

Abstract
Interstitial lung disease (ILD) is reported as a serious adverse event in lung cancer patients treated with gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR‐TKI). However, the mechanisms of ILD associated with gefitinib remain unknown. To address the molecular mechanisms of ILD‐associated gefitinib, we determined the effect of gefitinib treatment on surfactant protein expression in vitro and in vivo. Gefitinib treatment suppressed surfactant protein (SP)‐A expression in H441 human lung adenocarcinoma cells expressing SP‐A, ‐B, ‐C and ‐D by inhibiting epidermal growth factor signal. Next, gefitinib (200 mg/kg) was given p.o. to the mice daily for 1 week. Daily administration of gefitinib gradually reduced SP‐A level in the bronchoalveolar lavage fluid. When lipopolysaccharide (LPS) was instilled intratracheally to the mice pretreated with gefitinib for 1 week, lung inflammation by LPS was exacerbated and prolonged. This exacerbation of lung inflammation was rescued by intranasal administration of SP‐A. These results demonstrated that pretreatment with gefitinib exacerbated LPS‐induced lung inflammation by reducing SP‐A expression in the lung. This study suggests that epidermal growth factor receptor tyrosine kinase inhibitor may reduce SP‐A expression in the lungs of lung cancer patients and thus patients treated with epidermal growth factor receptor tyrosine kinase inhibitor may be susceptible to pathogens. (Cancer Sci 2008; 99: 1679–1684)
Gefitinib is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) that has antitumor activity in patients with advanced non‐small cell lung cancer (NSCLC). Two large phase II trials for previously treated advanced NSCLC revealed that gefitinib monotherapy was effective, with response rates of 18.4% and 12%.( 1 , 2 ) Higher response rates were achieved in Japanese, non‐smoking and adenocarcinoma patients,( 3 ) and subsequent reports revealed that some somatic mutations of the EGFR gene, frequently observed in these patients, are strongly correlated with sensitivity to gefitinib.( 4 , 5 ) The most common adverse effects associated with gefitinib are skin rash and diarrhea; these were generally mild in the phase II clinical trials and have been confirmed in extensive clinical practice.( 1 , 2 )
Interstitial lung disease (ILD) was reported as a serious adverse event associated with gefitinib after its approval in Japan.( 6 , 7 , 8 ) Retrospective analysis revealed that, in Japan, the incidence of ILD was 3–5% with a mortality rate of 2–3%.( 7 , 8 ) In contrast, outside of Japan, the rate of ILD was only 0.3%.( 9 ) These reports suggested that the risk factors of ILD associated with gefitinib were male gender, a smoking history and a coincidence of idiopathic pulmonary fibrosis. However, the molecular mechanisms of ILD associated with gefitinib remain largely unknown.
Epidermal growth factor receptor is expressed on the surface of various cells and activates gene transcription, thus regulating cellular growth and differentiation. Although expression of EGFR is sparse in healthy adult human airways, including bronchial epithelial cells and alveolar type II cells, EGF signaling upregulates various gene transcriptions in these cells.( 10 , 11 ) In alveolar type II cells, EGF signaling mediates cell differentiation and stimulates surfactant protein (SP)‐A production.( 12 ) Pulmonary surfactant secreted from alveolar type II cells is a mixture of lipids and proteins, and prevents the alveoli from collapsing at the end of expiration.( 13 , 14 ) Ten percent of surfactant is composed of proteins, including hydrophilic SP‐A and ‐D, and the hydrophobic proteins SP‐B and SP‐C.( 15 ) Although SP‐B and SP‐C are critical for reduction of alveolar surface tension, SP‐A and ‐D belong to the collectin subgroup of the C‐type lectin superfamily in which lectin domains are associated with collagenous structures along with SP‐D and mannose‐binding lectin.( 16 , 17 )
Surfactant protein A is a multifunctional protein involved in maintenance surfactant homeostasis, lipid sorting, tubular myelin formation and innate immune defense in the lung. SP‐A knockout mice reveal significant defects in host defense; SP‐A−/– mice show impaired microbial clearance after intratracheal administration of group B Streptococcus,( 18 ) Haemophilus influenza, ( 19 ) Pseudomonas aeruginosa, ( 20 ) Pneumocystis carinii, ( 21 ) and respiratory syncytial virus.( 22 ) In addition, SP‐A directly protects surfactant phospholipids and macrophages from oxidative damage.( 23 )
In this study, we hypothesized that the EGFR‐TKI gefitinib inhibits the expression of SP‐A on the alveolar type II cells and thus increases the susceptibility to pathogens.
Materials and Methods
Gefitinib treatment and Western blotting of EGFR on H441 cells. We used human lung adenocarcinoma H441 cells instead of alveolar type II cells because H441 cells stably express surfactant proteins in vitro and harbor wild‐type EGFR gene.( 24 , 25 ) H441 cells were cultured in RPMI‐1640 medium with 10% fetal bovine serum. H441 grown in serum‐free RPMI‐1640 medium ± 20 µg/mL recombinant epidermal growth factor (EGF; Biomedical Technologies, Stoughton, MA, USA) were treated for 48 h with 1 µM gefitinib (AstraZeneca, UK).
After 48 h treatment with 1 µM gefitinib, the H441 cells were lysed in RIPA buffer containing protease inhibitors (Roche Molecular Biochemicals, Indianapolis, IN, USA) and 1 mM NaVO3 (Sigma, St Louis, MO, USA). After determination of protein concentration using the Bio‐Rad Bradford protein assay (Bio‐Rad Laboratories, Hercules, CA, USA), 30 µg of total protein was loaded per lane on a 10% Bis‐Tris Gel (Invitrogen, Carlsbad, CA, USA), and transferred to a PVDF membrane (Invitrogen). The membrane was incubated with rabbit antihuman EGFR (1005) Antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or mouse antiphospho‐EGFR monoclonal antibody (YTyr1173; Upstate Biotechnologies, Lake Placid, NY, USA). The blots were stained with secondary antibody (goat antirat IgG‐HR or rat antimouse IgG‐HR; Santa Cruz Biotechnology) and the detection of specific signals was performed using the ECL Detection System (Amersham Pharmacia Biotech AB, Uppsala, Sweden).
Reverse transcription polymerase chain reaction of SP‐A, ‐B, ‐C and ‐D and immunohistochemistry of SP‐A on PC‐3 cells. The total RNA (2 µg) extracted from the H441 cells treated with 1 µM gefitinib was converted into cDNA by Oligo(dT)12–18 primers and Superscript II reverse transcription (Gibco BRL, Carlsbad, CA, USA) in a final volume of 20 µL. Of this cDNA, 1 µL was amplified with the following sense and antisense primers, respectively: SP‐A, 5′‐gAAggACgTTTgTgTTggAA‐3′ and 5′‐TggATTCCTTgggACAgCAA‐3′; SP‐B, 5′‐TACTCCgTCATCCTgCTCgA‐3′ and 5′‐gCTgCTCCACAAATTgCTTg‐3′; SP‐C, 5′‐AgCAAAgAggTCCTgATggA‐3′ and 5′‐CTATTgAgAgCCTCAAgACT‐3′; SP‐D, 5′‐AgCTgggCCCAAAggAgAAgTAgg‐3′ and 5′‐AgCggCAgAgCgTggAgAgg‐3′; EGFR, 5′‐CTTCTTgCAgCgATACAgCTC‐3′ and 5′‐ATgCTCCAATAAATTCACTgC‐3′; and b‐action, 5′‐CTCTTTgATgTCACgCACgATTTC‐3′ and 5′‐gTgggCCgCTCTAggCACCAA‐3′.
The amplification profile was 95°C for 5 min, 40 cycles of 94°C for 1 min, 60°C for 90 s and 72°C for 2 min.
H441 cells cultured on the slide glass with/without 1 µM gefitinib for 48 h were fixed with 4% buffered formaldehyde, and stained with the mouse antihuman SP‐A antibody (clone PE‐10; DAKO, San Diego, CA, USA) at ×50 dilution. The cells were then incubated with the avidin–biotin–peroxidase complex method Envision+System (DAKO).
In vivo experiments. All animal experiments were approved by the institutional review board for animal experiments of Tohoku University. Female C57BL/6 mice purchased from Japan Charles River (Atsugi, Japan) were given a daily dose of 200 µL 1% Tween 80 (Sigma) solution p.o. containing either gefitinib (200 mg/kg) or no drug (control).
After 1 week, mice treated with gefitinib were instilled intratracheally with 250 µg/kg (5 µg/mouse) Escherichia coli lipopolysaccharide (LPS) 055:B5 (Sigma). To determine the response of SP‐A to the lung injury, human SP‐A (3 µg in 50 µL of phosphate‐buffered saline/mouse), obtained from patients with alveolar proteinosis,( 26 ) was administered intranasally on days 1–3 following the intratracheal LPS administration (5 µg/mouse).( 27 )
Bronchoalveolar lavage and Western blotting of SP‐A and ‐D in the bronchoalveolar lavage fluid. Mice were subjected to brief anesthesia with i.p. injection of ketamine and xylazine. After loss of consciousness, the trachea was exposed with a midline incision and cannulated with a 24‐G catheter. After the mice were killed by exsanguination, the lungs were lavaged three times with sterile 0.9% NaCl at a volume of 0.7 mL/wash. The average fluid recovery was greater than 80%. The bronchoalveolar lavage (BAL) fluid was centrifuged at 500 g for 10 min at 4°C and the supernatants were stored at –20°C until analysis.
Cells from BAL samples were resuspended in 1 mL normal saline. The cell differentials were performed on slides prepared in a Cytospin 3 (Shandon, Pittsburgh, PA, USA) centrifuged at a speed of 150 g for 2 min and stained with a modified Wright‐Giemsa technique (Diff‐Quik; Dade Behring, Dudingen, Switzerland).
Western blots of SP‐A and ‐D in the BAL fluid were performed as described above using the rabbit antihuman SP‐A polyclonal antibody and the rabbit antimouse SP‐D polyclonal antibody Anti‐Surfactant D (Chemicon International). Loading of BAL fluid was normalized by an amount of protein (25 µg/lane). The relative amount of immunoreactive SP‐A present in each sample was quantitated by NIH Image (National Institute of Mental Health). The densitometric data from each blot were normalized to the control condition with the control value set equal to one for each experiment.
Enzyme‐linked immunosorbent assay for tumor necrosis factor‐α. Tumor necrosis factor (TNF)‐α concentrations in BAL fluid and plasma were determined using a specific enzyme‐linked immunosorbent assay TNF‐α ELISA Kit (BioSource International, Camarillo, CA, USA) in conjunction with a Bio‐Rad model 550 microplate reader with accompanying software (Bio‐Rad, Hercules, CA, USA) as directed by the manufacturer. The ELISA had a lower detection limit of 3 pg/mL.
Histopathology of the lung. After thoracotomy, the left lung was inflated with 4% phosphate‐buffered formalin (pH 7.4) at a pressure of 20 cm H2O through the trachea for 6 h and subsequently fixed in 15% phosphate‐buffered formalin for 24 h. After paraffin embedding, 4‐µm sections were cut and stained with hematoxylin–eosin (HE) for histological analysis.
Statistics. Data are expressed as means ± standard error (SE). Statistical analysis was performed with StatView (SAS Institute). anova was used to determine differences among experimental groups. The Student–Newman–Keuls test was used for a multiple comparison. P < 0.05 was considered significant.
Results
Suppression of SP‐A by gefitinib treatment on H441 cells. A dose–response curve to 72 h treatment of gefitinib on H441 cells revealed that IC50 was 20 µM. Treatment with 1 µM gefitinib did not significantly inhibit H441 cell growth (data not shown). Treatment with 1 µM gefitinib completely blocked the EGF‐induced autophosphorylation of EGFR in H441 cells (Fig. 1a, upper panel), whereas the level of whole EGFR protein was not changed by gefitinib treatment (Fig. 1a, lower panel). EGF increased autophosphorylation of EGFR. This result implied that treatment with 1 µM gefitinib completely blocked the EGF signaling in H441 cells. The expression level of surfactant proteins on H441 cells after gefitinib treatment was examined because previous reports had suggested a relationship between EGF signaling and surfactant proteins. The level of SP‐A mRNA was suppressed by gefitinib treatment in serum‐free medium plus 20 µg/mL EGF conditions, whereas SP‐B, ‐C and ‐D expression levels did not change after gefitinib treatment (Fig. 1b). Immunohistochemistry also revealed strong suppression of SP‐A protein after gefitinib treatment (Fig. 1c). SP‐A protein could not be detected by Western blotting, possibly because of the low expression of SP‐A in H441 cells.
Figure 1.
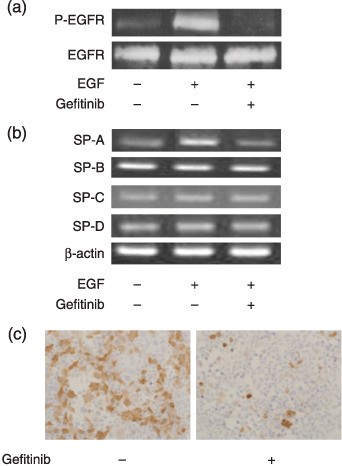
Autophosphorylation of epidermal growth factor receptor (EGFR) and expression levels of surfactant proteins (SP)‐A, ‐B, ‐C and ‐D in H441 cells. H441 cells cultured in serum‐free RPMI‐1640 medium plus 20 µg/mL EGF were treated with 1 µM gefitinib for 48 h. (a) Western blotting of whole EGFR protein and autophosphorylated EGFR protein in H441 cells after gefitinib treatment. (b) Reverse transcription polymerase chain reaction of SP‐A, ‐B, ‐C and ‐D in H441 cells after gefitinib treatment. (c) Immunohistochemistry of SP‐A on H441 cells after gefitinib treatment. P‐EGFR.
Suppression of SP‐A in BAL fluid of gefitinib‐treated mice. The examination of SP‐A and ‐D in BAL fluid revealed that gefitinib gradually suppressed the SP‐A level, and SP‐A was suppressed by 68% by day 8 (Fig. 2a,b) (P < 0.01). The level of SP‐D in BAL fluid did not change throughout the gefitinib treatment period (Fig. 2a,b). The intratracheal instillation of LPS to gefitinib‐treated mice was conducted to assess the effect of gefitinib on inflammation induced by bacterial infection. In control mice, LPS instillation increased the level of SP‐A in BAL fluid by 42% after 72 h and declined after peak, suggesting an anti‐inflammatory response of SP‐A. However, although the SP‐A level in BAL fluid in gefitinib‐treated mice was increased after LPS instillation, the level of SP‐A was significantly lower than control at 24 and 72 h after LPS instillation (Fig. 3a,b). Although we found lower levels of SP‐A in BAL fluid after gefitinib treatment, SP‐A immunohistochemistry of the lung did not show any change of staining intensity (data not shown). We suppose that expression level of SP‐A after gefitinib treatment was high enough for staining. In contrast, increases of SP‐D after LPS instillation were of similar levels in both control and gefitinib‐treated mice.
Figure 2.
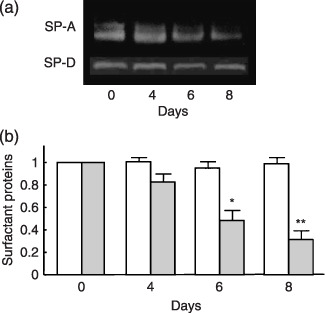
Contents of surfactant proteins (SP)‐A and ‐D in bronchoalveolar lavage (BAL) fluid after gefitinib treatment. (a) Western blotting and (b) densitometric data of SP‐A and ‐D in BAL fluid from mice at 0, 4, 6 and 8 days treatment with or without gefitinib (200 mg/kg). The values were normalized to the control condition for each experiment. ( ) SP‐D; (grey) SP‐A. Data presents means ± standard error in five mice. *P < 0.05, **P < 0.01.
) SP‐D; (grey) SP‐A. Data presents means ± standard error in five mice. *P < 0.05, **P < 0.01.
Figure 3.
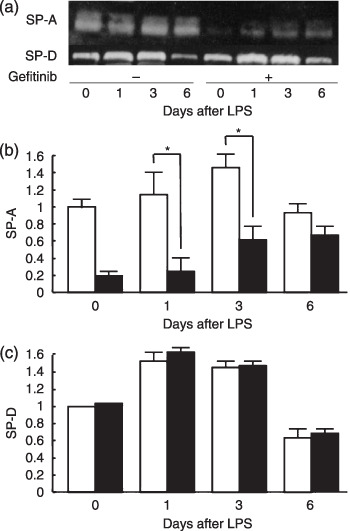
Surfactant protein‐A levels after lipopolysaccharide (LPS) instillation in the bronchoalveolar lavage (BAL) fluid. (a) Western blotting of SP‐A and ‐D and densitometric data of (b) SP‐A and (c) SP‐D in BAL fluid from mice with/without 7 days gefitinib pretreatment, 72 h after the intratracheal LPS instillation (250 µg/kg). () mice receiving LPS only; ( ) mice receiving gefitinib and LPS. Data presents means ± standard error in five mice. *P < 0.05.
) mice receiving gefitinib and LPS. Data presents means ± standard error in five mice. *P < 0.05.
Gefitinib treatment exacerbates LPS‐induced lung inflammation. LPS was administered intratracheally to mice following 1 week of gefitinib treatment in order to assess whether pretreatment with gefitinib increases the susceptibility of lung inflammation by LPS. The cell density of BAL fluid remained at a high level on day 6 in gefitinib‐pretreated mice, but improved to a normal level in control mice (Fig. 4a). The percentage of neutrophils in BAL fluid of gefitinib‐pretreated mice (13.5%) remained higher than control mice (6.0%) on day 6 (Table 1). Histopathological examination of the lung section also revealed that gefitinib pretreated mice showed stronger inflammatory infiltrates, mainly consisting of neutrophils (Fig. 4b). Neither gefitinib alone nor LPS alone induced the neutrophil accumulation in the lung 3 days after the treatment (Fig. 4b).
Figure 4.
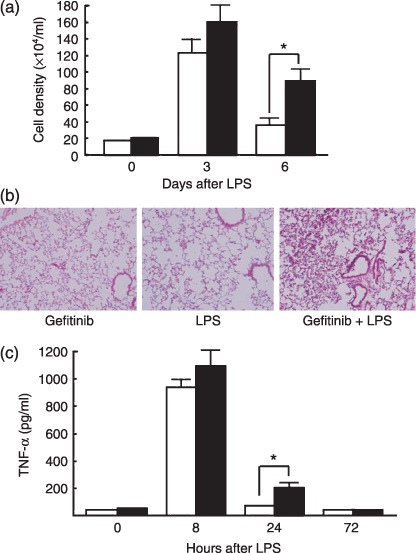
Lipopolysaccharide (LPS)‐induced inflammatory change in vivo. (a) Cell density in bronchoalveolar lavage (BAL) fluid from mice with/without gefitinib pretreatment, 6 days after intratracheal LPS instillation. (b) Infiltration of inflammatory cells 3 days after LPS instillation in the lungs of mice treated with gefitinib only (left), with LPS only (middle), or with gefitinib and LPS (right). (c) Tumor necrosis factor (TNF)‐α enzyme‐linked immunosorbent assay was performed on mouse BAL fluid 8, 24 and 72 h after the intratracheal LPS instillation (n = 6). () mice receiving LPS only; () mice receiving gefitinib and LPS. Data presents means ± standard error (SE) in five mice. *P < 0.05.
Table 1.
Percentage of alveolar macrophages and neutrophils in bronchoalveolar lavage fluid
| Days after LPS | Gefitinib | LPS | SP‐A | Macrophages (%) | Neutrophils (%) |
|---|---|---|---|---|---|
| 0 | – | – | – | 93.3 ± 0.3 | 2.1 ± 0.3 |
| 0 | + | – | – | 89.0 ± 1.5 | 6.9 ± 1.1 |
| 3 | – | + | – | 34.1 ± 5.5 | 61.0 ± 6.3 |
| 3 | + | + | – | 21.6 ± 2.4 | 72.5 ± 3.3 |
| 3 | + | + | + | 43.0 ± 1.6 | 40.7 ± 1.8** |
| 6 | – | + | – | 77.3 ± 2.8 | 6.0 ± 0.3 |
| 6 | + | + | – | 81.1 ± 0.6 | 13.5 ± 1.1 |
| 6 | + | + | + | 84.6 ± 3.6 | 1.3 ± 0.5* |
P < 0.05 (compared with gefitinib+/LPS + mice);
P < 0.01 (compared with gefitinib+/LPS + mice). BAL, bronchoalveolar lavage; LPS, lipopolysaccharide; SP, surfactant protein.
Although the level of inflammatory cytokine TNF‐α in BAL fluid was drastically increased 8 h after the administration of LPS in both groups of mice, gefitinib pretreated mice retained a detectable level of TNF‐α 24 h after LPS administration (Fig. 4c). TNF‐α could not be detected in the plasma of both control and gefitinib‐treated mice after LPS instillation (data not shown). These data suggest that gefitinib pretreatment prolonged the lung inflammatory response to LPS.
SP‐A rescue inhibits exacerbation of LPS‐induced inflammation in vivo. The effect of rescue SP‐A on lung inflammation induced by LPS in gefitinib‐pretreated mice was examined. The cell density of BAL fluid in the gefitinib pretreated mice was decreased by SP‐A rescue on both days 3 and 6 (Fig. 5a). SP‐A rescue also inhibited the increase of neutrophils induced by LPS in gefitinib‐treated mice on days 3 and 6 (Table 1). Histological assessment also showed that SP‐A administration significantly improved the neutrophil accumulation in the lung (Fig. 5b).
Figure 5.
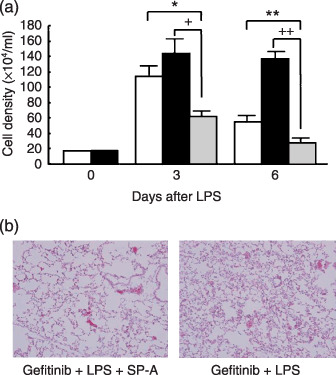
Effects of surfactant protein (SP)‐A rescue on lipopolysaccharide (LPS)‐induced lung inflammation. (a) Mice with/without gefitinib pretreatment received intratracheal LPS instillation on day 1 and SP‐A (150 µg/kg) on days 1–3. BAL was performed on days 3 and 6 after LPS instillation. () control mice; () mice treated with gefitinib only (grey) mice treated with gefitinib and rescued by SP‐A or gefitinib+/LPS + group. Data presents means ± standard error in three mice. *P < 0.05, **P < 0.01. (b) Lung tissue (HE) 3 days after LPS instillation in the SP‐A treated group (left) and control group (gefitinib + LPS) (right).
Discussion
The present study demonstrated that mice pretreated with gefitinib had exacerbated LPS‐induced lung inflammation, and that this exacerbation was due to the suppression of SP‐A expression in the lung. It was also demonstrated in vitro that gefitinib treatment of human lung adenocarcinoma H441 cells suppressed the expression of SP‐A. These results suggest that ILD observed in patients treated with gefitinib may be, at least in part, associated with the suppression of SP‐A expression by gefitinib.
Gefitinib, an EGFR‐TKI, has been shown to be effective for the treatment of advanced NSCLC with a favorable adverse event profile in phase I and II trials,( 1 , 2 , 28 ) Shortly after the approval of gefitinib for the treatment of inoperable and recurrent NSCLC, a number of cases were reported of ILD after gefitinib treatment.( 6 , 7 , 8 ) Major risk factors of ILD were a smoking history, pre‐existing or concurrent idiopathic pulmonary fibrosis, and poor performance status. Although the existence of ILD associated with gefitinib has been seen in clinics, the mechanism of ILD associated with gefitinib has not been fully elucidated.
This study has shown that the expression of SP‐A mRNA is regulated by EGF signaling in human adenocarcinoma H441 cells, whereas SP‐B, ‐C and ‐D are not. SP‐A levels in BAL fluid were also reduced in mice treated with gefitinib. These results revealed that SP‐A expression in human type II alveolar cells could be regulated by the EGF signal.
Pulmonary surfactant proteins are synthesized specifically by alveolar type II cells and bronchioalveolar Clara cells, and are positively or negatively regulated by various factors such as glucocorticoids, retinoids, insulin, growth factors and cytokines.( 29 ) Among these regulatory factors, EGF positively regulates SP‐A, a major constituent of the surfactant. EGF accelerates alveolarization and decreases the severity of respiratory syndromes in fetal lambs.( 30 ) Blockade of EGF signaling by the EGFR‐TKI, genistein and tyrphostin, inhibited SP‐A expression in cultured fetal lung explants.( 31 ) Antisense oligonucleotide against EGFR mRNA inhibited SP‐A expression in human fetal lung tissue during alveolar type II differentiation.( 32 ) Mechanisms of different effects of EGFR‐TKI on expression among SP‐A and ‐B, ‐C and SP‐D are still unclear. Although precise mechanisms of SP‐A expression have been studied,( 29 ) regulatory mechanisms of SP‐B, ‐C and ‐D are warranted. In this context, it could be suggested that treatment with EGFR‐TKI reduces SP‐A expression in the human adult lung.
Pathogen‐derived components, such as LPS derived from Gram‐negative bacteria and peptideglycan derived from Gram‐positive bacteria, are potent stimulators of inflammation. SP‐A interacts with CD14 on alveolar macrophages and inhibits the binding of smooth LPS to CD14 and reduces TNF‐α expression induced by LPS.( 33 ) Therefore, SP‐A−/– mice show significantly enhanced TNF‐α production induced by smooth LPS.( 34 ) TNF‐α production induced by peptideglycan was also inhibited by SP‐A.( 35 ) These previous studies are in agreement with our results. Pre‐treatment with gefitinib exacerbates LPS‐induced lung inflammation in mice, and this exacerbation could be rescued by the administration of SP‐A.
Among the several risk factors for ILD in clinical studies, smoking history and interstitial pulmonary fibrosis are linked to a reduced expression of SP‐A in the lung. Betsuyaku et al. reported that aging alone or combined with long‐term smoking leads to a decrease of SP‐A levels in the lungs of human subjects.( 36 ) The concentration of SP‐A in the BAL fluid of patients with idiopathic pulmonary fibrosis was lower than in control subjects.( 37 , 38 ) Tamura et al. retrospectively analyzed baseline SP‐A expression levels of cancer tissues and normal bronchial tissues by immunohistochemistry from 20 NSCLC patients treated with gefitinib; 10 patients developed ILD after gefitinib treatment and 10 patients did not.( 39 ) They found that baseline SP‐A expression levels in both cancer and normal tissues was significantly lower in patients with ILD than in patients without ILD. They concluded that baseline SP‐A expression could be a predictive marker for ILD by gefitinib. Although the present study and that by Suzuki et al. used a larger dose of gefitinib (200 mg/kg/day) than that used in humans (1–14 mg/kg/day [50–700 mg/body/day]),( 40 ) we observed that a lower dose of gefitinib (25 mg/kg) equally suppressed SP‐A expression in mice (data not shown). Combined with these clinical findings and this present study in mice, it could be speculated that human subjects with low expression levels of SP‐A due to smoking or idiopathic pulmonary fibrosis become susceptible to lung damage by pathogens, oxidative stress or other factors.
Suzuki et al. reported that, in mice, treatment with gefitinib exacerbated the pulmonary fibrosis induced by bleomycin because EGF and EGFR were upregulated early in the response to lung injury.( 40 , 41 ) These findings suggest that inhibition of EGFR signaling by gefitinib impairs the repair of and, thereby, exacerbates pulmonary injury, especially in patients with pulmonary comorbidities.
Although ILD associated with gefitinib in Japanese patients has been reported in Japan at a prevalence of 3–5%,( 7 , 8 ) the prevalence of ILD among gefitinib‐treated patients in the USA and Europe was less than 1%.( 42 , 43 ) The reason for the different prevalence of ILD associated with gefitinib between Japan and Western countries remains unclear. It might be attributable to Japanese patients having an increased genetic susceptibility to ILD.
In conclusion, we have demonstrated that pretreatment with gefitinib exacerbates LPS‐induced lung inflammation by reducing levels of SP‐A. These findings suggest that ILD after EGFR‐TKI treatment may be related in part to a reduction of SP‐A.
Acknowledgments
This study was supported by the Smoking Research Foundation in Japan (2003–2005) and the Public Trust Haraguchi Memorial Cancer Research Fund (2005).
References
- 1. Fukuoka M, Yano S, Giaccone G et al . Multi‐institutional randomized phase II trial of gefitinib for previously treated patients with advanced non‐small‐cell lung cancer (The IDEAL 1 Trial). J Clin Oncol 2003; 21: 2237–46. [DOI] [PubMed] [Google Scholar]
- 2. Kris MG, Natale RB, Herbst RS et al . Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non‐small cell lung cancer: a randomized trial. JAMA 2003; 290: 2149–58. [DOI] [PubMed] [Google Scholar]
- 3. Miller VA, Kris MG, Shah N et al . Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non‐small‐cell lung cancer. J Clin Oncol 2004; 22: 1103–9. [DOI] [PubMed] [Google Scholar]
- 4. Paez JG, Janne PA, Lee JC et al . EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 5. Lynch TJ, Bell DW, Sordella R et al . Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 6. Inoue A, Saijo Y, Maemondo M et al . Severe acute interstitial pneumonia and gefitinib. Lancet 2003; 11 (361): 137–9. [DOI] [PubMed] [Google Scholar]
- 7. Ando M, Okamoto I, Yamamoto N et al . Predictive factors for interstitial lung disease, antitumor response, and survival in non‐small‐cell lung cancer patients treated with gefitinib. J Clin Oncol 2006; 24 (16): 2549–56. [DOI] [PubMed] [Google Scholar]
- 8. Takano T, Ohe Y, Kusumoto M et al . Risk factors for interstitial lung disease and predictive factors for tumor response in patients with advanced non‐small cell lung cancer treated with gefitinib. Lung Cancer 2004; 45 (1): 93–104. [DOI] [PubMed] [Google Scholar]
- 9. Forsythe B, Faulkner K. Overview of the tolerability of gefitinib (IRESSA®) monotherapy. Clinical experience in non‐small cell lung cancer. Drug Saf 2004; 27: 1081–92. [DOI] [PubMed] [Google Scholar]
- 10. Takeyama K, Dabbagh K, Lee HM et al . Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci USA 1999; 96 (6): 3081–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeyama K, Fahy JV, Nadel JA. Relationship of epidermal growth factor receptors to goblet cell production in human bronchi. Am J Respir Crit Care Med 2001 Feb; 163 (2): 511–16. [DOI] [PubMed] [Google Scholar]
- 12. Whitsett JA, Weaver TE, Lieberman MA, Clark JC, Daugherty C. Differential effects of epidermal growth factor and transforming growth factor‐beta on synthesis of Mr = 35 000 surfactant‐associated protein in fetal lung. J Biol Chem 1987; 262: 7908–13. [PubMed] [Google Scholar]
- 13. King RJ, Clements JA. Surface active materials from dog lung. I. Method of isolation. Am J Physiol 1972; 223: 707–14. [DOI] [PubMed] [Google Scholar]
- 14. King RJ, Clements JA. Surface active materials from dog lung. II. Composition and physiological correlations. Am J Physiol 1972; 223: 715–26. [DOI] [PubMed] [Google Scholar]
- 15. Kuroki Y, Voelker DR. Pulmonary surfactant proteins. J Biol Chem 1994; 269: 25943–6. [PubMed] [Google Scholar]
- 16. Sano H, Kuroki Y. The lung collectins, SP‐A and SP‐D, modulate pulmonary innate immunity. Mol Immunol 2005; 42: 279–87. [DOI] [PubMed] [Google Scholar]
- 17. Wright JR, Borron P, Brinker KG, Folz RJ. Surfactant protein A regulation of innate and adaptive immune responses in lung inflammation. Am J Respir Cell Mol Biol 2001; 24: 513–7. [DOI] [PubMed] [Google Scholar]
- 18. LeVine AM, Bruno MD, Huelsman KM, Ross GF, Whitsett JA, Korfhagen TR. Surfactant protein A‐deficient mice are susceptible to group B streptococcal infection. J Immunol 1997; 1 (158): 4336–40. [PubMed] [Google Scholar]
- 19. LeVine AM, Whitsett JA, Gwozdz JA et al . Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol 2000; 165: 3934–40. [DOI] [PubMed] [Google Scholar]
- 20. LeVine AM, Kurak KE, Bruno MD, Stark JM, Whitsett JA, Korfhagen TR. Surfactant protein‐A‐deficient mice are susceptible to Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol 1998; 19: 700–8. [DOI] [PubMed] [Google Scholar]
- 21. Linke MJ, Harris CE, Korfhagen TR et al . Immunosuppressed surfactant protein A‐deficient mice have increased susceptibility to Pneumocystis carinii infection. J Infect Dis 2001; 183: 943–52. [DOI] [PubMed] [Google Scholar]
- 22. LeVine AM, Gwozdz J, Stark J, Bruno M, Whitsett J, Korfhagen T. Surfactant protein‐A enhances respiratory syncytial virus clearance in vivo . J Clin Invest 1999; 103: 1015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bridges JP, Davis HW, Damodarasamy M et al . Pulmonary surfactant proteins A and D are potent endogenous inhibitors of lipid peroxidation and oxidative cellular injury. J Biol Chem 2000; 275: 38848–55. [DOI] [PubMed] [Google Scholar]
- 24. Fukazawa T, Maeda Y, Durbin ML et al . Pulmonary adenocarcinoma‐targeted gene therapy by a cancer‐ and tissue‐specific promoter system. Mol Cancer Ther 2007; 6: 244–52. [DOI] [PubMed] [Google Scholar]
- 25. Mukohara T, Engelman JA, Hanna NH et al . Differential effects of gefitinib and cetuximab on non‐small‐cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst 2005; 97: 1185–94. [DOI] [PubMed] [Google Scholar]
- 26. Kuroki Y, Tsutahara S, Shijubo N et al . Elevated levels of lung surfactant protein A in sera from patients with idiopathic pulmonary fibrosis and pulmonary alveolar proteinosis. Am Rev Respir Dis 1993; 147: 723–9. [DOI] [PubMed] [Google Scholar]
- 27. Madan T, Kishore U, Singh M et al . Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest 2001; 107: 467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakagawa K, Tamura T, Negoro S et al . Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (‘Iressa’, ZD1839) in Japanese patients with solid malignant tumors. Ann Oncol 2003; 14: 922–30. [DOI] [PubMed] [Google Scholar]
- 29. Mendelson CR. Role of transcription factors in fetal lung development and surfactant protein gene expression. Annu Rev Physiol 2000; 62: 875–915. [DOI] [PubMed] [Google Scholar]
- 30. Sundell HW, Gray ME, Serenius FS, Escobedo MB, Stahlman MT. Effects of epidermal growth factor on lung maturation in fetal lambs. Am J Pathol 1980; 100: 707–25. [PMC free article] [PubMed] [Google Scholar]
- 31. Klein JM, DeWild LJ, McCarthy TA. Effect of tyrosine kinase inhibition on surfactant protein A gene expression during human lung development. Am J Physiol 1998; 274 (4 Part 1): L542–51. [DOI] [PubMed] [Google Scholar]
- 32. Klein JM, McCarthy TA, Dagle JM, Snyder JM. Antisense inhibition of epidermal growth factor receptor decreases expression of human surfactant protein A. Am J Respir Cell Mol Biol 2000; 22: 676–84. [DOI] [PubMed] [Google Scholar]
- 33. Sano H, Sohma H, Muta T, Nomura S, Voelker DR, Kuroki Y. Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J Immunol 1999; 163: 387–95. [PubMed] [Google Scholar]
- 34. Borron P, McIntosh JC, Korfhagen TR, Whitsett JA, Taylor J, Wright JR. Surfactant‐associated protein A inhibits LPS‐induced cytokine and nitric oxide production in vivo . Am J Physiol Lung Cell Mol Physiol 2000; 278: L840–7. [DOI] [PubMed] [Google Scholar]
- 35. Murakami S, Iwaki D, Mitsuzawa H et al . Surfactant protein A inhibits peptidoglycan‐induced tumor necrosis factor‐alpha secretion in U937 cells and alveolar macrophages by direct interaction with toll‐like receptor 2. J Biol Chem 2002; 277: 6830–7. [DOI] [PubMed] [Google Scholar]
- 36. Betsuyaku T, Kuroki Y, Nagai K, Nasuhara Y, Nishimura M. Effects of ageing and smoking on SP‐A and SP‐D levels in bronchoalveolar lavage fluid. Eur Respir J 2004; 24: 964–70. [DOI] [PubMed] [Google Scholar]
- 37. McCormack FX, King TE Jr, Voelker DR, Robinson PC, Mason RJ. Idiopathic pulmonary fibrosis. Abnormalities in the bronchoalveolar lavage content of surfactant protein A. Am Rev Respir Dis 1991; 144: 160–6. [DOI] [PubMed] [Google Scholar]
- 38. McCormack FX, King TE Jr, Bucher BL, Nielsen L, Mason RJ. Surfactant protein A predicts survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1995; 152: 751–9. [DOI] [PubMed] [Google Scholar]
- 39. Tamura K, Okamoto I, Kurata T, Satoh T, Nakagawa K, Fukuoka M. Low expressions of surfactant‐associated protein (SP‐A) in cancer tissues or in normal bronchial epithelial cells by immuno‐histochemistry predict interstitial lung diseases induced by gefitinib in patients with advanced non‐small cell lung cancer. Proc Am Soc Clin Oncol 2005; 23: 667s. [Google Scholar]
- 40. Suzuki H, Aoshiba K, Yokohori N, Nagai A. Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosis. Cancer Res 2003; 63: 5054–9. [PubMed] [Google Scholar]
- 41. Madtes DK, Busby HK, Strandjord TP, Clark JG. Expression of transforming growth factor‐alpha and epidermal growth factor receptor is increased following bleomycin‐induced lung injury in rats. Am J Respir Cell Mol Biol 1994; 11: 540–51. [DOI] [PubMed] [Google Scholar]
- 42. Giaccone G, Herbst RS, Manegold C et al . Gefitinib in combination with gemcitabine and cisplatin in advanced non‐small‐cell lung cancer: a phase III trial – INTACT 1. J Clin Oncol 2004; 22: 777–84. [DOI] [PubMed] [Google Scholar]
- 43. Herbst RS, Giaccone G, Schiller JH et al . Gefitinib in combination with paclitaxel and carboplatin in advanced non‐small‐cell lung cancer: a phase III trial – INTACT 2. J Clin Oncol 2004; 22: 785–94. [DOI] [PubMed] [Google Scholar]