

Abstract
The authors investigated the DNA methylation patterns of the E‐cadherin, Connexin 26 (Cx26), Rassf1a and c‐fos genes in the early phase of rat hepatocarcinogenesis induced by a choline‐deficient L‐amino acid‐defined (CDAA) diet. Six‐week‐old F344 male rats were continuously fed with the CDAA diet, and three animals were then killed at each of 4 and 8 days and 3 weeks. Genomic DNA was extracted from livers for assessment of methylation status in the 5′ upstream regions of E‐cadherin, Cx26, Rassf1a and c‐fos genes by bisulfite sequencing, compared with normal livers. The livers of rats fed the CDAA diet for 4 and 8 days and 3 weeks were methylated in E‐cadherin, Cx26 and Rassf1a genes, while normal livers were all unmethylated. In contrast, normal livers were highly methylated in c‐fos gene. Although the livers at 4 days were weakly methylated, those at 8 days and 3 weeks were markedly unmethylated. Methylation patterns of CpG sites in E‐cadherin, Cx26 and Rassf1a were sparse and the methylation was not associated with gene repression. These results indicate that gene‐specific DNA methylation patterns were found in livers of rats after short‐term feeding of the CDAA diet, suggesting gene‐specific hypermethylation might be involved in the early phase of rat hepatocarcinogenesis induced by the CDAA diet. (Cancer Sci 2007; 98: 1318–1322)
Abbreviations:
- 8‐OHdG
8‐hydroxydeoxyguanosine
- CD diet
choline‐deficient diet
- CDAA diet
choline‐deficient L‐amino acid defined diet
- Cx26
Connexin 26
- Dnmt
DNA methyltransferase
- HCC
hepatocellular carcinoma
- PCR
polymerase chain reaction
- RT
reverse transcription
It is well known that unequivocal liver tumors can be induced by prolonged feeding of rats with a CD diet.( 1 , 2 , 3 ) The choline deficiency causes fatty liver, cirrhosis and HCC in rats.( 1 , 2 , 3 ) Possible mechanisms underlying liver carcinogenesis from the CD diet have been proposed to include the following: liver necrosis associated with subsequent regeneration;( 4 , 5 ) induction of oxidative DNA damage and lipid peroxidation;( 6 , 7 , 8 , 9 ) and generation of genetic alterations.( 10 , 11 ) It has also been considered that DNA hypomethylation might play an important role in liver carcinogenesis induced by methyl donor deficiency.( 12 , 13 ) So far, hypomethylation of the c‐fos, c‐myc, and c‐Ha‐ras genes has been detected in the livers of rats fed with the CD diet.( 14 , 15 )
The CDAA diet used in the present study is semi‐synthetic, and provides stronger carcinogenic effects than the CD diet in rats.( 16 , 17 ) The authors have previously reported the hypomethylation of c‐myc in HCC resulting from the CDAA diet in rats.( 18 ) In another study, hypermethylation of the E‐cadherin and Cx26 genes was also detected in those tumors.( 19 ) While genome‐wide hypomethylation occurs in several human cancer cells, site‐specific hypermethylation such as CpG islands of tumor suppressor genes, is also found.( 20 ) It has been suggested that aberrant DNA methylation of promoter regions of genes is the major mechanism of gene silencing in the development of tumors.( 21 , 22 ) In fact, aberrant DNA methylation has been found in a variety of human cancers, including liver tumors.( 23 , 24 , 25 , 26 ) However, it is unclear why DNA hypermethylation occurs in rat HCC induced by the CDAA diet despite methyl donor deficiency. Therefore, to better understand the disturbance of DNA methylation under methyl donor deficiency, the authors investigated DNA methylation status in the E‐cadherin, Cx26, Rassf1a and c‐fos genes, and measured expression levels of the Dnmt1 gene in livers with short‐term feeding of the CDAA diet in rats.
Materials and Methods
Animals and treatment. A total of 12 F344 male rats, 5 weeks old, were purchased from Japan SLC Inc. (Shizuoka, Japan), and were housed three per plastic cage containing white flake bedding, in an air‐conditioned room, at a constant temperature of 25°C, and a 12‐h light–dark cycle. Food and water were available ad libitum throughout the study. After a 1‐week acclimation period on basal diet in pellet form (CF‐2 Diet; Clea Japan, Tokyo, Japan), nine animals received the CDAA diet (product number 518753; Dyets Inc., Bethlehem, PA, USA), consisting of ingredients as previously described.( 16 , 17 ) Subgroups of three rats were killed by exsanguination from the abdominal aorta, under light ether anesthesia, at 4 and 8 days and 3 weeks after the beginning of the experiment. To obtain normal liver tissues, three rats were also killed at 6 weeks of age without the CDAA diet feeding.
Tissue preparation. Upon killing, whole livers were immediately excised and frozen in liquid nitrogen, and stored at –80°C until analysis. Part of the livers was fixed in 10% neutral buffered formalin at 4°C, routinely processed for HE staining, and histopathologically evaluated according to diagnostic criteria as previously described.( 16 , 17 )
Bisulfite sequencing. Bisulfite treatment of genomic DNA was performed as previously described.( 19 , 27 ) Briefly, genomic DNA was extracted from pooled liver samples of three rats in each subgroup, using a DNeasy tissue kit (Qiagen, Hilden, Germany), and 500 ng of each sample was denatured in 0.3 M NaOH. Then, 2.9 M sodium bisulfite (Sigma, St Louis, MO, USA) and 0.5 mM hydroquinone (Sigma) were added, and the mixture underwent 15 cycles of 30‐s denaturation at 95°C, and 15‐min incubation at 50°C. Samples were then desalted with a Wizard DNA cleanup system (Promega, Madison, WI, USA), and desulfonated by treatment with 0.3 M NaOH at room temperature for 5 min. After ethanol precipitation with ammonium acetate, DNA was dissolved in distilled water.
For bisulfite sequencing, PCR was performed with the primer sets of rat E‐cadherin and Rassf1a genes as described previously.( 19 , 27 ) The primer sets for rat Cx26 and c‐fos genes were also designed for rat c‐fos gene (NCBI accession number NW_047454 and GenBank accession number AF126534, respectively). All primer sets are listed in Table 1. The PCR amplification was performed as described previously.( 19 , 27 ) PCR products were subcloned using a TOPO TA cloning kit (Invitrogen Corporation), and were sequenced with a BigDye terminator v3.0 cycle sequencing ready reaction kit (Applied Biosystems Japan Ltd) and an ABI PRISM 310 genetic analyzer (Applied Biosystems Japan Ltd). For each sample, 10 clones were sequenced.
Table 1.
The primer sequence used in the present study
| Gene | Primer sequence | Annealing temperature (°C) |
|---|---|---|
| Bisulfite sequencing | ||
| E‐cadherin | F: 5′‐GGAATAAGGAAGTAAGGAAGTT‐3′ | 56 |
| R: 5′‐CCACATACCTACAACAAAAACA‐3′ | ||
| Connexin 26 | F: 5′‐GGAGTGATTTAGGTTTTAGGAGAG‐3′ | 62 |
| R: 5′‐TCCCCACAAATCCTAATAAAAACTAC‐3′ | ||
| Rassf1a | F: 5′‐GGATTAGGTTATAGTATTAGTAAATTAG‐3′ | 62 |
| R: 5′‐TCATAATTCAATAAATTCTAACTCC‐3′ | ||
| c‐fos | F: 5′‐TATTTATAGGTGAAAGTTATAGATTG‐3′ | 54 |
| R: 5′‐CACTAATAAAAACTACAAAACAAAACT‐3′ | ||
| Reverse transcription–polymerase chain reaction | ||
| E‐cadherin | F: 5′‐CTCCCTGAGCTCGCTGAAC‐3′ | 65 |
| R: 5′‐GTGCCACACAGGAACGACTC‐3′ | ||
| Connexin 26 | F: 5′‐ACGTTGGCCTTTTGGTTATG‐3′ | 63 |
| R: 5′‐TGTTGCGGGCTGTACTCAG‐3′ | ||
| Rassf1a | F: 5′‐GCTTCATCAAGGTTCAGCTGA‐3′ | 64 |
| R: 5′‐TCAAAGAGTGCAAACTTGCG‐3′ | ||
| c‐fos | F: 5′‐TTGCGCAGATCTGTCCGTCT‐3′ | 65 |
| R: 5′‐GTTGATCTGTCTCCGCTTGG‐3′ | ||
| Dnmt1 | F: 5′‐AGAAAGCCAACGGTTGTCCT‐3′ | 64 |
| R: 5′‐GTCTCACTGTCCGACTTGCTC‐3′ | ||
Semi‐quantitative RT‐PCR amplification for expression of E‐cadherin, Cx26, Rassf1a, c‐fos and Dnmt1. Total RNA was extracted from pooled liver samples of three rats in each subgroup, using ISOGEN (Nippon Gene, Inc., Toyama, Japan) and first‐strand cDNA was synthesized from 0.2‐µg samples with Ready‐To‐Go Your‐Prime First‐Strand Beads (Pharmacia Co. Ltd, Tokyo, Japan). To eliminate possible false‐positives caused by residual genomic DNA, all samples were treated with DNase.
Semi‐quantitative RT‐PCR analysis was performed with the primer sets of rat E‐cadherin, Cx26, and Rassf1a genes as described previously.( 19 , 27 ) Primer pairs for rat c‐fos and Dnmt1 genes were also designed against rat c‐fos and Dnmt1 sequences (GenBank accession numbers X06769 and AF116344, respectively; Table 1). The rat Gapdh gene was used as an internal control gene.( 27 ) PCR amplification was carried out in a reaction volume of 20 µL containing 1 µM of each gene primer, 200 µM of each dNTP, 1 × PCR buffer (Perkin Elmer, Applied Biosystems Division, Foster City, CA, USA), 0.5 U of AmpliTaq Gold (Perkin Elmer), and 0.5 µL of synthesized cDNA mixture. For each gene, multiple cycles of PCR amplification were tested. The cycle at which a sample having the highest expression reached an amplification plateau was determined, and a cycle number smaller than this was adopted for the analysis. The amplified products were then separated on 2% agarose gels containing 0.05 µg/mL ethidium bromide.
Results
Histologically, in the livers of rats fed the CDAA diet, fat deposits occurred in hepatocytes at 4 days and diffuse fatty changes were observed at 8 days. Fatty change widely expanded in all areas of livers at 3 weeks and the extension of collagen fibers was observed from Glisson's sheaths.
Results of the bisulfite sequencing analysis for the E‐cadherin, Cx26 and Rassf1a genes are shown in 1, 2, 3, respectively. Methylation status was measured in the 5′ upstream regions of E‐cadherin (between nt –183 and 182), containing 26 CpG sites; of Cx26 (between nt –206 and 85), containing 29 CpG sites; and of Rassf1a (between nt –389 and 48), containing 28 CpG sites. The livers of rats fed the CDAA diet for 4 and 8 days and 3 weeks were methylated in E‐cadherin, Cx26 and Rassf1a genes, while normal livers were all unmethylated. The methylation status in the 5′ upstream region of c‐fos (between nt –202 and 90), containing 19 CpG sites, is shown in Fig. 4. Normal livers were methylated in c‐fos gene. Although the livers at 4 days were weakly methylated, those at 8 days and 3 weeks were gradually unmethylated.
Figure 1.
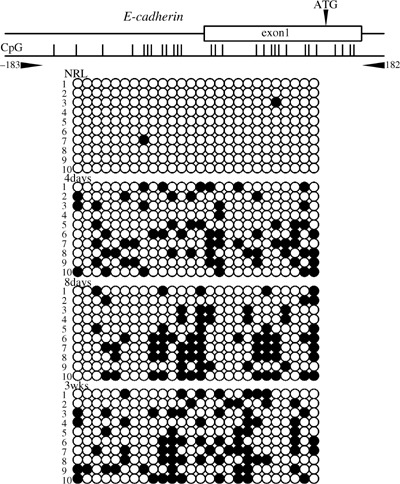
Methylation analysis of the 5′ upstream region of the E‐cadherin gene in rat by bisulfite sequencing. The primer pair used for bisulfite sequencing is shown. The transcription start site was defined as +1. Methylated CpG sites are represented by closed circles and unmethylated CpG sites are represented by open circles. NRL, normal liver tissue.
Figure 2.

Methylation analysis of the 5′ upstream region of the Cx26 gene in rat by bisulfite sequencing. The primer pair used for bisulfite sequencing is shown. The transcription start site was defined as +1. Methylated CpG sites are represented by closed circles and unmethylated CpG sites are represented by open circles. NRL, normal liver tissue.
Figure 3.

Methylation analysis of the 5′ upstream region of the Rassf1a gene in rat by bisulfite sequencing. The primer pair used for bisulfite sequencing is shown. The transcription start site was defined as +1. Methylated CpG sites are represented by closed circles and unmethylated CpG sites are represented by open circles. NRL, normal liver tissue.
Figure 4.
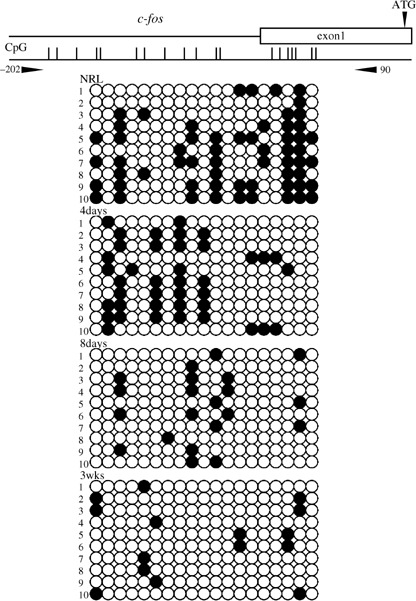
Methylation analysis of the 5′ upstream region of the c‐fos gene in rat by bisulfite sequencing. The primer pair used for bisulfite sequencing is shown. The transcription start site was defined as +1. Methylated CpG sites are represented by closed circles and unmethylated CpG sites are represented by open circles. NRL, normal liver tissue.
Expression levels of the E‐cadherin, Cx26, Rassf1a, c‐fos and Dnmt1 genes in the livers of rats fed the CDAA diet were measured using semiquantitative RT‐PCR analysis. Representative results are shown in Fig. 5. No changes in the expression level of E‐cadherin, Cx26 and Rassf1a were found in the livers of rats fed the CDAA diet. By contrast, while c‐fos and Dnmt1 genes weakly expressed in normal livers, these expressions were elevated after 8 or 4 days feeding of the CDAA diet, respectively.
Figure 5.
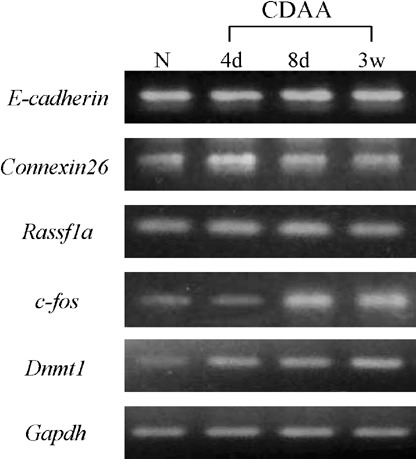
Expression levels of E‐cadherin, Cx26, Rassf1a, c‐fos and Dnmt1 mRNAs relative to Gapdh mRNA using semiquantitative reverse transcription–polymerase chain reaction analysis. N, normal liver tissue.
Discussion
The present study indicated that gene‐specific changes of DNA methylation patterns occurred in livers of rats after short‐term feeding of the CDAA diet, suggesting that gene‐specific hypermethylation might be involved in the early phase of rat hepatocarcinogenesis induced by the CDAA diet.
DNA hypomethylation is well known as one of the mechanisms underlying rat hepatocarcinogenesis resulting from a CD diet, because of a multiple methyl group donor‐deficiency.( 12 , 13 ) Indeed, genome‐wide hypomethylation of liver DNA has been demonstrated during hepatocarcinogenesis induced by the CD diet.( 28 , 29 ) Regional hypomethylation of growth‐related genes such as c‐fos, c‐myc, and c‐Ha‐ras has also been detected in the livers of rats after short‐term feeding with the CD diet.( 14 , 15 ) Moreover, hypomethylation of the c‐myc gene in rat HCC induced by the CDAA diet has been reported.( 18 ) In contrast, site‐specific hypermethylation of the p16 gene in pre‐neoplastic lesions and tumors has been reported to occur from folate/methyl deficiency in rat livers.( 30 ) Recently, the authors showed regional methylation in the 5′ upstream regions of the E‐cadherin and Cx26 genes in rat HCC as a result of the CDAA diet,( 19 ) and another group reported hypermethylation of intron 1 of neutral endopeptidase 24.11 in rat HCC induced by the same model.( 31 ) Despite methyl‐donor deficiency, it is unclear why site‐specific hypermethylation occurs in rat hepatocarcinogenesis as a result of the CDAA diet. Although the elevated expression of the c‐fos, c‐myc and c‐Ha‐ras genes was returned to control levels after ending the methyl‐deficient diet feeding, hypomethylation of specific sites in these genes was irreversible.( 14 , 15 ) In the present study, methylation of CpG sites within the CpG island observed was sparse, not dense, and the methylation was not associated with gene repression. Therefore, gene‐specific hyper‐ and hypomethylation changes might persist and accelerate until the development of HCC under the feeding of the CDAA diet in rats.
Increased expression levels of Dnmt1 were detected in livers of rats fed with a folate/methyl‐deficient diet as well as in HCC.( 29 ) In the present study, the authors also observed elevated Dnmt1 expression in the livers of rats fed the CDAA diet. Dnmt1 can bind with high affinity to DNA‐damaged lesions such as strand breaks, gaps and abasic sites, induced by folate/methyl‐deficiency.( 29 ) It is suggested that DNA methylation efficiency due to sequestration from the replication fork might be relative low in DNA containing damaged lesions, resulting in promotion of passive replication‐dependent demethylation.( 29 ) In promoter regions of tumor suppressor genes, inappropriate binding of Dnmt1 may also induce local histone deacetylation, methylation, and regional hypermethylation.( 29 ) In addition, a common oxygen radical‐induced guanine derivative, 8‐OHdG, could inhibit DNA methylation, resulting in reduced gene expression.( 32 ) 8‐OHdG is significantly detectable after only 1 day and progressively accumulates for at least up to 12 weeks in the livers of rats fed the CDAA diet.( 16 , 33 ) Therefore, it is possible that the aberrant DNA methylation pattern might be due to inappropriate binding of Dnmt1 to DNA containing damaged lesions, or the accumulation of 8‐OHdG in livers of rats fed the CDAA diet.
In conclusion, the present investigation demonstrated that gene‐specific methylation changes were found in livers of rats fed the CDAA diet. While global hypomethylation generally occurs in several human cancer cells, CpG islands of tumor suppressor genes are regionally hypermethylated.( 20 ) Despite methyl group donor‐deficiency, gene‐specific hypermethylation occurs during rat hepatocarcinogenesis induced by the CDAA diet.
Acknowledgments
This study was supported in part by the Foundation for Promotion of Cancer Research in Japan, and by a grant (RK17‐027) from the Faculty of Science and Engineering, Kinki University.
References
- 1. Mikol YB, Hoover K, Crasis D, Poirier LA. Hepatocarcinogenesis in rats fed methyl‐deficient, amino acid defined diets. Carcinogenesis 1983; 4: 1619–29. [DOI] [PubMed] [Google Scholar]
- 2. Ghoshal AK, Farber E. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis 1984; 5: 1367–70. [DOI] [PubMed] [Google Scholar]
- 3. Lombardi B. The choline‐devoid diet model of hepatocarcinogenesis in rats. In: Feo F, Pani P, Columbano A, Grecea R, eds. Chemical Carcinogenesis. New York: Plenum Press, 1988: 569–81. [Google Scholar]
- 4. Chandar N, Lombardi B. Liver cell proliferation and incidence of hepatocellular carcinomas in rats fed consecutively a choline‐devoid and a choline‐supplemented diet. Carcinogenesis 1988; 9: 259–63. [DOI] [PubMed] [Google Scholar]
- 5. Giambarresi LI, Katyal SL, Lombardi B. Promotion of liver carcinogenesis in the rat by a choline‐devoid diet. Role of liver cell necrosis and regeneration. Br J Cancer 1982; 46: 825–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shinozuka H, Katyal SL, Perera MIR. Choline deficiency and chemical carcinogenesis. In: Poirier LA, Newberne PM, Pariza MW, eds. Essential Nutrients in Carcinogenesis. New York: Plenum Press, 1986: 253–67. [DOI] [PubMed] [Google Scholar]
- 7. Rushmore TH, Farber E, Ghoshal AK et al . A choline‐devoid diet, carcinogenic in the rat, induces DNA damage and repair. Carcinogenesis 1986; 7: 1677–80. [DOI] [PubMed] [Google Scholar]
- 8. Rushmore TH, Lim YP, Farber E, Ghoshal AK. Rapid lipid peroxidation in the nuclear fraction of rat liver induced by a diet deficient in choline and methionine. Cancer Lett 1984; 24: 251–5. [DOI] [PubMed] [Google Scholar]
- 9. Perera MIR, Demetris AJ, Katyal SL, Shinozuka H. Lipid peroxidation of liver microsome membranes induced by choline‐deficient diets and its relationship to the diet‐induced promotion of the induction of gamma‐glutamyltranspeptidase‐positive foci. Cancer Res 1985; 5: 2533–8. [PubMed] [Google Scholar]
- 10. Chandar N, Lombardi B, Locker J. c‐myc gene amplification during hepatocarcinogenesis by a choline‐devoid diet. Proc Natl Acad Sci USA 1989; 86: 2703–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith ML, Yeleswarapu L, Scalamogna P et al . p53 mutations in hepatocellular carcinomas induced by a choline‐devoid diet in male Fischer 344 rats. Carcinogenesis 1993; 14: 503–10. [DOI] [PubMed] [Google Scholar]
- 12. Poirier LA. Methyl group deficiency in hepatocarcinogenesis. Drug Metab Rev 1994; 26: 185–99. [DOI] [PubMed] [Google Scholar]
- 13. Christman JK. Lipotrope deficiency and persistent changes in DNA methylation: lipotrope deficiency and DNA methylation. Adv Exp Med Biol 1995; 375: 97–106. [DOI] [PubMed] [Google Scholar]
- 14. Christman JK, Chen ML, Sheiknejad G et al . Methyl deficiency, DNA methylation, and cancer: studies on the reversibility of the effects of a lipotrope‐deficient diet. J Nutr Biochem 1993; 4: 672–80. [Google Scholar]
- 15. Christman JK, Sheiknejad G, Dizik M et al . Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe methyl deficiency. Carcinogenesis 1993; 14: 551–7. [DOI] [PubMed] [Google Scholar]
- 16. Nakae D, Yoshiji H, Maruyama H et al . Production of both 8‐hydroxydeoxyguanosine in liver DNA and γ‐glutamyltransferase‐positive hepatocellular lesions in rats given a choline‐deficient, 1‐amino acid‐defined diet. Jpn J Cancer Res 1990; 81: 1081–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakae D, Yoshiji H, Mizumoto Y et al . High incidence of hepatocellular carcinomas induced by a choline deficient 1‐amino acid defined diet in rats. Cancer Res 1992; 52: 5042–5. [PubMed] [Google Scholar]
- 18. Tsujiuchi T, Tsutsumi M, Sasaki Y et al . Hypomethylation of CpG sites and c‐myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline‐deficient 1‐amino acid‐defined diet in rats. Jpn J Cancer Res 1999; 90: 909–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsujiuchi T, Shimizu K, Itsuzaki Y et al . CpG site hypermethylation of E‐cadherin and Connexin26 genes in hepatocellular carcinomas induced by a choline‐deficient 1‐amino acid‐defined diet in rats. Mol Carcinog 2006; 46: 269–74. [DOI] [PubMed] [Google Scholar]
- 20. Laird PW, Jaenisch R. DNA methylation and cancer. Hum Mol Genet 1994; 3: 1487–95. [DOI] [PubMed] [Google Scholar]
- 21. Zingg JM, Jones PA. Genetic and epigenetic aspects of mutation and carcinogenesis. Carcinogenesis 1997; 18: 869–82. [DOI] [PubMed] [Google Scholar]
- 22. Jones PA, Laird PW. Cancer epigenetic comes of age. Nat Genet 1999; 21: 163–7. [DOI] [PubMed] [Google Scholar]
- 23. Kondo Y, Kanai Y, Sakamoto M et al . Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis: a comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology 2000; 32: 970–9. [DOI] [PubMed] [Google Scholar]
- 24. Saito Y, Kanai Y, Sakamoto M et al . Expression of mRNA for DNA methyltransferases and methyl‐CpG binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology 2001; 33: 561–8. [DOI] [PubMed] [Google Scholar]
- 25. Shen L, Ahuja N, Shen Y et al . DNA methylation and environmental exposures in human hepatocellular carcinoma. J Natl Cancer Inst 2002; 94: 755–61. [DOI] [PubMed] [Google Scholar]
- 26. Lee S, Lee HJ, Kim JH et al . Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol 2003; 163: 1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shimizu K, Kato A, Shigemura M et al . Aberrant methylation patterns of the Rassf1a gene in rat lung adenocarcinomas induced by N‐nitrosobis (2‐hydroxypropyl) amine. Mol Carcinog 2006; 45: 112–17. [DOI] [PubMed] [Google Scholar]
- 28. Locker J, Reddy TV, Lombardi B. DNA methylation and hepatocarcinogenesis in rats fed a choline‐devoid diet. Carcinogenesis 1986; 8: 1309–12. [DOI] [PubMed] [Google Scholar]
- 29. James SJ, Pogribny IP, Pogribna M et al . Mechanisms of DNA damage, DNA hypomethylation, and tumor progression in the folate/methyl‐deficient rat model of hepatocarcinogenesis. J Nutr 2003; 133: 3740–7. [DOI] [PubMed] [Google Scholar]
- 30. Pogribny IP, Lames SJ. De novo methylation of the p16 INK4A gene in early preneoplastic liver and tumors induced by folate/methyl deficiency in rats. Cancer Lett 2002; 187: 69–75. [DOI] [PubMed] [Google Scholar]
- 31. Uematsu F, Takahashi M, Yoshida M et al . Methylation of neutral endopeptidase 24.11 promoter in rat hepatocellular carcinoma. Cancer Sci 2006; 97: 611–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weitzman SA, Turk PW, Milkowski DH, Kozlowski K. Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci USA 1994; 91: 1261–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshiji H, Nakae D, Mizumoto Y et al . Inhibitory effect of dietary iron deficiency on inductions of putative preneoplastic lesions as well as 8‐hydroxydeoxyguanosine in DNA and lipid peroxidation in the livers of rats caused by exposure to a choline‐deficient 1‐amino acid defined diet. Carcinogenesis 1992; 13: 1227–33. [DOI] [PubMed] [Google Scholar]