

Abstract
Werner syndrome (WS) is an autosomal recessive genetic disorder causing premature aging, and WRN has been identified as the causative gene of WS. The product of the WRN gene (WRN) acts as a DNA helicase with exonuclease activity, and data have accumulated showing that the WRN gene strongly participates in carcinogenesis: (1) the normal WRN gene likely participates in the immortalization of B‐lymphoblastoid cell lines through telomeric crisis caused by telomere shortening, (2) a much higher incidence of rare cancers occurs in WS patients than in other kinds of patients, and (3) levels of WRN expressed in virus‐transformed cells and cancer cells are usually markedly up‐regulated and are inversely correlated with the sensitivity of these cells against various genotoxins, including camptothecin. In this paper, we review the events that show a close correlation of the WRN gene and WRN with carcinogenesis and their underlying molecular mechanisms. (Cancer Sci 2008; 99: 843–848)
Werner syndrome (WS) is a rare autosomal recessive disorder causing symptoms of premature aging.( 1 ) In 1996 the Werner syndrome gene (WRN) responsible for WS was identified,( 2 ) and the gene product (WRN) was subsequently shown to act as a DNA helicase with exonuclease activity.( 1 , 3 ) All the mutations of WRN genes thus far found in WS cause an early termination of protein synthesis resulting in defective WRN. The WRN gene consists of 34 exons, and the mRNA codes for WRN helicase, which consists of 1432 amino acids.( 2 ) WRN has several functional domains: N‐terminal exonuclease domains, central helicase domains, and C‐terminal nuclear and nucleolar localization signals.( 3 ) Our previous study on the structure of the promoter region of the WRN gene strongly suggested that WRN expression is under Sp1 control of the transcriptional control system.( 4 )
In addition to premature aging, WS patients with a mutation in the WRN gene have a much higher incidence of rare cancers,( 5 ) than the other kinds of patients, suggesting that the lack of normal function of WRN strongly affects carcinogenesis. An intriguing relationship exists between the sensitivity of virus‐transformed cells and cancer cells against genotoxins and the expression levels of WRN in these cells; that is, cells with higher levels of WRN expression are less sensitive to genotoxins than cells with no expression of WRN or with poor expression of WRN.( 6 , 7 , 8 )
This review does not comprehensively describe the role of WRN. It focuses on describing the role and underlying mechanisms of WRN in carcinogenesis and in drug resistance to tumor cells.
Failure to Immortalize by B‐lymphoblastoid Cell Lines from WS
One key event in carcinogenesis is the immortalization of cancer cells to obtain infinite proliferating capability, in most cases by activation of telomerase to maintain telomere length.( 9 )
Recent studies,( 10 , 11 , 12 , 13 , 14 ) proposed a new in vitro model regarding senescence, immortalization and carcinogenesis of lymphoblastoid cell lines (LCLs) transformed by Epstein–Barr virus (EBV). LCLs have long been believed to be immortal, but we disproved this commonly held view and showed that LCLs are mortal: after repeated replication, they become senescent due to the shortening of telomeres, enter the so‐called ‘telomeric crisis’ or ‘proliferation crisis’, and then most LCLs end their lifespan. Figure 1 summarizes the cumulative data of the lifespan of LCLs from normal individuals, WS patients, and a family with a tendency towards hereditary type 2 diabetes mellitus (DM).( 15 ) These LCLs were continuously cultured for a long time, even up to 8 years in some cases, and their lifespan was determined by recording population doubling levels (PDLs) when they stopped proliferating. Among 50 LCLs from normal individuals, five LCLs (10.0%) were immortalized and the remaining 45 LCLs were mortal. Immortal LCL cells are characterized by markedly up‐regulated telomerase activity, extremely shortened telomeres (usually shorter than 5 Kbp), abnormal karyotypes, and unlimited cell proliferation.( 12 , 15 ) Because all cells in one immortal LCL shared at least partly abnormal karyotypes, they were assumed to be derived from one cell (one cell in every 108 cells). After prolonged culture of these immortal LCLs, some of them were tumorigenic in nude mice.( 16 ) None of 44 LCLs (0%; P < 0.031 against normal individuals by the χ2‐test) from WS patients were immortalized. Among 11 LCLs from the hereditary DM family, five LCLs (45.5%; P < 0.0040 against normal individuals; P < 0.00001 against WS patients) were immortalized. Why LCLs from the hereditary DM family showed a significantly higher incidence of immortalization remains to be clarified. Interestingly, there have been few clinical case reports of B‐cell lymphoma, an in vitro counterpart of LCL cells, for WS patients,( 5 , 17 ) suggesting an intimate correlation with the in vitro results of LCLs from WS patients that show failure of immortalization.
Figure 1.
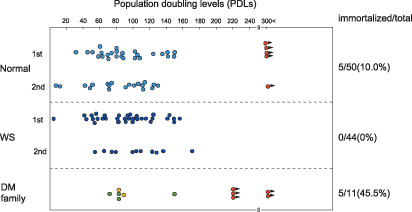
Lifespan expressed PDLs or LCL from normal individuals (Normal), Werner syndrome patients, and a DM family. Circles without an arrow indicate the PDL when the LCLs ended their lifespan. Red circles with an arrow indicate a postimmortal LCL that continues proliferation. Normal and WS individuals show the PDLs at the first and second measurements, and the DM family has two orange circles that indicate repeatedly measured PDLs of the same mortal LCL (green circles). From Sugimoto et al.( 15 )
Figure 2 schematically depicts the hypothetical process of immortalization of LCLs from non‐WS individuals,( 11 ) and failure of immortalization of LCLs from WS individuals. In non‐WS individuals, LCL cells senesce during repeated cell proliferation accompanied by shortening of telomeres, and then finally enter the telomeric crisis, during which chromosomes become extremely unstable and most cells die. Ironically, a few cells have an opportunity to rearrange chromosomes so that they obtain a new ability to immortalize, for instance, through the up‐regulation of telomerase. The schematic process of immortalization of LCLs (Fig. 2) agrees well with the telomere theory of carcinogenesis proposed initially by Harley et al.( 9 ) and later modified by Artandi and DePinho.( 18 )
Figure 2.
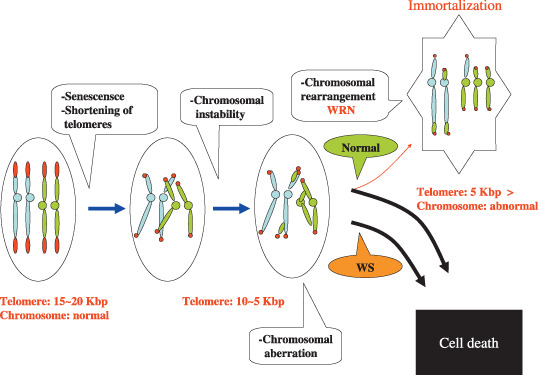
Scheme to show the assumed rearrangement of unstable chromosomes at the telomere crisis of cells for immortalization of LCL cells. During prolonged culture of LCL cells from non‐WS patients (Normal), telomere gradually shortens and stops cell proliferating at a telomere length of between 5 and 10 Kbp, culminating in cell death due to cellular senescence accompanied by chromosomal instability (telomeric crisis). However, rare cells overcome this telomeric crisis by means of chromosomal rearrangement to become immortalized with a shortened telomere (usually, less than 5 Kbp) which length is maintained by simultaneously increased telomerase activity.( 19 ) WRN product is assumed to participate in the above‐mentioned chromosomal arrangement. However, LCL cells from WS patients with WRN dysfunction show abnormal variation of telomere length during the prolonged cell culture, finally resulting cell death without exception due to the failure of appropriate chromosomal rearrangement. From Sugimoto et al.( 11 )
However, although LCLs from WS follow similar cellular senescence during repeated cell proliferation, they fail to immortalize and all LCL cells of WS die after telomeric crisis. Why do LCLs of WS fail to immortalize? WRN may be essential to maintain normal telomere dynamics,( 19 ) as we discuss later in detail. WRN also participates in suppression of hyper‐recombination of genes, as shown in yeast: partial complementation of the hyper‐recombination phenotype in yeast Sgs 1 mutant supports the role of WRN as a suppressor of recombination.( 20 ) Thus we argue that WRN has a role in properly rearranging chromosomes so that cells can obtain new functions essential for immortalization and maintain stably the chromosomes newly rearranged at telomeric crisis. This view is consistent with every immortalized LCL having a set of abnormal chromosomes that is clonal and unique to each LCL and is maintained throughout their cellular proliferating.
The unique role of telomere crisis accompanied by telomere shortening for carcinogenesis of various cancers (telomere theory) has been emphasized in studies( 18 , 21 ) that noted the additional role of a few oncogenic mutations, and overlapping tumor suppressor mechanisms, including p16 (INK4a)‐Rb and ARF‐p53, for immortalization and carcinogenesis. WRN in LCLs from WS that is essential for immortalization of cells through telomeric crisis supports the idea that WS patients may have a lower incidence of cancers generated through this mechanism resulting in a high incidence of rare nonepithelial cancers, as described in the next section. Indeed, recent evidence reveals that telomere dysfunction caused by telomere shortening is likely a tumor initiation mechanism in sporadic cancers, including breast, colorectal, and hepatocellular carcinomas.( 22 )
High Incidence of Rare Cancers in WS Patients
Soft‐tissue sarcoma (STS) and benign meningioma are associated with WS, as shown by Goto et al.( 5 ) for 124 case reports of neoplasia of WS patients from Japan and 34 case reports from outside Japan between 1939 and August 1995. Noticeably, the ratio of epithelial to nonepithelial cancers was about 1:1 for Japanese and for non‐Japanese instead of the usual 10:1. Goto and Ishikawa( 17 ) published data with additional patients (196 tumors from 160 WS patients). In WS patients, STS, osteosarcoma, myeloid disorders, and benign meningioma were excessive. Japanese patients included a higher number of thyroid cancer cases (27 cases) and melanoma cases (27 cases), including eight intranasal cases and 15 cases of the feet. STS, osteosarcoma, melanoma, and thyroid carcinoma comprised 59% of all cancers in WS patients compared with the expected proportion of 2% in the Osaka population at 25–64 years of age.( 5 ) These events strongly suggest that the defect in the WRN gene stimulates the incidence of rare cancers compared with usual cancers, including STS.
Expression Levels of WRN in Transformed and Cancer cells
WRN is weakly expressed in normal fibroblasts and is barely detectable in resting B‐cells, but is markedly up‐regulated after transformation by SV40 T‐antigen or EBV, respectively.( 23 , 24 ) In tumor cell lines (Fig. 3), WRN is markedly up‐regulated in MOLT4 (T‐cell leukemia), K562 (erythroleukemia), A2182 (fibrosarcoma), and PA‐1 (ovary cancer), and is moderately up‐regulated in A‐172 (glioma), T98G (glioblastoma), U25IMG (glioma), SK‐N‐MC (neuroepithelioma), HTB 82 (rhabdomyosarcoma), HT1080 (fibrosarcoma), YCR (renal cell carcinoma), SW620 (colon cancer), COLO320DM (colon cancer), C33A (cervical carcinoma), HeLa S3 (cervical cancer), and T24 (bladder primary cancer). Exceptionally, the expression level of KMST‐6 (fibroblast transformed by irradiation with Co‐60) is little up‐regulated. Notably, the transformed cell line KMST‐6 is immortalized by a telomerase‐independent mechanism consisting of alternative lengthening of telomeres (ALT), as we describe later.
Figure 3.

Expression of WRN in various human cells and cell lines. TIG‐3, human fibroblast cell line; TIG‐3 + SV40, TIG‐3 transformed with SV40 T antigen. See text for other cancer cell lines. Cell lysates of 60 µg per lane were separated by polyacrylamide gel electrophoresis, and were analyzed by Western blotting using anti‐WRN monoclonal antibodies.( 24 )
Correlation Between Resistance Against Genotoxins and Expression Levels of WRN in Transformed and Tumor Cells
Okada et al.( 8 ) studied the effect of camptothecin (CPT), the DNA topoisomerase I‐trapping agent, on 14 LCLs from non‐WS individuals and on 13 LCLs from WS patients. They found that CPT was significantly more toxic to LCLs from WS individuals lacking normal WRN than LCLs from non‐WS individuals (P < 0.05) (Fig. 4a). The expression levels of WRN in B‐cells are up‐regulated during transformation and immortalization: WRN increases markedly by transformation from resting B‐cells to LCLs, and then is further moderately increased by immortalization of LCLs.( 23 , 24 ) Interestingly, postimmortal LCLs expressing a higher level of WRN are more resistant to CPT than preimmortal LCLs expressing a lower level of WRN (Fig. 4b).( 25 ) These results support the view that levels of WRN are correlated with the resistance of cells to CPT.
Figure 4.
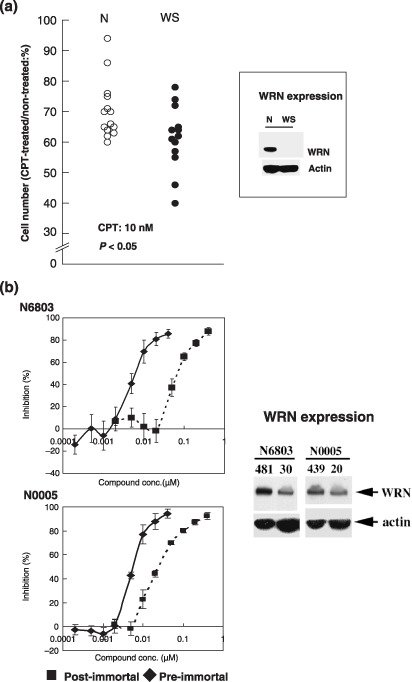
(a) Effect of CPT on the relative proliferation of LCL cells from WS patients and non‐WS individuals (N), or (b) of preimmortal N6803 and N0005 LCL cells from non‐WS patients and corresponding postimmortal N6803 and N0005 LCL cells. Numbers 481 and 30 under N6903 and numbers 439 and 20 under N0005 indicate PDLs. Cells with 481 and 439 PDLs were postimmortal and cells with 30 and 20 PDLs were preimmortal. Cell proliferation was assayed by using the monotetrazolium method. (a) Based on data in Okada et al.( 8 ) and (b) on data in Sawada et al.( 25 )
Our original observation( 8 ) of the effect of CPT on WS LCL cells was confirmed and extended by various studies. Poot et al.( 26 ) presented data indicating that CPT stimulates death of WS cells through apoptosis. The above results for human LCLs were also supported by a study by Lebel and Leder,( 27 ) who deleted a segment of the WRN gene and created WRN‐deficient (WS) mouse embryonic stem cells. Although several DNA repair systems were apparently intact in homozygous WS embryonic stem cells, such cells were markedly more sensitive to CPT than were wild‐type embryonic stem cells.
These results for human and mouse cells strongly support the idea that CPT increases toxicity in WRN‐deficient cells due to failure in the WRN‐mediated repair of DNA damage caused by CPT. LCLs from WS patients are also hypersensitive to the DNA topoisomerase II inhibitor VP16.( 28 )
The above results at cellular levels are consistent with the following clinical results to treat colorectal cancer patients by the CPT analog irinotecan. The WRN function is eliminated in human cancer cells by transcriptional silencing associated with CpG island‐promoter hypermethylation.( 6 ) Importantly, WRN gene hypermethylation in colorectal cancers predicts a good clinical response to the CPT analog irinotecan commonly used in the clinical setting to treat this tumor type. These findings highlight the importance of WRN epigenetic inactivation in human cancer, leading to hypersensitivity to chemotherapeutic drugs.
We investigated the in vitro effect of WRN‐siRNA‐induced gene‐silencing that specifically down‐regulates WRN expression:( 29 ) WRN silencing increased markedly the chemotherapeutic activity of CPT on cancer cells in terms of extent of efficacy and lowering effective drug dosage. WRN protein localizes mainly in nucleoli, but some of it localizes in the nucleoplasm.( 24 , 30 ) When cells are treated with CPT, WRN protein moves from the nucleoli to form distinct nuclear foci (Fig. 5a–c).( 31 ) After further culturing CTP‐treated cells in the absence of CPT, WRN protein returns to the nucleoli and most foci disappear (Fig. 5d). However, in WRN‐silenced cells this recovery process is retarded (Fig. 5e): CPT‐treated HeLa cells maintain many and markedly large replication protein A (RPA) foci in the nucleoplasm even after removal of CPT. These WRN foci overlap with foci of RPA almost entirely, implicating cooperative functions by the WRN protein and RPA in response to DNA damage. These results suggest that the WRN protein and RPA function together in vivo in a pathway of DNA metabolism, such as replication, recombination, or repair. WRN interacts directly with RPA by coimmunoprecipitation of the purified proteins.( 32 ) RPA increases WRN helicase activity by adhering to single‐strand DNAs and thereby preventing their reannealing. From this finding, we proposed a potential combination therapy of WRN‐siRNA‐induced silencing and CPT in anticipation of minimizing the inevitable adverse effects associated with CPT administration.
Figure 5.
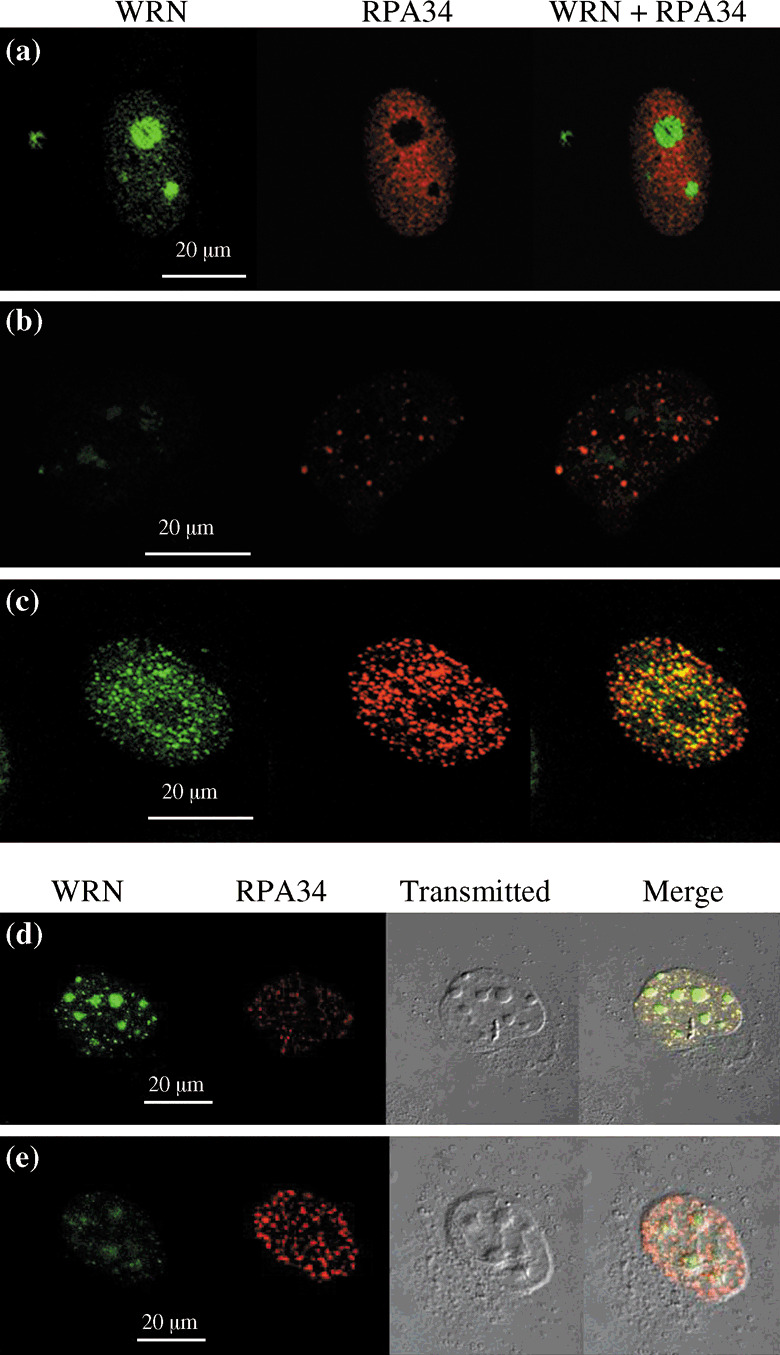
Induction and recovery of DNA damage by CPT in HeLa cells assessed by immunofluorescene. (a) Most WRN was in the nucleolus and replication protein A was dispersed in the nucleoplasm in the absence of CPT. (b) Down‐regulation of WRN by WRN‐siRNA caused some replication protein A to form nuclear foci in the nucleoplasm. (c) DNA damage was induced by adding 10 nM of CPT during 6 h in HeLa cells. The nuclear foci at DNA‐damaged sites stained with anti‐WRN and anti‐replication protein A antibodies were obvious and were colocalized. (d,e) At 30 h after transfection with WRN‐siRNA (e) or NS‐siRNA (d), cells were cultured in the presence of 10 nM of CPT for 6 h. Then, the cells were further cultured for 3 h in the absence of CPT to recover DNA damage. From Futami et al.( 29 )
Genotoxin 4‐nitroquinoline‐1‐oxide (4NQO) induces a more distinct increase in break and interchange aberrations of chromosomes in mitogen‐stimulated peripheral T‐lymphocytes from WS patients than in control lymphocytes or those from patients with other diseases.( 7 )
Consideration of the Role of WRN Function at Molecular Levels
Chromosomal stability. As described thus far, WS is associated with a high incidence of nonepithelial tumors, including STS, with fewer epithelial tumors (carcinomas); whether the net incidence of carcinoma in WS is lower than in non‐WS is unknown. Classical cytogenetic studies of WS skin fibroblasts and LCL cells provided evidence that WS accompanies genomic instability, called ‘variegated translocation mosaicism’, including many different translocations, as well as inversions and deletions of chromosomes.( 33 ) This chromosomal instability caused by the lack of normal WRN is implicated as a participant in the high incidence of nonepithelial tumors in WS. Then, why is the incidence of epithelial tumors low compared with nonepithelial tumors in WS? In this regard, we argue the possibility that the route of carcinogenesis by means of telomere crisis is suppressed in WS by the lack of normal WRN function (see also Fig. 2), as discussed above.
The following functions of WRN are considered to be associated with chromosomal stability: (1) suppressing hyper‐recombination of genes as shown in yeast Sgs 1 mutant,( 20 ) (2) maintaining normal telomere dynamics, and (3) repairing DNA. The ability of WRN to repair DNA is also intimately associated with the resistance of cells expressing high levels of WRN against genotoxin as well as with the high sensitivity of cells lacking normal WRN against genotoxins (Fig. 4).
Telomere maintenance. The following evidence supports the idea that WRN contributes to the maintenance of telomeres. Statistical evidence indicated an accelerated shortening of the length of telomere restriction fragments (TRF) in serially passaged fibroblast cultures from WS patients, but the mean TRF length of WS cultures that had stopped replicating was significantly longer than for senescent controls.( 34 ) Monitoring of the telomere length of both normal and WS LCL cells during a long culture period over 160 PDLs suggested that WRN‐gene mutation causes abnormal dynamics of the telomere: (1) a markedly higher proportion of LCLs from WS showed marked shortening or lengthening of TRF length during cell passages compared with normal cell lines, and (2) LCLs from WS ended their lifespan at a wide TRF length range (between 3.5 and 18.5 Kbp), but normal LCLs ended their lifespan within a narrow TRF length range (between 5.5 and 9 Kbp).( 19 )
In 1999 Griffith et al.( 35 ) clarified the end structure of telomeres. Mammalian telomeres contain a duplex array of telomeric repeats bound to the telomeric repeat‐binding factors TRF1 and TRF2. Inhibition of TRF2 results in immediate deprotection of chromosome ends, shown by loss of the telomeric‐3′ overhang. Electron microscopy showed that TRF2 can remodel linear telomeric DNA into large duplex loops (t‐loops) in vitro. Binding of TRF1 and single‐strand binding protein suggests that t‐loops are formed by invasion of the 3′ telomeric overhang into the duplex telomeric repeat array. Greider( 36 ) speculated that with t‐loops, the three‐stranded DNA displacement loop (D‐loop) structure must be unwound, perhaps by a specific helicase, to allow access by telomerase. WRN associates with telomeres when dissociation of telomeric D‐loops is likely during replication and recombination. These events support the idea that WRN participates in telomere dynamics by its function to unwind t‐loops.
A possible relationship may also be between an alternative immortalization mechanism and tumors in WS. Immortalization of human cells is associated with acquisition of a telomere maintenance mechanism that is usually dependent on the expression of the enzyme telomerase. However, about one‐third of in vitro immortalized human cell lines have no detectable telomerase, but contain telomeres that are abnormally long. Bryan et al.( 37 ) showed an ALT in these telomerase‐negative cells. They found that two ALT cell lines have no detectable expression of the human telomerase RNA (hTR) gene, showing that the ALT mechanism in these cell lines is not dependent on hTR. Notably, tumor cells immortalized by means of the ALT mechanism include many nonepithelial tumors, that is, fibroblast‐derived tumros.( 38 ) In this context, whether nonepithelial tumors in WS patients, including STS, are immortalized by up‐regulation of telomerase activity or by ALT is an intriguing question to be answered.
DNA repair. As described above, the inversed correlation between the expression levels of WRN and sensitivity against genotoxins both in vitro (Fig. 4) and in vivo, ( 6 ) strongly suggests that WRN participates in DNA repair. In fact, the helicase activity and exonuclease activity of WRN are implicated to participate in DNA repair.( 3 ) Therefore, many tumor cell lines expressing high levels of WRN (Fig. 3) are assumed to be less sensitive (that is, more resistant) to genotoxins than cells with lower levels of WRN or without normal WRN.
Interaction of WRN with DNA‐PKcs and the KU70‐80 complex may participate in double‐strand break DNA repair through the non‐homologous end‐joining pathway.( 39 , 40 ) WRN is modified by phosphorylation( 41 ) and acetylation.( 42 ) WRN is also reported to interact with human 5′flap endonuclease/5′‐3′ exonuclease,( 43 ) and tyrosine kinase.( 44 ) The interaction of WRN with several components of the DNA replication fork, including proliferating cell nuclear antigen,( 45 ) and the DNA polymerase delta,( 46 ) is suggested. This evidence together supports the idea that WRN participates in DNA transaction during replication and consequently contributes to the stability of chromosomes.
In context of the roles of WRN in DNA repair, Weinstein( 47 ) proposed the intriguing idea that cancer cells are often ‘addicted to’ (that is, physiologically dependent on) the continued activity of specific activated or over‐expressed oncogenes to maintain their malignant phenotype; these kind of genes and gene products have been studied as ideal chemotherapeutic targets as anticancer agents with few adverse effects.( 48 ) WRN itself has never been assumed to be an oncogene‐related product, but assuming that cancer cells may also be addicted to greater copy numbers of DNA repair enzyme, such as WRN, in addition to the above‐mentioned oncogenes, is possible.
Conclusion
We propose apparently inconsistent dual mechanisms of WRN participating in carcinogenesis. Chromosomal instability caused by WRN‐deficiency in WS patients is probably the major reason for a much higher incidence of rare nonepithelial tumors, including STS. We propose also the novel hypothesis that the failure of epithelial carcinoma through telomeric crisis may be an additional reason for dominance of rare nonepithelial tumors in WS patients. An abundant expression of WRN in cancer cells is implicated to participate in sensitivity to genotoxins by means of augmented DNA repair function: tumor cells expressing high levels of WRN are assumed to be more resistant to genotoxins. In this regard, we propose a potential combination cancer therapy that uses WRN‐siRNA causing silencing of WRN and CPT that aims to minimize inevitable adverse effects associated with CPT administration.
References
- 1. Goto M, Miller RW, eds. From Premature Gray Hair to Helicase – Werner Syndrome: Implications for Aging and Cancer. Tokyo: Scientific Societies Press & Karger, 2001. [Google Scholar]
- 2. Yu CE, Oshima J, Fu YH et al . Positional cloning of the Werner's syndrome gene. Science 1996; 272 (5259): 258–62. [DOI] [PubMed] [Google Scholar]
- 3. Shimamoto A, Sugimoto M, Furuichi Y. Molecular biology of Werner syndrome. Int J Clin Oncol 2004 Aug; 9 (4): 288–98. [DOI] [PubMed] [Google Scholar]
- 4. Yamabe Y, Shimamoto A, Goto M, Yokota J, Sugawara M, Furuichi Y. Sp1‐mediated transcription of the Werner helicase gene is modulated by Rb and p53. Mol Cell Biol 1998; 18 (11): 6191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goto M, Miller RW, Ishikawa Y, Sugano H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 1996; 5 (4): 239–46. [PubMed] [Google Scholar]
- 6. Agrelo R, Cheng WH, Setien F et al . Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci USA 2006. (Jun 6); 103 (23): 8822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gebhart E, Bauer R, Raub U, Schinzel M, Ruprecht KW, Jonas JB. Spontaneous and induced chromosomal instability in Werner syndrome. Hum Genet 1988; 80 (2): 135–9. [DOI] [PubMed] [Google Scholar]
- 8. Okada M, Goto M, Furuichi Y, Sugimoto M. Differential effects of cytotoxic drugs on mortal and immortalized B‐lymphoblastoid cell lines from normal and Werner's syndrome patients. Biol Pharm Bull 1998; 21 (3): 235–9. [DOI] [PubMed] [Google Scholar]
- 9. Harley CB, Kim NW, Prowse KR et al . Telomerase, cell immortality, and cancer. Cold Spring Harb Symp Quant Biol 1994; 59: 307–15. [DOI] [PubMed] [Google Scholar]
- 10. Counter CM, Botelho FM, Wang P, Harley CB, Bacchetti S. Stabilization of short telomeres and telomerase activity accompany immortalization of Epstein‐Barr virus‐transformed human B lymphocytes. J Virol 1994; 68 (5): 3410–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugimoto M, Furuichi Y, Goto M, Tahara H, Ide T. Senecence, immortalization and tumorigenesis of B‐lymphoblastoid cell lines established by transformation with Epstein‐Barr virus: disproof of the commonly‐held view. In: Umar CS, ed. New Developments in Epstein‐Barr Virus Research. New York: Nova Science publishers, Inc., 2006. [Google Scholar]
- 12. Sugimoto M, Tahara H, Ide T, Furuichi Y. Steps involved in immortalization and tumorigenesis in human B‐lymphoblastoid cell lines transformed by Epstein‐Barr virus. Cancer Res 2004. (May 15); 64 (10): 3361–4. [DOI] [PubMed] [Google Scholar]
- 13. Sugimoto M, Ide T, Goto M, Furuichi Y. Reconsideration of senescence, immortalization and telomere maintenance of Epstein‐Barr virus‐transformed human B‐lymphoblastoid cell lines. Mech Ageing Dev 1999; 107 (1): 51–60. [DOI] [PubMed] [Google Scholar]
- 14. Sugimoto M, Ide T, Goto M, Furuichi Y. Incorrect use of ‘immortalization’ for B‐lymphoblastoid cell lines transformed by Epstein‐Barr virus. J Virol 1999; 73: 9690–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sugimoto M, Tahara H, Okubo M et al . WRN gene and other genetic factors affecting immortalization of human B‐lymphoblastoid cell lines transformed by Epstein‐Barr virus. Cancer Genet Cytogenet 2004. (Jul 15); 152 (2): 95–100. [DOI] [PubMed] [Google Scholar]
- 16. Takahashi T, Kawabe T, Okazaki Y et al . In vitro establishment of tumorigenic human B‐lymphoblastoid cell lines transformed by Epstein‐Barr virus. DNA Cell Biol 2003 Nov; 22 (11): 727–35. [DOI] [PubMed] [Google Scholar]
- 17. Goto M, Ishikawa Y. [Werner syndrome]. Nippon Rinsho 2000 Jul; 58 (7): 1490–5 (In Japanese.) [PubMed] [Google Scholar]
- 18. Artandi SE, DePinho RA. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr Opin Genet Dev 2000 Feb; 10 (1): 39–46. [DOI] [PubMed] [Google Scholar]
- 19. Tahara H, Tokutake Y, Maeda S et al . Abnormal telomere dynamics of B‐lymphoblastoid cell strains from Werner's syndrome patients transformed by Epstein‐Barr virus. Oncogene 1997; 15 (16): 1911–20. [DOI] [PubMed] [Google Scholar]
- 20. Yamagata K, Kato J, Shimamoto A, Goto M, Furuichi Y, Ikeda H. Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc Natl Acad Sci USA 1998; 95 (15): 8733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest 2004 Jan; 113 (2): 160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greenberg RA. Telomeres, crisis and cancer. Curr Mol Med 2005 Mar; 5 (2): 213–18. [DOI] [PubMed] [Google Scholar]
- 23. Kawabe T, Tsuyama N, Kitao S et al . Differential regulation of human RecQ family helicases in cell transformation and cell cycle [In Process Citation]. Oncogene 2000; 19 (41): 4764–72. [DOI] [PubMed] [Google Scholar]
- 24. Shiratori M, Sakamoto S, Suzuki N et al . Detection by epitope‐defined monoclonal antibodies of Werner DNA helicases in the nucleoplasm and their upregulation by cell transformation and immortalization. J Cell Biol 1999; 144 (1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sawada K, Noda K, Nakajima H, Shimbara N, Furuichi Y, Sugimoto M. Differential cytotoxicity of anticancer agents in pre‐ and post‐immortal lymphoblastoid cell lines. Biol Pharm Bull 2005 July; 28 (7): 1202–7. [DOI] [PubMed] [Google Scholar]
- 26. Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin‐induced apoptosis in S‐phase. Hum Genet 1999 Jan; 104 (1): 10–14. [DOI] [PubMed] [Google Scholar]
- 27. Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci USA 1998; 95 (22): 13 097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elli R, Chessa L, Antonelli A, Petrinelli P, Ambra R, Marcucci L. Effects of topoisomerase II inhibition in lymphoblasts from patients with progeroid and ‘chromosome instability’ syndromes. Cancer Genet Cytogenet 1996 Apr; 87 (2): 112–16. [DOI] [PubMed] [Google Scholar]
- 29. Futami K, Takagi M, Shimamoto A, Sugimoto M, Furuichi Y. Increased chemotherapeutic activity of camptothecin in cancer cells by siRNA‐induced silencing of WRN helicase. Biol Pharm Bull 2007; 30 (10): 1958–61. [DOI] [PubMed] [Google Scholar]
- 30. Marciniak RA, Lombard DB, Johnson FB, Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proc Natl Acad Sci USA 1998; 95 (12): 6887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sakamoto S, Nishikawa K, Heo SJ, Goto M, Furuichi Y, Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co‐localizes with RPA and Rad51. Genes Cells 2001; 6 (5): 421–30. [DOI] [PubMed] [Google Scholar]
- 32. Brosh RM Jr, Orren DK, Nehlin JO, Ravn PH, Kenny MK, Machwe A et al . Functional and physical interaction between WRN helicase and human replication protein A. J Biol Chem 1999. (Jun 25); 274 (26): 18341–50. [DOI] [PubMed] [Google Scholar]
- 33. Martin GM. A brief history of research on the Werner syndrome. In: Goto M, Miller RW, eds. From Premature Gray Hair to Helicase – Werner Syndrome: Implications for Aging and Cancer. Tokyo: Science Societies Press & Kagrer, 2001. [Google Scholar]
- 34. Schulz VP, Zakian VA, Ogburn CE et al . Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum Genet 1996; 97 (6): 750–4. [DOI] [PubMed] [Google Scholar]
- 35. Griffith JD, Comeau L, Rosenfield S et al . Mammalian telomeres end in a large duplex loop. Cell 1999; 97 (4): 503–14. [DOI] [PubMed] [Google Scholar]
- 36. Greider CW. Telomeres do d‐loop‐T‐loop [comment]. Cell 1999; 97 (4): 419–22. [DOI] [PubMed] [Google Scholar]
- 37. Bryan TM, Marusic L, Bacchetti S, Namba M, Reddel RR. The telomere lengthening mechanism in telomerase‐negative immortal human cells does not involve the telomerase RNA subunit. Hum Mol Genet 1997; 6 (6): 921–6. [DOI] [PubMed] [Google Scholar]
- 38. Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. Telomere elongation in immortal human cells without detectable telomerase activity. Embo J 1995; 14 (17): 4240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li B, Comai L. Functional interaction between Ku and the werner syndrome protein in DNA end processing. J Biol Chem 2000; 275 (37): 28 349–52. [DOI] [PubMed] [Google Scholar]
- 40. Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev 2000; 14 (8): 907–12. [PMC free article] [PubMed] [Google Scholar]
- 41. Pichierri P, Rosselli F, Franchitto A. Werner's syndrome protein is phosphorylated in an ATR/ATM‐dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene 2003. (Mar 13); 22 (10): 1491–500. [DOI] [PubMed] [Google Scholar]
- 42. Blander G, Zalle N, Daniely Y, Taplick J, Gray MD, Oren M. DNA damage‐induced translocation of the Werner helicase is regulated by acetylation. J Biol Chem 2002. (Dec 27); 277 (52): 50 934–40. [DOI] [PubMed] [Google Scholar]
- 43. Brosh RM Jr, Von Kobbe C, Sommers JA et al . Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. Embo J 2001. (Oct 15); 20 (20): 5791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng WH, Von Kobbe C, Opresko PL et al . Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol Cell Biol 2003 Sept; 23 (18): 6385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lebel M, Spillare EA, Harris CC, Leder P. The Werner syndrome gene product co‐purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J Biol Chem 1999. (Dec 31); 274 (53): 37 795–9. [DOI] [PubMed] [Google Scholar]
- 46. Kamath‐Loeb AS, Loeb LA, Johansson E, Burgers PM, Fry M. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d (CGG) n trinucleotide repeat sequence. J Biol Chem 2001. (May 11); 276 (19): 16 439–46. [DOI] [PubMed] [Google Scholar]
- 47. Weinstein IB. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science 2002. (Jul 5); 297 (5578): 63–4. [DOI] [PubMed] [Google Scholar]
- 48. Sharma SV, Gajowniczek P, Way IP et al . A common signaling cascade may underlie ‘addiction’ to the Src, BCR‐ABL, and EGF receptor oncogenes. Cancer Cell 2006 Nov; 10 (5): 425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]