

Abstract
To isolate novel diagnostic markers and drug targets for pancreatic ductal adenocarcinoma (PDAC), we previously performed expression profile analysis of PDAC cells using a genome‐wide cDNA microarray combined with laser microdissection. Among dozens of up‐regulated genes identified in PDAC cells, we herein focused on one tyrosine kinase receptor, Eph receptor A4 (EphA4), as a molecular target for PDAC therapy. Immunohistochemical analysis validated EphA4 overexpression in approximately half of the PDAC tissues. To investigate its biological function in PDAC cells, we knocked down EphA4 expression by siRNA, which drastically attenuated PDAC cell viability. In concordance with the siRNA experiment, PDAC‐derivative cells that were designed to constitutively express exogenous EphA4 showed a more rapid growth rate than cells transfected with mock vector, suggesting a growth‐promoting effect of EphA4 on PDAC cells. Furthermore, the expression analysis for ephrin ligand family members indicated the coexistence of ephrinA3 ligand in PDAC cells with EphA4 receptor, and knockdown of ephrinA3 by siRNA also attenuated PDAC cell viability. These results suggest that the EphA4–ephrinA3 pathway is likely to be a promising molecular target for pancreatic cancer therapy. (Cancer Sci 2006; 97: 1211–1216)
Pancreatic ductal adenocarcinoma (PDAC) is the fourth lleading cause of cancer death in the developed world and has one of the worst mortality rates among the common malignancies, with a 5‐year survival rate of only 4%.( 1 , 2 ) Approximately 33 730 new patients are expected to be diagnosed with pancreatic cancer in 2006 in the USA, and nearly 32 300 to die of the disease;( 3 ) in Japan nearly 18 000 PDAC patients are expected to die each year. The great majority of PDAC patients are diagnosed at an advanced stage where no effective therapy is available. Surgical resection offers the only possibility for cure at present, but 80–90% of patients who undergo curative surgery suffer from relapse and die due to the metastatic or disseminated disease.( 1 , 2 ) Some approaches that combine surgery and chemotherapy based on gemcitabine or 5‐fluorouracil, with or without radiation therapy, can improve the quality of life of patients.( 1 , 2 ) However, such treatments have a very limited effect on long‐term survival because PDAC is biologically extremely aggressive, and usually chemo‐ and radiation‐resistant. Hence, the management of most PDAC patients is now focused on palliative measures.
To overcome this dismal situation, development of novel molecular‐targeted therapies for PDAC through identification of cancer‐specific drug‐amenable molecules is urgently desired. We previously performed detailed and accurate expression profile analysis of PDAC using a genome‐wide cDNA microarray consisting of approximately 27 000 genes, in combination with laser microdissection to purify the cancer cell population.( 4 ) Among the genes we identified as being trans‐activated in PDAC cells, we herein investigated one of the Eph receptors, EphA4, as a possible molecular target for this disease.
The Eph receptor family constitutes one of the largest groups of transmembrane receptor tyrosine kinases.( 5 ) They are activated by a second family of cell surface‐anchored ligands, the ephrins, that are attached to the plasma membrane via either a glycosylphosphatidylinositol (GPI) linkage (type A) or a transmembrane sequence (type B). The Eph receptors are also divided into type A or type B according to their ligand‐binding specificities. In general, type A receptors bind type A ephrin ligands and type B ephrin ligands stimulate type B receptors. One molecule that shows an exception to this rule is EphA4, which can bind and respond to type B as well as type A ephrin ligands.( 6 ) These Eph receptors and their ligands have been implicated in playing important roles in a variety of biological activities including axon guidance and migration of neural crest cells in the nervous system, establishment of segmental boundaries, and formation of angiogenic capillary plexi.( 7 , 8 , 9 , 10 , 11 ) Among the Eph receptor family members, EphA2 and EphB2 were shown to be frequently overexpressed or functionally altered in many types of cancers, and to be involved in tumor progression or angiogenesis.( 12 , 13 , 14 , 15 , 16 , 17 , 18 )
The aim of this study was to determine the role of EphA4 in pancreatic carcinogenesis. We identified a possible candidate for a ligand to the EphA4 receptor, ephrinA3, which coexisted in PDAC cells with EphA4, and our second aim was to determine the effects of EphA4 and ephrinA3 on the viability of PDAC cells.
Materials and Methods
Cell lines. Pancreatic ductal adenocarcinoma cell lines MIA‐PaCa2 and Panc‐1 were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA), and were grown in Delbecco's modified Eagle's medium or RPMI1640 (Sigma‐Aldrich, St Louis, MO, USA). PK‐59, KLM‐1, PK‐45P, and PK‐1 were provided by the Cell Resource Center for Biomedical Research, Tohoku University (Sendai, Japan) and maintained in RPMI1640; both media were supplemented with 10% fetal bovine serum (Cansera, Ontario, Canada) and 1% antibiotic/antimycotic solution (Sigma‐Aldrich). Cells were maintained at 37°C in an atmosphere of humidified air with 5% CO2.
Semi‐quantitative RT‐PCR. Microdissection of PDAC cells and normal pancreatic ductal cells were as described previously.( 4 ) RNAs from the PDAC cells and normal pancreatic ductal cells were subjected to two rounds of T7‐based RNA amplification (Epicentre Technologies, Madison, WI, USA) and subsequent synthesis of single‐strand cDNA. For reverse transcription‐polymerase chain reaction (RT‐PCR), total RNA from human PDAC cell lines was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Extracted RNA was treated with DNase I (Roche, Mannheim, Germany) and reverse‐transcribed to single‐stranded cDNA using oligo (dT) primer with Superscript II reverse transcriptase (Invitrogen). The primer sequences used were 5′‐CATCCACGAAACTACC TTCAACT‐3′ and 5′‐TCTCCTTAGAGAGAAGTGGGGTG‐3′ for β‐actin (ACTB), 5′‐GAAGGCGTGGTCACTAAATGTAA‐3′ and 5′‐TTTAATTTCAGAGGGCGAAGAC‐3′ for EphA4, 5′‐GAGT CCCTTCCCTCTTTAACC‐3′ and 5′‐TATGAAAGTCACAGCCA AAGC‐3′ for ephrinA3. The primer sequences for other ephrin ligands are available on request.
The RT‐PCR exponential phase was determined to allow semiquantitative comparisons among cDNAs developed from identical reactions. Each PCR regime involved a 95°C, 5‐min initial denaturation step followed by 23 cycles (for ACTB), 28 cycles (for EphA4), or 30 cycles (for ephrinA3) at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, on a Gene Amp PCR system 9600 (PE Applied Biosystems, Foster, CA, USA).
Northern blot analysis. We extracted total RNA from several PDAC cell lines using Trizol reagent and performed Northern blot analysis. After treatment with DNase I (Nippon Gene, Osaka, Japan), mRNA was purified with Micro‐FastTrack (Invitrogen), according to the manufacturer's protocols. A 1‐µg aliquot of each mRNA from PDAC cell lines, as well as those isolated from normal human heart, lung, liver, kidney, brain, and pancreas (BD Biosciences, Palo Alto, CA, USA), were separated on 1% denaturing agarose gels and transferred onto nylon membranes. The 1014‐bp probe specific to EphA4 was prepared by PCR using the following primer set: forward 5′‐GAAGGCGTGGTCACTAAA TGTAA‐3′ and reverse 5′‐CTTTAATTTCAGAGGGCGAAGAC‐3′.
Hybridization with a random‐primed, α32P‐dCTP‐labeled probe was carried out according to the instructions for Megaprime DNA labeling system (Amersham Biosciences, Buckinghamshire, UK). Prehybridization, hybridization and washing were performed according to the supplier's recommendations. The blots were auto‐radiographed with intensifying screens at −80°C for 10 days.
Immunohistochemical staining. Conventional sections from PDAC tissues were obtained from surgical specimens that were resected in Osaka Medical Center for Cancer and Cardiovascular Diseases under the appropriate informed consent. Sections from normal pancreas were purchased from Biochain (Hayward, CA, USA). Tissue microarray sections of pancreatic carcinoma (AccuMax Array) were purchased from ISU ABXIS (Seoul, Korea), where 31 PDAC tissues, two endocrine tumor tissues, and two normal pancreas tissues were spotted duplicated.
The sections were deparaffinized and autoclaved at 108°C in Dako Cytomation Target Retrieval Solution High pH (Dako, Carpinteria, CA, USA) for 15 min. After blocking of endogenous peroxidase and proteins, the sections were incubated with anti‐EphA4 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA, diluted by 1:200) or anti‐ephrinA3 antibody (Santa Cruz Biotechnology, diluted by 1:150) at room temperature for 30 min. After washing with PBS, immunodetection was performed with peroxidase labeled antirabbit immunoglobulin (Envision kit, Dako). Finally, the reactants were developed with 3,3′‐diaminobenzidine (Dako). Counterstaining was performed using hematoxylin.
siRNA‐expressing constructs and transfection. To knock down endogenous EphA4 or ephrinA3 expression in PDAC cells, we used psiU6BX3.0 vector for expression of short hairpin RNA against a target gene as described previously.( 19 ) The target sequences of the synthetic oligonucleotides for small interfering RNA (siRNA) for EphA4 were as follows: EphA4‐198si, 5′‐TCCGAACCTACCAA GTGTG‐3′; EphA4‐486si, 5′‐TCATGAAGCTGAACACCGA‐3′; EphA4‐1313si, 5′‐GCAGCACCATCATCCATTG‐3′; and EGFPsi, 5′‐GAAGCAGCACGACTTCTTC‐3′ (as a negative control). The target sequences for siRNA for ephrinA3 were: ephrinA3‐539si, 5′‐GTGTCTGAGGATGAAGGTG‐3′; ephrinA3‐689si, 5′‐GCTTG AGAAGAGCATCAGC‐3′; and ephrinA3‐1584si, 5′‐CGCACAG ACACTTTTGGAG‐3′.
The PDAC cell lines MIA‐PaCa2 and PK‐59, which expressed both EphA4 and ephrinA3, were plated onto six‐well plates, and transfected with plasmid designed to express siRNA to either EphA4 or ephrinA3 (10 µg or 20 µg) using FuGENE6 (Roche) or Lipofectamine reagent (Invitrogen) according to the manufacturer's instructions. Cells were selected by 0.15 mg/mL (for PK‐59) or 0.8 mg/mL (for MIA‐PaCa2) of Geneticin (Sigma‐Aldrich) for 5 days, and then harvested to analyze the knockdown effect on EphA4 or ephrinA3 expression.
RT‐PCR for EphA4 or ephrinA3 knockdown was performed using the primers described above, and Western blot analysis for EphA4 or ephrinA3 knockdown was performed by using the antibodies to EphA4 or ephrinA3 described above and anti‐β‐actin (ACTB) antibody (Sigma‐Aldrich) as a loading control.
For colony formation assay, transfectants expressing siRNA were grown for 14 days in media containing Geneticin. After fixation with 4% paraformaldehyde, transfected cells were stained with Giemsa solution to assess colony formation. Cell viability was quantified using Cell counting kit‐8 (Dojindo, Kumamoto, Japan). After 14 days of culture in the Geneticin‐containing medium, the solution was added at a final concentration of 10%. Following incubation at 37°C for 3 h, absorbance at 450 nm was measured with a Microplate Reader 550 (Bio‐Rad, Hercules, CA, USA).
Establishment of EphA4‐expressing cells and their growth. EphA4 cDNA was prepared by PCR amplification using the forward primer that included the Kozak sequence and EcoRI linker, and the reverse primer including an XhoI linker. The PCR product was inserted into the EcoRI and XhoI sites of the mammalian expression vector, pCAGGS/FLAG for expressing a FLAG‐tagged protein.( 20 ) The pCAGGS‐EphA4‐FLAG or empty pCAGGS/FLAG mock vector was transfected into Panc‐1 cell line, which exhibited barely detectable expression of EphA4 among pancreatic cancer cell lines we examined, by FuGENE6 (Roche) according to the manufacturer's protocol. Then, the Geneticin‐resistant clones were selected in the culture medium containing 1.0 mg/mL of Geneticin.
The exogenous EphA4 expression in each clone was confirmed by Western blot analysis using anti‐FLAG tag and anti‐β‐actin antibodies (Sigma‐Aldrich). For growth assay, 50 000 cells of each of EphA4 expressing clone (Panc1‐EphA4) or control clone (Panc1‐Mock) was seeded into each well of a six‐well culture dish and incubated in the medium containing 10% fetal bovine serum. Cell viability was quantified with MTT assay every day. The experiment was repeated at least three times.
Results
EphA4 overexpressed in PDAC cells. Among the dozens of up‐regulated genes in PDAC identified previously through detailed expression profile analysis,( 4 ) we here focused on further expressional and functional analysis of EphA4. The semiquantitative RT‐PCR analysis using RNA from microdissected PDAC cells and normal pancreatic ductal cells demonstrated that expression of EphA4 was significantly up‐regulated in PDAC cells, compared with that of normal pancreatic ductal cells that were believed to be the origins of PDAC cells (Fig. 1A). Further comparisons of EphA4 expression patterns in PDAC cells and normal tissues by Northern blot analysis revealed apparently strong and specific expression of EphA4 in PDAC cells although its expression was observed in normal brain (Fig. 1B). Two bands (about 6.5 kb and 3 kb) were observed, which are differential splicing patterns of 3′ UTR of human EphA4.
Figure 1.
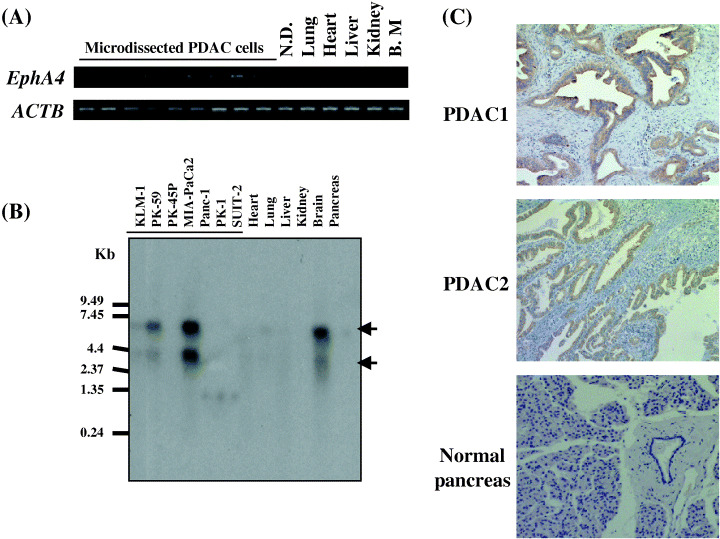
Overexpression of EphA4 in pancreatic ductal adenocarcinoma (PDAC) cells. (A) Semi‐quantitative RT‐PCR validated that EphA4 expression was up‐regulated in the microdissected PDAC cells compared with normal pancreatic duct cell (ND) that were also microdissected and several vital organs (lung, heart, liver, kidney and bone marrow). Expression of ACTB served as the quantitative control. (B) Northern blot analysis showed the strong expression of EphA4 in PDAC cell lines, MIA‐PaCa2 and PK‐59, and the normal brain, while no expression was observed in vital organs including heart, lung, liver, and kidney. (C) Immunohistochemical study using anti‐EphA4 antibody. Intense staining was observed in PDAC cells in two representative specimens (PDAC1 and PDAC2), while acinar cells and normal ductal epithelium in normal pancreatic tissue showed no staining (original magnification × 200).
To confirm overexpression of EphA4 protein in PDAC, we performed immunohistochemical staining using anti‐EphA4 antibody and validated overexpression of EphA4 protein in PDAC cells, but no staining in normal pancreas (Fig. 1C). The immunohistochemical analysis on the tissue microarray combined with conventional tissue sections showed that EphA4 protein was overexpressed in 48% (22/46) of PDAC specimens we examined. There was no significant correlation between EphA4 expression and differentiation of PDAC in our analysis.
Knockdown of EphA4 by siRNA on PDAC cells. To investigate a potential growth‐promoting role of EphA4 aberrant expression, we constructed several siRNA‐expression vectors to examine their knockdown effects on two EphA4‐overexpressing PDAC cell lines, MIA‐PaCa2 and PK‐59. After transfection of each of these siRNA‐expressing constructs into them, semiquantitative RT‐PCR (data not shown) and Western blot revealed that 1313si construct, but no other constructs, significantly knocked down EphA4 expression (Figs 2 A,D). After 14‐day selection in culture medium containing Geneticin, MTT assay (Figs 2B,E) and colony formation assay (Figs 2C,F) demonstrated that introduction of 1313si in MIA‐PaCa2 and PK‐59 cells drastically attenuated their cell growth or viability; however, other siRNAs that could not affect EphA4 expression did not affect cell growth.
Figure 2.
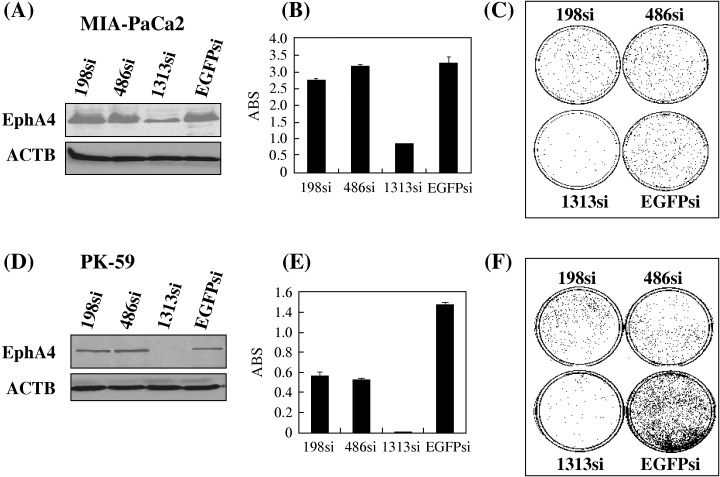
Knockdown effect on EphA4 by siRNA attenuated pancreatic ductal adenocarcinoma (PDAC) cell viability. Three EphA4 siRNA expression vectors (EphA4‐198si, ‐486si, and ‐1313si) and an EGFP siRNA expression vector (EGFPsi) as a negative control were transfected into (A, B, C) MIA‐PaCa2 and (D, E, F) PK‐59 cells. (A and D) Knockdown effect on EphA4 was validated by Western blot analysis, with ACTB level as a quantitative control. EphA4‐1313si revealed a strong knockdown effect, while EphA4‐198si, EphA4‐486si and EGFPsi did not show any effect on the level of EphA4. Transfection with EphA4‐1313si vector resulted in drastic reduction of the numbers of viable cells measured by (B and E) MTT assay and (C and F) the number of colonies formed, compared with the cells transfected with other siRNA expression vectors that did not showed their knockdown effect on EphA4. ABS, absorbance at 490 nm (630 nm reference), measured with a microplate reader.
Growth promoting effect of EphA4 expression on PDAC cells. To further explore the potential oncogenic property of EphA4, we established the Panc1‐derivative cell line Panc1‐EphA4, in which exogenous EphA4 expressed constitutively. We also prepared control Panc‐1 cells transfected with the mock vector (Panc1‐Mock) and compared their growth rates. Western blot analysis (Fig. 3A) validated exogenous EphA4 expression in three Panc1‐derivate clones. The growth curve measured by MTT assay demonstrated that the three Panc1‐EphA4 clones (#9, #13 and #16) grew significantly more rapidly than the two Panc1‐mock clones (#6, and #25; P = 0.0358) (Fig. 3B), indicating the EphA4 expression enhanced proliferation of PDAC cells.
Figure 3.
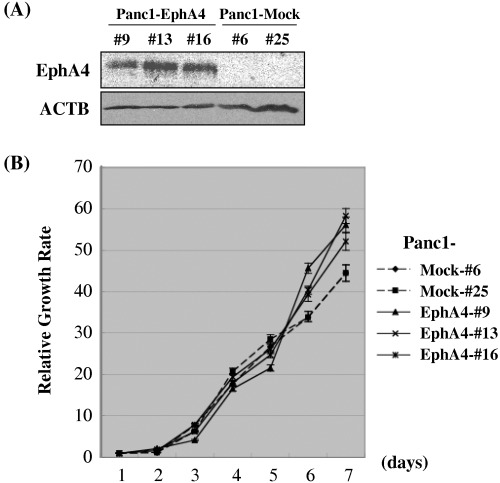
Expression of exogenous EphA4 promoted pancreatic ductal adenocarcinoma (PDAC) cell growth. (A) Western blot analysis of three Panc‐1 derivative cells (Panc1‐EphA4 #9, #13, and #16) expressing exogenous EphA4 constitutively and those transfected with mock vector (Panc1‐Mock #6 and #25). Exogenous introduction of EphA4 expression was validated with anti‐FLAG tag antibody. ACTB served as a loading control. (B) The growth measurement by MTT assay demonstrates that the three Panc1‐EphA4 clones (#9, #13 and #16, solid lines) grew significantly more rapidly than the two Panc1‐mock clones (#6, and #25, dashed lines) (P = 0.0358). X‐axis represents day point after seeding and Y‐axis represents relative growth rate that was calculated in absorbance of the diameter by comparison with the absorbance value of day 1 as a control. Each average is plotted with bars representing standard error. These experiments were performed in triplicate.
Expression pattern of ephrin ligands in PDAC cells. Type A Eph receptors usually bind to type A ligands that are known to be GPI‐anchored to the plasma membrane, and type B Eph receptors bind to type B ligands that have a transmembrane domain. However, there is one exception to this rule; the EphA4 receptor has been shown to bind to both type A and type B ephrin ligands.( 6 ) The ligand‐receptor combinations for the Eph family are considered to occur in a cancer‐type or tissue‐type specific manner.( 16 , 18 )
To search for potential candidate ligands for the EphA4 receptor in PDAC cells, we examined the expression patterns of members in the ephrin A (A1–5) and B (B1–3) ligand families by semiquantitative RT‐PCR using RNA from microdissected PDAC cells, and found that the expression patterns of ephrinA1 and ephrinA3 were concordant with that of EphA4 (Fig. 4A). However, the expression of ephrinA1 was not concordant with that of EphA4 in PDAC cell lines. As better concordance between EphA4 and ephrinA3 expression was observed in PDAC cell lines than between EphA4 and ephrinA1 (Fig. 4B), we considered that ephrinA3 might be a better candidate as a ligand for EphA4. Immunohistochemical staining in 31 PDAC samples spotted on tissue microarray and five conventional sections of PDAC also showed concordant expression of ephrinA3 and EphA4 (Fig. 4C) (P > 0.001 by χ2 test).
Figure 4.
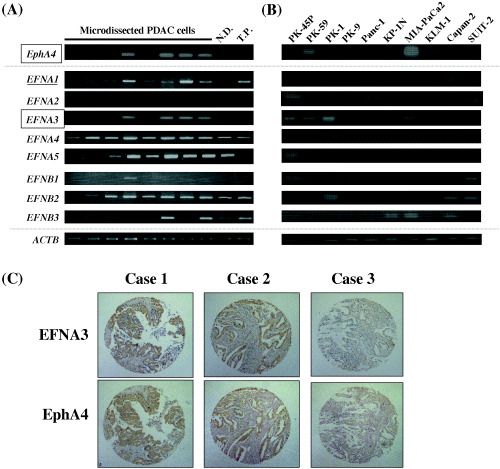
Expression pattern of ephrin ligands in pancreatic ductal adenocarcinoma (PDAC) cells. Semi‐quantitative RT‐PCR in (A) clinical PDAC cells and (B) PDAC cell lines demonstrated that, among ephrin ligands A1‐5 and B1‐3, ephrinA3 (EFNA3) expression pattern was relatively correlated with that of EphA4. ACTB served as a quantitative control. ND, normal pancreatic ductal cells; TP, total pancreas. (C) Immunohistochemical staining in PDAC tissue microarray demonstrated that ephrinA3 (EFNA3) expression (upper panel) was correlated with EphA4 expression (lower panel). Three representative specimens with both EFNA3 and EphA4 positive (Cases 1 and 2) and with both negative (Case 3) are shown (original magnification × 200).
Knockdown of ephrinA3 by siRNA attenuated PDAC cell growth. To investigate possible roles of ephrinA3 in PDAC, we constructed several expression vectors that were designed to express siRNA to ephrinA3 and examined knockdown effects in MIA‐PaCa2 cells, in which both EphA4 and ephrinA3 ligand were expressed at a high level. Semi‐quantitative RT‐PCR (data not shown) and Western blot analysis revealed that 539si and 689si, but no other siRNAs, clearly knocked down ephrinA3 expression (Fig. 5A). After 14‐day culture in the medium containing Geneticin, we observed that introduction of 539si or 689si into MIA‐PaCa2 cells drastically attenuated cell viability measured by colony formation assay (Fig. 5B) and by MTT assay (Fig. 5C), while no growth‐suppressive effect was observed by either 1584si or EGFPsi, which showed no knockdown effect on ephrinA3 expression. Similar results were also obtained when we used PK‐59 cells, in which EphA4 and ephrinA3 were co‐expressed (data not shown). These findings indicate that, in addition to the EphA4 receptor, the ephrinA3 ligand could also play an important role in cell viability of PDAC cells.
Figure 5.
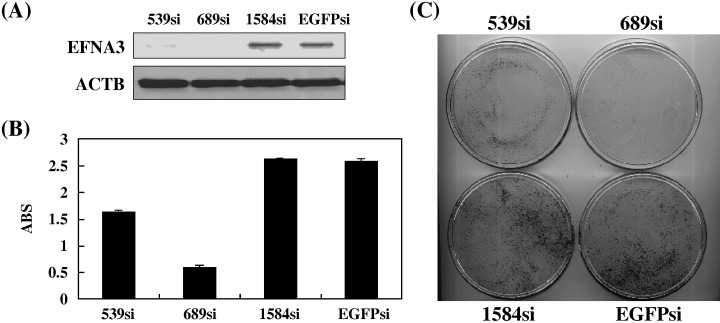
Knockdown effect on ephrinA3 (EFNA3) by siRNA attenuated pancreatic ductal adenocarcinoma (PDAC) cell viability. Three EFNA3 siRNA expression vectors (EFNA3‐539si, ‐689si, and ‐1584si) and an EGFP siRNA expression vector (EGFPsi) as a negative control were transfected into MIA‐PaCa2 cells. (A) Knockdown effect on EFNA3 was validated by Western blot analysis with ACTB level as a quantitative control. EFNA3‐689si and EFNA3‐539si revealed a strong knockdown effect on EFNA3 expression, while EFNA3‐1584si and EGFPsi did not show any effect on EFNA3 expression level. Transfection with EFNA3‐689si and EFNA3‐539si resulted in a drastic reduction of (B) the numbers of viable cells measured by MTT assay and (C) the number of colonies formed, compared with the cells transfected with siRNA expression vectors that did not show a knockdown effect on EFNA3. ABS, absorbance at 490 nm (630 nm reference), measured with a microplate reader.
Discussion
In this study, we found overexpression of EphA4, a member of the Eph receptor kinase family, in PDAC cells, which indicates that molecules involved in the EphA4‐signaling pathway might be very promising molecular targets for the development of anticancer drugs. In general, receptor tyrosine kinases and their ligands play critical roles in the regulation of a variety of cell activities including cellular survival, proliferation, differentiation, and tissue organization.( 21 ) Eph receptors and their ligands, ephrins, are indeed involved in several cell processes during embryonic development, including pattern formation, cell aggregation and migration, segmentation, neural development, angiogenesis, and vascular hierarchical remodeling.( 7 , 8 , 9 , 10 , 11 ) The overexpression of some members of the Eph receptor family in various cancers was found to play an important role in the development and progression of cancer cells. In particular, EphA2 and EphB2 overexpression was found frequently in human invasive cancers including PDAC( 12 , 13 , 14 , 15 , 16 , 17 , 18 ) and they are considered to be good targets for antibody treatment.( 22 , 23 ) In addition to these two members, we here demonstrated a critical role of one member of the Eph receptor family, EphA4, in pancreatic tumorigenesis. We also identified ephrinA3 as a candidate ligand for EphA4 in PDAC cells. In our gene‐expression profile studies for other types of cancer, we found that EphA4 was also overexpressed in a subset of prostate cancer and soft tissue sarcomas.( 24 , 25 ) Hence, we suspect it has a carcinogenic role in a wide range of malignancies like other Eph family members.
A unique feature of the signaling of the Eph receptors and ephrin ligands is its bi‐directionality.( 26 , 27 ) In contrast to other receptor tyrosine kinases, Eph receptors are activated by interaction with membrane‐attached ephrin ligands; interaction between cells expressing Eph receptors and those expressing ephrin ligand is required for activation of the Eph receptor signaling pathway. In addition, ephrin ligands on plasma membrane can also be activated and lead to separate signaling into ephrin‐expressing cells. In this case, the ephrin signaling in ephrin‐expressing cells is activated by the phosphorylation of tyrosine residing in the cytoplasmic tail of ephrins B or by ephrin A interaction with integrins or other membrane proteins.( 27 , 28 ) Our expression data by RT‐PCR and immunohistochemical analysis indicated that ephrinA3 ligand co‐expressed with EphA4 in PDAC cells, and that ephrinA3 is likely to be a possible candidate for the ligand of EphA4 receptor in PDAC cells. Furthermore, experiments using siRNA to EphA4 and ephrinA3 have indicated that both EphA4 and ephrinA3 could play essential roles in PDAC cell viability, and EphA4 introduction into PDAC cells lacking in EphA4 expression resulted in growth promotion. These findings suggest that interactions between the EphA4 receptor and ephrinA3 in PDAC cells function as growth‐promoting factors for PDAC cells in an autocrine/paracrine manner and probably through the EphA4–ephrinA3 bi‐directional signaling in PDAC cells. Indeed EphA4 has been studied for its association in axon guidance or repulsion and regeneration in the central nervous system,( 27 , 29 ) and the interaction between EphA4 and ephrinA3 was implicated to be essential in spinal morphology and synaptic connections at neural development.( 30 ) Our results are the first evidence indicating that this specific interaction between EphA4 and ephrinA3 might play a critical role in promoting cancer cell proliferation in PDAC carcinogenesis or development.
We demonstrated that overexpression of ephrinA3 was involved in PDAC cell viability as well as EphA4. However, other Eph receptors and ephrin ligands have been implicated in altering cellular environment; in particular, neovascularization in fetal tissues and pathogenic angiogenesis in tumor are well known.( 31 , 32 ) Our results do not exclude the possibility that other ephrin ligands expressed in vascular cells or mesenchymal cells as well as cancer cells in pancreatic cancer tissues could promote tumor growth by cross‐communication of these cells, and further in vitro and in vivo investigations are required to make clear the interaction between overexpressing EphA4 and ephrin ligands in PDAC.
Our previous microarray data obtained by gene expression profile analysis of 29 normal human tissues( 33 ) as well as our Northern blot analysis in Fig. 1B clearly indicate that EphA4 expression in normal adult tissues is very restricted. EphA4 was found to be expressed in the adult brain, but it has been suggested that therapeutic antibody targeting membrane molecules could not pass through the blood–brain barrier and were unlikely to affect the central nervous system.( 34 ) Considering the expression pattern of EphA4 together with its oncogenic function, EphA4 receptor could be an attractive and promising target for drug design by targeting its kinase property and also by an antibody‐based strategy.
Acknowledgments
This work was supported in part by Research for the Future Program Grant #00L01402 from the Japan Society for the Promotion of Science (Y. Nakamura). We thank Ms Satomi Uchida and Ms Ryo Ishimine for their technical assistance.
References
- 1. DiMagno EP, Reber HA, Tempero MA. AGA technical review on the epidemiology, diagnosis, and treatment of pancreatic ductal adenocarcinoma. Gastroenterology 1999; 117: 1464–84. [DOI] [PubMed] [Google Scholar]
- 2. Zervos EE, Rosemurgy AS, Al‐Saif O, Durkin AJ. Surgical management of early‐stage pancreatic cancer. Cancer Control 2004; 11: 23–31. [DOI] [PubMed] [Google Scholar]
- 3. Jemal A, Siegel R, Ward E et al. Cancer statistics, 2006. CA Cancer J Clin 2006; 56: 106–30. [DOI] [PubMed] [Google Scholar]
- 4. Nakamura T, Furukawa Y, Nakagawa H et al. Genome‐wide cDNA microarray analysis of gene‐expression profiles in pancreatic cancers using populations of tumor cells and normal ductal epithelial cells selected for purity by laser microdissection. Oncogene 2004; 23: 2385–400. [DOI] [PubMed] [Google Scholar]
- 5. Eph Nomenclature Committee. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 1997; 90: 403–4. [DOI] [PubMed] [Google Scholar]
- 6. Gale NW, Holland SJ, Valenzuela DM et al. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron 1996; 17: 9–19. [DOI] [PubMed] [Google Scholar]
- 7. Flanagan JG, Vanderhaeghen P. The ephrins and Eph receptors in neural development. Ann Rev Neurosci 1998; 21: 309–45. [DOI] [PubMed] [Google Scholar]
- 8. Wang HU, Anderson DJ. Eph family transmembrane ligands can mediate repulsive guidance of trunk neural crest migration and motor axon outgrowth. Neuron 1997; 18: 383–96. [DOI] [PubMed] [Google Scholar]
- 9. Xu Q, Mellitzer G, Robinson V, Wilkinson DG. In vivo cell sorting in complementary segmental domains mediated by Eph receptors and ephrins. Nature 1999; 399: 267–71. [DOI] [PubMed] [Google Scholar]
- 10. Palmer A, Klein R. Multiple roles of ephrins in morphogenesis, neuronal networking, and brain function. Genes Dev 2003; 17: 1429–50. [DOI] [PubMed] [Google Scholar]
- 11. Gale NW, Yancopoulos GD. Growth factors acting via endothelial cell‐specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev 1999; 13: 1055–66. [DOI] [PubMed] [Google Scholar]
- 12. Walker‐Daniels J, Hess AR, Hendrix MJ, Kinch MS. Differential regulation of EphA2 in normal and malignant cells. Am J Pathol 2003; 162: 1037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. EphA2: A determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene 2004; 23: 1448–56. [DOI] [PubMed] [Google Scholar]
- 14. Jubb AM, Zhong F, Bheddah S et al. EphB2 is a prognostic factor in colorectal cancer. Clin Cancer Res 2005; 11: 5181–7. [DOI] [PubMed] [Google Scholar]
- 15. Nakada M, Niska JA, Miyamori H et al. The phosphorylation of EphB2 receptor regulates migration and invasion of human glioma cells. Cancer Res 2004; 64: 3179–85. [DOI] [PubMed] [Google Scholar]
- 16. Hafner C, Schmitz G, Meyer S et al. Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin Chem 2004; 50: 490–9. [DOI] [PubMed] [Google Scholar]
- 17. Kataoka H, Igarashi H, Kanamori M et al. Correlation of EphA2 overexpression with high microvessel count in human primary colorectal cancer. Cancer Sci 2004; 95: 136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakamura R, Kataoka H, Sato N et al. EPHA2/EFNA1 expression in human gastric cancer. Cancer Sci 2005; 96: 42–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taniuchi K, Nakagawa H, Nakamura T et al. Down‐regulation of RAB6KIFL/KIF20A, a kinesin involved with membrane trafficking of discs large homologue 5, can attenuate growth of pancreatic cancer cell. Cancer Res 2005; 65: 105–12. [PubMed] [Google Scholar]
- 20. Niwa H, Yamamura K, Miyazaki J. Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene 1991; 108: 193–9. [DOI] [PubMed] [Google Scholar]
- 21. Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron 1992; 9: 383–91. [DOI] [PubMed] [Google Scholar]
- 22. Kinch‐Carles K, Kilpatrick KE, Stewart JC, Kinch MS. Antibody targeting of the EphA2 tyrosine kinase inhibits malignant cell behavior. Cancer Res 2002; 62: 2840–7. [PubMed] [Google Scholar]
- 23. Mao W, Luis E, Ross S et al. EphB2 as a therapeutic antibody drug target for the treatment of colorectal cancer. Cancer Res 2004; 64: 781–8. [DOI] [PubMed] [Google Scholar]
- 24. Ashida S, Nakagawa H, Katagiri T et al. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: genome‐wide gene‐expression profiles of prostate cancers and PINs. Cancer Res 2004; 64: 5963–72. [DOI] [PubMed] [Google Scholar]
- 25. Nagayama S, Katagiri T, Tsunoda T et al. Genome‐wide analysis of gene expression in synovial sarcomas using a cDNA microarray. Cancer Res 2002; 62: 5859–66. [PubMed] [Google Scholar]
- 26. Murai KK, Pasquale EB. ‘Eph’ective signaling: forward, reverse and crosstalk. J Cell Sci 2003; 116: 2823–32. [DOI] [PubMed] [Google Scholar]
- 27. Klein R, Kullander K. Mechanisms and functions of EPH and Ephrin signaling. Nature Rev Mol Cell Biol 2002; 3: 475–86. [DOI] [PubMed] [Google Scholar]
- 28. Davy A, Robbins SM. Ephrin‐A5 modulates cell adhesion and morphology in an integrin‐dependent manner. EMBO J 2000; 19: 5396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goldshmit Y, Galea MP, Wese G, Bartlett PF, Turnley AM. Axonal regeneration and lack of astrocytic gliosis in EphA4‐deficient mice. J Neurosci 2004; 24: 10 064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrinA3/EphA4 signaling. Nature Neurosci 2003; 6: 153–60. [DOI] [PubMed] [Google Scholar]
- 31. Ogawa K, Pasqualini R, Lindberg RA et al. The ephrin‐A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene 2000; 19: 6043–52. [DOI] [PubMed] [Google Scholar]
- 32. Dobrzanski P, Hunter K, Jones‐Bolin S et al. Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist. Cancer Res 2004; 64: 910–9. [DOI] [PubMed] [Google Scholar]
- 33. Saito‐Hisaminato A, Katagiri T, Kakiuchi S et al. Genome‐wide profiling of gene expression in 29 normal human tissues with a cDNA microarray. DNA Res 2002; 9: 35–45. [DOI] [PubMed] [Google Scholar]
- 34. Bendell JC, Domchek SM, Burstein HJ et al. Central nervous system metastases in women who receive trastuzumab‐based therapy for metastatic breast carcinoma. Cancer 2003; 97: 2972–7. [DOI] [PubMed] [Google Scholar]