA. Tumor necrosis factor, lymphotoxin and cancer. IUBMB Life 2010; 62: 283–9. [DOI] [PubMed] [Google Scholar]
- 14. Fisher WE. The promise of a personalized genomic approach to pancreatic cancer and why targeted therapies have missed the mark. World J Surg 2011; 35: 1766–9. [DOI] [PubMed] [Google Scholar]
- 15. Ito A, Shinkai M, Honda H, Kobayashi T. Heat‐inducible TNF‐alpha gene therapy combined with hyperthermia using magnetic nanoparticles as a novel tumor‐targeted therapy. Cancer Gene Ther 2001; 8: 649–54. [DOI] [PubMed] [Google Scholar]
- 16. Kang J‐H, Toita R, Katayama Y. Bio and nanotechnological strategies for tumor‐targeted gene therapy. Biotechnol Adv 2010; 28: 757–63. [DOI] [PubMed] [Google Scholar]
- 17. Chen R, Chiba M, Mori S, Fukumoto M, Kodama T. Periodontal gene transfer by ultrasound and nano/microbubbles. J Dent Res 2009; 88: 1008–13. [DOI] [PubMed] [Google Scholar]
- 18. Horie S, Watanabe Y, Chen R, Mori S, Matsumura Y, Kodama T. Development of localized gene delivery using a dual‐intensity ultrasound system in the bladder. Ultrasound Med Biol 2010; 36: 1867–75. [DOI] [PubMed] [Google Scholar]
- 19. Kodama T, Aoi A, Watanabe Y et al. Evaluation of transfection efficiency in skeletal muscle using nano/microbubbles and ultrasound. Ultrasound Med Biol 2010; 36: 1196–205. [DOI] [PubMed] [Google Scholar]
- 20. Watanabe Y, Horie S, Funaki Y et al. Delivery of Na/I symporter gene into skeletal muscle using nanobubbles and ultrasound: visualization of gene expression by PET. J Nucl Med 2010; 51: 951–8. [DOI] [PubMed] [Google Scholar]
- 21. Guzman HR, McNamara AJ, Nguyen DX, Prausnitz MR. Bioeffects caused by changes in acoustic cavitation bubble density and cell concentration: a unified explanation based on cell‐to‐bubble ratio and blast radius. Ultrasound Med Biol 2003; 29: 1211–22. [DOI] [PubMed] [Google Scholar]
- 22. Kodama T, Tomita N, Horie S et al. Morphological study of acoustic liposomes using transmission electron microscopy. J Electron Microsc (Tokyo) 2009; 59: 187–96. [DOI] [PubMed] [Google Scholar]
- 23. Aoi A, Watanabe Y, Mori S, Takahashi M, Vassaux G, Kodama T. Herpes simplex virus thymidine kinase‐mediated suicide gene therapy using nano/microbubbles and ultrasound. Ultrasound Med Biol 2008; 34: 425–34. [DOI] [PubMed] [Google Scholar]
- 24. Suzuki R, Takizawa T, Negishi Y, Utoguchi N, Maruyama K. Effective gene delivery with novel liposomal bubbles and ultrasonic destruction technology. Int J Pharm 2008; 354: 49–55. [DOI] [PubMed] [Google Scholar]
- 25. Watanabe Y, Aoi A, Horie S et al. Low‐intensity ultrasound and microbubbles enhance the antitumor effect of cisplatin. Cancer Sci 2008; 99: 2525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheung AM, Brown AS, Hastie LA et al. Three‐dimensional ultrasound biomicroscopy for xenograft growth analysis. Ultrasound Med Biol 2005; 31: 865–70. [DOI] [PubMed] [Google Scholar]
- 27. Kim JB, Urban K, Cochran E et al. Non‐invasive detection of a small number of bioluminescent cancer cells in vivo. PLoS ONE 2010; 5: e9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suzuki R, Namai E, Oda Y et al. Cancer gene therapy by IL‐12 gene delivery using liposomal bubbles and tumoral ultrasound exposure. J Control Release 2010; 142: 245–50. [DOI] [PubMed] [Google Scholar]
- 29. Walther W, Arlt F, Fichtner I, Aumann J, Stein U, Schlag PM. Heat‐inducible in vivo gene therapy of colon carcinoma by human mdr1 promoter‐regulated tumor necrosis factor‐alpha expression. Mol Cancer Ther 2007; 6: 236–43. [DOI] [PubMed] [Google Scholar]
- 30. Araujo RP, McElwain DL. A history of the study of solid tumour growth: the contribution of mathematical modelling. Bull Math Biol 2004; 66: 1039–91. [DOI] [PubMed] [Google Scholar]
- 31. Singh N, Khanna N, Sharma H, Kundu S, Azmi S. Insights into the molecular mechanism of apoptosis induced by TNF‐alpha in mouse epidermal JB6‐derived RT‐101 cells. Biochem Biophys Res Commun 2002; 295: 24–30. [DOI] [PubMed] [Google Scholar]
- 32. Olin MR, Andersen BM, Zellmer DM et al. Superior efficacy of tumor cell vaccines grown in physiologic oxygen. Clin Cancer Res 2010; 16: 4800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ameyar M, Shatrov V, Bouquet C et al. Adenovirus‐mediated transfer of wild‐type p53 gene sensitizes TNF resistant MCF7 derivatives to the cytotoxic effect of this cytokine: relationship with c‐myc and Rb. Oncogene 1999; 18: 5464–72. [DOI] [PubMed] [Google Scholar]
- 34. Donato NJ, Perez M. Tumor necrosis factor‐induced apoptosis stimulates p53 accumulation and p21WAF1 proteolysis in ME‐180 cells. J Biol Chem 1998; 273: 5067–72. [DOI] [PubMed] [Google Scholar]
- 35. Buchanan KD, Huang SL, Kim H, McPherson DD, MacDonald RC. Encapsulation of NF‐kappaB decoy oligonucleotides within echogenic liposomes and ultrasound‐triggered release. J Control Release 2010; 141: 193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]