

Abstract
Chronic myelogenous leukemia (CML) is a hematological malignancy that begins as indolent chronic phase (CP) but inevitably progresses to fatal blast crisis (BC). p210BCR/ABL, a chimeric protein with enhanced kinase activity, initiates CML CP, and additional genetic alterations account for progression to BC, but the precise mechanisms underlying disease evolution are not fully understood. In the present study, we investigated the possible contribution of dysfunction of Bcl11b, a zinc‐finger protein required for thymocyte differentiation, and of H2AX, a histone protein involved in DNA repair, to the transition from CML CP to BC. For this purpose, we crossed CML CP‐exhibiting p210BCR/ABL transgenic (BA tg/–) mice with Bcl11b heterozygous (Bcl11b +/–) mice and H2AX heterozygous (H2AX +/–) mice. Interestingly, p210BCR/ABL transgenic, Bcl11b heterozygous (BA tg/– Bcl11b +/–) mice and p210BCR/ABL transgenic, H2AX heterozygous (BA tg/– H2AX +/–) mice frequently developed CML BC with T‐cell phenotype and died in a short period. In addition, whereas p210BCR/ABL was expressed in all of the leukemic tissues, the expression of Bcl11b and H2AX was undetectable in several tumors, which was attributed to the loss of the residual normal allele or the lack of mRNA expression. These results indicate that Bcl11b and H2AX function as tumor suppressor and that haploinsufficiency and acquired loss of these gene products cooperate with p210BCR/ABL to develop CML BC. (Cancer Sci 2009; 100: 1219–1226)
Chronic myelogenous leukemia (CML) is a disorder of hematopoietic stem cells, characterized by excessive and uncontrolled proliferation of differentiated myeloid cells.( 1 , 2 , 3 ) Clinically, CML undergoes two different stages.( 1 , 2 , 3 ) In the initial stage, chronic phase (CP), the leukemic cells retain the ability to differentiate into mature granulocytes and are sensitive to conventional therapies. However, after several years’ duration of CP, the disease inevitably accelerates and ultimately progresses to the terminal stage, blast crisis (BC), which exhibits aggressive proliferation of immature blast cells and is resistant to intensive therapies.( 1 , 2 , 3 )
The cytogenetic hallmark of CML CP is t(9;22)(q34;q11) (known as Philadelphia chromosome, Ph), which generates a BCR–ABL fusion gene encoding a 210‐kDa chimeric protein (p210BCR/ABL).( 1 , 2 , 3 ) p210BCR/ABL possesses a constitutively active tyrosine kinase activity, which plays an essential role in the initiation of the disease.( 1 , 2 , 3 ) Although Ph is the unique and sole chromosomal abnormality in CP, additional and non‐random chromosomal abnormalities are frequently observed in BC, indicating that secondary genetic events account for the disease progression.( 1 , 2 , 3 )
To understand the pathogenesis of the disease, it is necessary to establish animal models that express p210BCR/ABL and recapitulate the clinical course of CML. For this purpose, we generated transgenic mice expressing p210BCR/ABL under the control of the mouse TEC promoter.( 4 ) The p210BCR/ABL transgenic (hereafter, designated as BA tg/–) mice reproducibly exhibited a myeloproliferative disorder closely resembling human CML CP.( 4 ) In addition, by crossing BA tg/– mice with p53 heterozygous mice and Dok‐1/Dok‐2 knockout mice, we showed that the loss of p53 and absence of Dok‐1/Dok‐2 accelerated the disease and caused CML BC.( 5 , 6 ) Furthermore, by applying retroviral insertional mutagenesis to BA tg/– mice, we demonstrated that overexpression and enhanced kinase activity of p210BCR/ABL and altered expression of Notch1 contribute to CML BC.( 7 ) These results demonstrated that the BA tg/– mouse is not only regarded as a model for CML CP, but is also useful for investigating the molecular mechanisms underlying the progression from CP to BC.
Chromosomal and molecular analyses have revealed that several mechanisms are implicated in this process, such as: (i) loss of tumor suppressor; (ii) differentiation arrest; and (iii) chromosomal instability.( 3 ) Indeed, as an example of (i), we demonstrated that loss of p53 cooperates with p210BCR/ABL and induces CML BC.( 5 , 6 ) In the present report, as candidate genes for (ii) and (iii), we chose Bcl11b (also known as Rit1 and Ctip2), encoding a transcription factor required for thymocyte differentiation,( 8 ) and H2AX, encoding a histone protein involved in DNA repair,( 9 ) and examined the possible contribution that dysfunction of these gene produces for the disease progression of CML. For this purpose, we crossed BA tg/– mice with mice heterozygous for Bcl11b (Bcl11b +/–) or H2AX (H2AX +/–) and generated BA tg/– Bcl11b +/– mice and BA tg/– H2AX +/– mice. Interestingly, both types of double transgenic mouse frequently developed CML BC and died in a short period. The pathological, flow cytometric, molecular, and chromosomal analyses of the diseased mice are described.
Materials and Methods
Mice. p210BCR/ABL transgenic, Bcl11b heterozygous, and H2AX heterozygous mice were generated as described previously.( 4 , 8 , 10 ) Crossing and genotyping of the mice were carried out as described previously.( 5 ) All of the mice were kept according to the guidelines of the Institute of Laboratory Animal Science, Hiroshima University.
Pathological analysis. Autopsies were carried out on dead or moribund animals. Peripheral blood smears were stained with Wight‐Giemsa. After gross examination, tissues were fixed in 10% neutral buffered formaldehyde and representative slices were stained with hematoxylin–eosin (HE).
Western blot analysis. Proteins were extracted from tissues, separated by SDS‐PAGE, transferred to a nitrocellulose membrane, and blotted with appropriate antibodies as described previously.( 4 , 8 ) The antibodies used in this study were: anti‐ABL monoclonal antibody, Ab3 (Oncogene Science, Cambridge, MA, USA); an anti‐Bcl11b polyclonal antibody;( 8 ) and an antihistone H2AX antibody (Millipore, Bedford, MA, USA). Positive signals were detected with the enhanced chemiluminescence system.
Southern blot analysis and genomic PCR. For Southern blotting, DNA was digested with restriction enzymes, separated in an agarose gel, blotted to a nylon membrane, and hybridized with a 32P‐dCTP‐labeled TCRβ probe. Genomic PCR was carried out using the following primers as described previously:( 11 ) P1 (5′‐TGCAGCTTTCCGGGCGATGCCA‐3′), P2 (5′‐ACTTTCCCAGAACCCCACGC‐3′), and P3 (5′‐CCTGCTTGCCGAATATCATGGTGG‐3′) for Bcl11b; and P1 (5′‐TCACATTGTTTCCTTCGGTGTCAC‐3′), P2 (5′‐AAGTGTTGTGATTGGGAAGCGTAG‐3′), P3 (5′‐AGATCCCGTTGACTGAACACAGG‐3′), P4 (5′‐TTCAGGTTTTGTTGTCGCGCCGTAG‐3′), and P5 (5′‐TCAGCTCTTTCTGTGAGGGAGGTGG‐3′) for H2AX.
Northern blot analysis and RT‐PCR. Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA), separated in 1.2% formaldehyde gel, blotted to a nylon membrane, and hybridized with a 32P‐dCTP‐labeled H2AX probe. RT‐PCR was carried out using the following primers as described previously:( 11 ) 5′‐CGAGCTCAGGAAAGTGTCCGAG‐3′ and 5′‐GGAAATTCATGAGCGGGGACTG‐3′ for Bcl11b; 5′‐CCTTCTGGAAGACTTGGCCTTC‐3′ and 5′‐GAGGAAGATGTGCCTGTTACC‐3′ for H2AX; and 5′‐TTCAACACCCCAGCCATGTA‐3′ and 5′‐CTCAGGAGGAGCAATGATCT‐3′ for β‐actin.
Flow cytometric analysis. Cells were stained with FITC‐ or phycoerythrin (PE)‐conjugated anti‐Thy‐1.2, anti‐B220, anti‐Mac1, and anti‐Gr1 monoclonal antibodies (Pharmingen, San Diego, CA, USA), as described previously.( 5 )
Chromosomal analysis. Chromosomes were prepared by means of standard culture procedures for tumor cells and treated with trypsin‐Giemsa as described previously.( 12 )
Patient samples and normal bone marrow cells. Patient samples were taken after obtaining informed consent and approval from the institutional review board at Hiroshima University.( 13 ) Diagnosis of CML CP or CML BC (myeloid or B‐lymphoid lineage) was carried out based on morphological, cytogenetic, and immunophenotypic analyses. Normal bone marrow cells were obtained from a healthy volunteer.
Results
BA tg/– Bcl11b +/– and BA tg/– H2AX +/– mice developed acute leukemia and died in a short period. To investigate the contribution of haploinsufficiency of Bcl11b and H2AX to the disease progression of CML, we crossed CML‐exhibiting BA tg/– mice with Bcl11b +/– mice and H2AX +/– mice. Mice with four different genotypes were generated by each crossing: BA tg/– × Bcl11b +/– created BA −/– Bcl11b +/+ (wild type), BA tg/– Bcl11b +/+ (p210BCR/ABL transgenic), BA −/– Bcl11b +/– (Bcl11b heterozygous), and BA tg/– Bcl11b +/– (p210BCR/ABL transgenic, Bcl11b heterozygous); and BA tg/– × H2AX +/– produced BA −/– H2AX +/+ (wild type), BA tg/– H2AX +/+ (p210BCR/ABL transgenic), BA −/– H2AX +/– (H2AX heterozygous), and BA tg/– H2AX +/– (p210BCR/ABL transgenic, H2AX heterozygous). Mice with these genotypes were normally born approximately at the expected Mendelian ratio (see the mouse number shown in parentheses in Fig. 1), indicating that the crossing did not affect the embryonic development of the mice.
Figure 1.
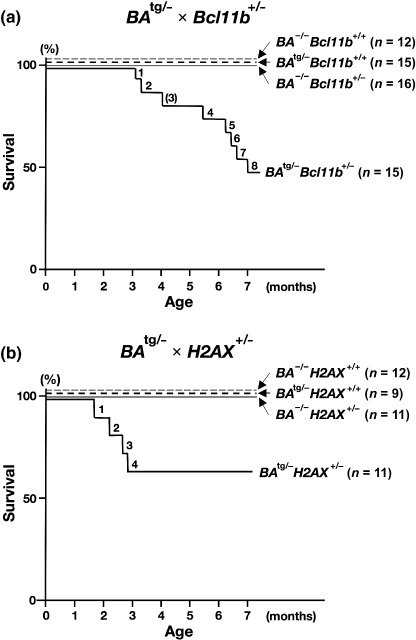
Survival curves of mice generated by (a) BA tg/– × Bcl11b +/– and (b) BA tg/– × H2AX +/–. The survival curves of BA −/– Bcl11b +/+ and BA −/– HX2A +/+, BA tg/– Bcl11b +/+ and BA tg/– H2AX +/+, BA −/– Bcl11b +/– and BA −/– H2AX +/–, and BA tg/– Bcl11b +/– and BA tg/– H2AX +/– mice are shown as thin dotted, thick dotted, thin continuous, and thick continuous lines respectively. In the BA tg/– × Bcl11b +/– group, 8 of 15 BA tg/– Bcl11b +/– mice died within 7 months of age and in the BA tg/– × H2AX +/– group, 4 of 11 BA tg/– H2AXb +/– died within 3 months of age. The number of an unanalyzable BA tg/– Bcl11b +/– mouse due to death (no. 3) is shown in parentheses.
All of the mice were observed continuously and peripheral blood parameters were counted routinely. The genotype‐based survival curves of the mice in each crossing are shown in Figure 1. During a 7‐month observation period, in the BA tg/– × Bcl11b +/– group, 8 of 15 BA tg/– Bcl11b +/– died of acute leukemia, in contrast BA −/– Bcl11b +/+, BA tg/– Bcl11b +/+, and BA −/– Bcl11b +/– littermates did not show any disorders (Fig. 1a). As for the BA tg/– × H2AX +/– group (lower panel), 4 of 11 BA tg/– H2AX +/– mice exhibited proliferation of blast cells and died within 3 months of birth, whereas no disease was observed in BA −/– H2AX +/+, BA tg/– H2AX +/+, and BA −/– H2AX +/– littermates (Fig. 1b).
The representative results of pathological analysis of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice are shown in Figure 2. Macroscopically, both leukemic mice exhibited marked thymic enlargement with splenomegaly, which were occasionally associated with lymph node swelling or pleural effusion (data not shown). The peripheral blood smears exhibited proliferation of blast cells morphologically resembling lymphoblasts (upper panels of Fig. 2). Tissue sections showed that the blast cells caused destruction of the basic structure of the thymus (second panels of Fig. 2) and infiltrated in non‐hematopoietic tissues, such as liver (third panels of Fig. 2). In contrast, the bone marrow showed a predominance of myeloid cells with differentiation and proliferation of megakaryocytes (bottom panels of Fig. 2). These results demonstrated that haploinsufficiency of Bcl11b and H2AX cooperated with p210BCR/ABL, transformed p210BCR/ABL‐expressing hematopoietic cells, and caused CML BC. The characteristics of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice are summarized in Table 1 and Table 2, respectively.
Figure 2.
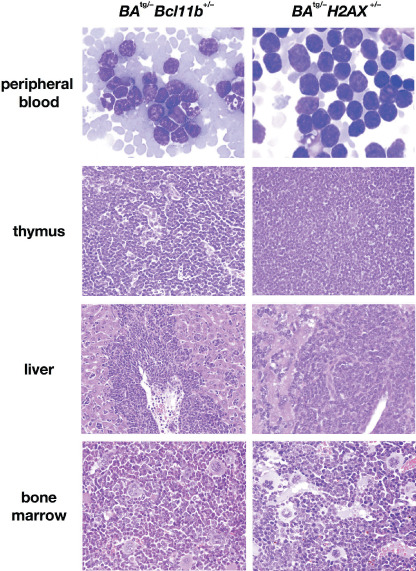
Representative results of pathological analysis of BA tg/– Bcl11b +/– (left panels) and BA tg/– H2AX +/– (right panels) leukemic mice. Wight‐Giemsa‐stained peripheral blood smears and HE‐stained tissue slices are shown. In both leukemic mice, blast cells proliferated in the peripheral blood (upper panels), caused destruction of the basal structure of the thymus (second panels), and infiltrated around the vessel and in the sinusoids in the liver (third panels). In contrast, bone marrow exhibited myeloid cell hyperplasia with differentiation and proliferation of megakaryocytes (bottom panels).
Table 1.
Characteristics of p210BCR/ABL tg/– Bcl11b +/– leukemic mice
| Mouse no. | Age at disease (months) | PB parameters | Macroscopic tumor sites | TCRβ status | p210BCR/ABL expression | Bcl11b expression | Bcl11b status | ||
|---|---|---|---|---|---|---|---|---|---|
| WBC (× 103/µL) | Hb (g/dL) | Plt (× 104/µL) | |||||||
| 1 | 3.1 | 35.0 | 12.5 | 65.6 | Thy, Spl | G/R | + | + | G/T |
| 2 | 3.3 | 5.0 | 10.5 | 64.8 | Thy, Spl | G/loss | + | + | G/T |
| 3 | 4.0 † | ND | ND | ND | Thy | ND | ND | ND | ND |
| 4 | 5.3 | 2.3 | 7.1 | 44.2 | Thy | G/loss | + | + | G/T |
| 5 | 6.0 | 12.0 | 12.3 | 35.5 | Thy, PE | G/loss | + | – | T/loss |
| 6 | 6.1 | 6.6 | 13.9 | 53.5 | Thy | G/R | + | – | T/loss |
| 7 | 6.4 | 14.6 | 15.5 | 47.1 | Thy, Spl | G/R | + | + | G/T |
| 8 | 6.9 | 1.5 | 14.1 | 74.9 | Thy, PE | G/R | + | – | T/loss |
Found dead. G, germline; Hb, hemoglobin; ND, not done; PB, peripheral blood; PE, pleural effusion; Plt, platelet; R, rearranged; Spl, spleen; T, targeted; Thy, thymus; WBC, white blood cell.
Table 2.
Characteristics of p210BCR/ABLtg/– H2AX+/– leukemic mice
| Mouse no. | Age at disease (months) | PB parameters | Macroscopic tumor sites | TCRβ status | p210BCR/ABL expression | H2AX expression | H2AX status | ||
|---|---|---|---|---|---|---|---|---|---|
| WBC (× 103/µL) | Hb (g/dL) | Plt (× 104/µL) | |||||||
| 1 | 1.8 | 15.3 | 16.1 | 56.4 | Thy, Spl | G/R | + | + | G/T |
| 2 | 2.2 | 160.8 | 10.4 | 53.4 | Thy, Spl, LN | G/R | + | – | G/T |
| 3 | 2.5 | 128.4 | 12.0 | 90.9 | Thy, Spl, LN | G/loss | + | – | G/T |
| 4 | 2.8 | 84.7 | 12.4 | 36.0 | Thy, Spl, LN | G/loss | + | + | G/T |
G, germline; LN, lymph node; R, rearranged; Spl, spleen; T, targeted; Thy, thymus.
Leukemias that developed in BA tg/– Bcl11b +/– and BA tg/– H2AX +/– mice were of T‐cell lineage and were mostly clonal in origin. To determine the cell lineage and clonality of the leukemias that developed in BA tg/– Bcl11b +/– and BA tg/– H2AX +/– mice, blast cells were subjected to flow cytometric and Southern blot analyses.
The representative results of flow cytometric analysis of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic cells are shown in Figure 3(a). In both types of mice, leukemic cells were highly positive for Thy1.2, the antigen specific for T lymphocytes, but were negative for CD19, Gr1, and Mac1, the markers for B lymphocytes, granulocytes, and macrophages respectively.
Figure 3.
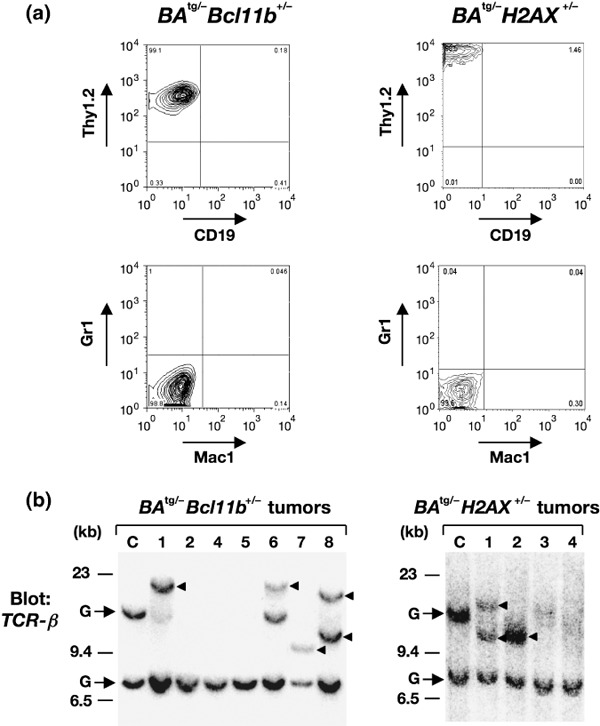
Results of flow cytrometric and Southern blot analyses of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice. (a) Representative results of flow cytometry of leukemic cells that developed in BA tg/– Bcl11b +/– (left panel) and BA tg/– H2AX +/– (right panel) mice. In both samples, blast cells were positive for Thy1.2 but negative for CD19, Gr1, and Mac1, indicating that they were of T‐cell phenotype. (b) Results of gene rearrangement analysis in tumors that developed in BA tg/– Bcl11b +/– (left panel) and BA tg/– H2AX +/– (right panel) mice. (c) DNA extracted from control thymus and thymomas that developed in BA tg/– Bcl11b +/– (left panel) and BA tg/– H2AX +/– (right panel) mice were digested with BamHI and blotted with TCR‐β probe. Germline and rearranged bands are indicated by arrows and arrowheads respectively. Molecular markers are shown on the left.
The clonality of the leukemic cells was examined by gene rearrangement analysis. DNA extracted from a control thymus and tumor tissues of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice were digested with a restriction enzyme and blotted with the T‐cell receptor β (TCR‐β) gene. As shown in Figure 3(b), more than half of the samples (no. 1 and no. 6–8 in BA tg/– Bcl11b +/– and no. 1 and 2 in BA tg/– H2AX +/–) showed rearranged bands, and in the remaining samples (no. 2, 4, and 5 in BA tg/– Bcl11b +/– and no. 3 and 4 in BA tg/– H2AX +/–), loss of the upper germline band was observed (the positions of germline bands are indicated by arrows and shown as ‘G’). These results demonstrated that the blast cells of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice were committed to the T‐cell lineage and most of the tumors were clonal in origin.
Frequent and acquired loss of Bcl11b and H2AX protein expression in the tumor tissues of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice. We then investigated protein expression in the tumor tissues of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice. Proteins extracted from a control thymus and tumor tissues of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice were blotted with antibodies against c‐ABL, Bcl11b, and H2AX.
The results of p210BCR/ABL expression in these tumors are shown in the upper panels of Figure 4(a,b). As shown in both panels, the 210‐kDa band was detected in all of the tumor samples, indicating that the blast cells originated from p210BCR/ABL‐expressing hematopoietic precursors. We next examined the expression of Bcl11b and H2AX proteins in BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic samples respectively. Interestingly, in the anti‐Bcl11b western blot, the expression of Bcl11b was found to be lost in three of seven samples (no. 5, 6, and 8, middle panel of Fig. 4a). In addition, in the anti‐H2AX blot, the expression of H2AX was undetectable in two of four samples (no. 2 and 3, middle panel of Fig. 4b). These results indicated that the protein expression of Bcl11b and H2AX was lost in several samples of BA tg/– Bcl11b +/– and BA tg/– H2AX +/– leukemic mice.
Figure 4.
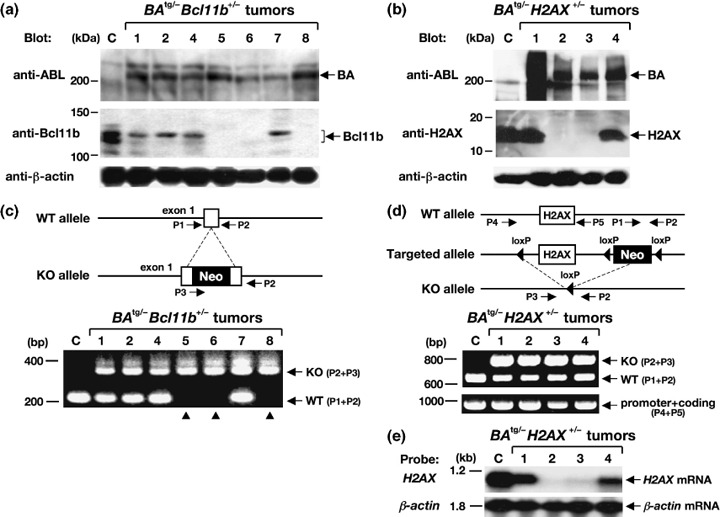
Gene expression and PCR analyses of the tumors that developed in BA tg/– Bcl11b +/– (left panels) and BA tg/– H2AX +/– (right panels) mice. (a,b) Western blot analysis for the expression of p210BCR/ABL, Bcl11b, and H2AX proteins. Proteins extracted from a control (C) thymus and tumor tissues of BA tg/– Bcl11b +/– (no. 1, 2, and 4–8) and BA tg/– H2AX +/– mice (no. 1–4) were blotted with an anti‐ABL antibody (upper panels) and anti‐Bcl11b or anti‐H2AX antibody (middle panels). The positions of p210BCR/ABL (BA), Bcl11b, and H2AX proteins are indicated by arrows. An anti‐β‐actin blot was carried out as an internal control (bottom panels). Protein markers are shown on the left. (c,d) Schematic illustrations of wild‐type and targeted alleles for Bcl11b and H2AX genes (upper panels) and the resultant genomic PCR products (lower panels). DNA extracted from a control (C) thymus and tumor tissues of BA tg/– Bcl11b +/– (no. 1, 2 and 4–8) and BA tg/– H2AX +/– mice (no. 1–4) were amplified with sets of primers (P1 and P2 for wild‐type [WT] alleles, P2 and P3 for knockout [KO] alleles, and P4 and P5 for a part of the promoter and the whole coding region of H2AX). The positions of primers are shown in the upper panels and WT‐ and KO‐derived PCR products are indicated by arrows in the lower panels. Molecular markers are shown on the left. Samples without Bcl11b expression are indicated by arrowheads. Neo, neomycin resistance gene. (e) Expression of H2AX mRNA in BA tg/– H2AX +/– tumors. RNA extracted from a control thymus (C) and tumor tissues of BA tg/– H2AX +/– mice (no. 1–4) were hybridized with H2AX cDNA probe. β‐Actin hybridization was carried out as an internal control. Molecular markers are shown on the left.
To investigate the molecular mechanism underlying the loss of Bcl11b and H2AX expression, DNA extracted from tumor tissues was subjected to genomic PCR that distinguished the PCR product of the wild‐type allele from that of the knockout allele (upper panels of Fig. 4c,d). The results showed that the wild‐type Bcl11b allele‐derived band was not amplified in the three samples without Bcl11b expression (no. 5, 6, and 8, lower panel of Fig. 4c), indicating that the absence of Bcl11b protein was attributed to the loss of the residual wild‐type Bcl11b allele. In contrast, the PCR product from the wild‐type H2AX allele was retained in the two samples lacking H2AX expression (no. 2 and 3 in the lower panel of Fig. 4d). Because the PCR primer set detecting the wild‐type allele (P1 + P2) did not amplify the coding region of the H2AX gene (upper panel of Fig. 4d), we designed another primer set encompassing the H2AX exon. As H2AX is a single‐exon gene,( 10 ) this primer set (shown as P4 and P5 in the upper panel of Fig. 4d) amplified a part of the promoter and the whole coding region. The results showed that a PCR product of expected size was detected in all of the BA tg/– H2AX +/– tumors (lower panel of Fig. 4d). To examine the possibility that subtle deletion and/or base substitution had occurred in this region, we sequenced the whole PCR product but could not detect any mutation (data not shown). In addition, Southern blotting using a 5′ external probe for the H2AX gene( 10 ) did not show any gross rearrangement (data not shown). These results indicated that the structure of the H2AX gene was largely unaffected. We next examined H2AX mRNA expression in the BA tg/– H2AX +/– tumors by northern blotting. Interestingly, as shown in Figure 4(e), no H2AX mRNA was detected in tumors lacking H2AX protein expression (no. 2 and 3). These results indicated that the absence of H2AX protein was not due to deletion or mutation in the H2AX gene but to a lack of mRNA expression.
Chromosomal abnormalities in the leukemic cells developed in BA tg/– H2AX +/– mice. We finally examined the chromosomal status of the leukemic cells developed in BA tg/– H2AX +/– mice, as previous reports demonstrated that haploinsufficiency and absence of H2AX led to increased incidence of chromosomal abnormalities.( 14 , 15 ) In the four tumors that arose from BA tg/– H2AX +/– mice, although two samples showed a normal karyotype (no. 1 and 4, data not shown), the other two samples (no. 2 and 3) that did not express H2AX protein (Fig. 4b) exhibited chromosomal aberrations. As shown in the left panel of Figure 5, sample no. 2 contained an additional chromosome (indicated by an arrowhead). In addition, as shown in the right panel of Figure 5, sample no. 3 exhibited deletions in the long arm of chromosome 6 and in the short arm of chromosome 13, and a breakage in chromosome 11 (indicated by arrows). These results suggested the possibility that the acquired loss of H2AX induced chromosomal instability and resulted in the chromosomal abnormalities observed in samples no. 2 and 3.
Figure 5.
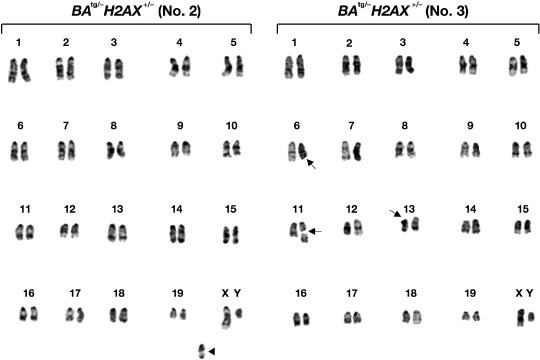
Chromosomal abnormalities observed in two tumors (no. 2 and 3) that developed in BA tg/– H2AX +/– mice. The additional chromosome in tumor no. 2 is indicated by an arrowhead, and deletion and breakage of the chromosomes in tumor no. 3 are indicated by arrows.
Discussion
Chronic myelogenous leukemia presents a paradigm for cancers that evolve through accumulation of genetic alterations. Generation of p210BCR/ABL initiates CML CP and additional genetic events progress the disease and develop CML BC.( 1 , 2 , 3 ) Although chromosomal and molecular analyses revealed that various mechanisms are involved in the transition from CP to BC,( 1 , 2 , 3 ) genes responsible for the evolution to BC have not fully been identified.
To elucidate the mechanisms underlying the disease evolution of CML, we have developed an in vivo model for CML in which expression of p210BCR/ABL induces CML CP, and additional genetic alterations cooperate with p210BCR/ABL to progress the disease to CML BC.( 4 , 5 , 6 , 7 ) Using this as a model system, we examined the possible contribution of haploisufficiency of Bcl11b and H2AX to CML BC, by crossing p210BCR/ABL transgenic mice (BA tg/–) with Bcl11b heterozygotes (Bcl11b +/–) and H2AX heterozygotes (H2AX +/–).
Bcl11b encodes a zinc finger protein involved in thymocyte development and differentiation.( 8 ) Bcl11b was originally identified as a gene homologous to Bcl11a, that was cloned from t(2;14)(p13;q32.3)‐carrying malignant lymphomas,( 16 ) and subsequently shown to be frequently deleted or mutated in radiation‐induced thymoma in mice.( 17 ) Conditional knockout analysis showed that acquired ablation of Bcl11b in thymocytes resulted in impaired positive selection, altered T‐cell receptor signaling, and reduced survival.( 18 ) In addition, a recent study revealed that Bcl11b is involved in human leukemia carrying inv(14)(q11.2q32.31), which resulted in generation of the Bcl11b–TRDC fusion transcript.( 19 ) On the other hand, H2AX is a member of the histone H2A family and a constituent of the nucleosome, the basic subunit of chromatin.( 9 , 20 , 21 ) In response to the DNA double‐strand break, H2AX rapidly becomes phosphorylated on the serine residue located at the C‐terminus to form γH2AX at the DNA double‐strand break sites.( 9 , 20 , 21 ) This event creates a focus in the nucleus, where DNA repair and chromatin remodeling proteins are recruited.( 9 , 20 , 21 ) In human hematopoietic malignancies, a single nucleotide polymorphism upstream of the H2AX gene was found to be tightly associated with susceptibility to non‐Hodgkin lymphoma.( 22 ) These results indicated that Bcl11b and H2AX are functionally implicated in cell differentiation and chromosomal stability, respectively, and are involved in subsets of hematopoietic malignancies.
We found that 8 of 15 BA tg/– Bcl11b +/– mice and 4 of 11 BA tg/– H2AX +/– mice developed acute leukemia and died in a short period (Fig. 1). These results indicated that haploinsufficiency of Bcl11b and H2AX conferred a growth advantage to p210BCR/ABL‐expressing hematopoietic cells and consequently induced acute leukemia. The blast cells were highly malignant, as evidenced by massive proliferation in the peripheral blood, destruction of the basic structure of the thymus, and marked infiltration in non‐hematopoietic tissues (upper 3 panels of Fig. 2). Surface marker analysis showed that the leukemic cells were of T‐cell phenotype and Southern blot analysis demonstrated that most of the tumors were clonal in origin (Fig. 3). As the bone marrow showed the typical picture of CML CP (bottom panels of Fig. 2), the leukemias that developed in BA tg/– Bcl11b +/– and BA tg/– H2AX +/– mice were considered to be CML T‐cell BC rather than de novo T‐cell malignancy.
Interestingly, protein analysis revealed that the expression of Bcl11b and H2AX was lost in several tumors that developed in the BA tg/– Bcl11b +/– and BA tg/– H2AX +/– mice (Fig. 4a,b, middle panels). These results strongly suggested that the expression of p210BCR/ABL rendered genetic instability in the hematopoietic cells and consequently lost the normal residual allele of Bcl11b and H2AX, as reported in our previous study.( 5 ) Indeed, in BA tg/– Bcl11b +/– tumors, genomic PCR analysis revealed that the wild‐type Bcl11b‐derived band was not amplified in tumors lacking Bcl11b expression (no. 5, 6, and 8 in the lower panel of Fig. 4c), indicating that loss of the normal Bcl11b allele was responsible for the lack of the protein product. In contrast, the tumor tissues with no H2AX expression in BA tg/– H2AX +/– mice retained the normal H2AX allele, including the 3′ region, a part of the promoter region, and the whole coding region (no. 2 and 3 in the lower panels of Fig. 4d). Instead, we found that no H2AX mRNA was expressed in tumors lacking H2AX protein (no. 2 and 3 in the upper panel of Fig. 4e), which indicated that the absence of H2AX protein was due to the lack of H2AX mRNA expression. Although the mechanism underlying loss of the H2AX message in these tumors remains unclear, one possibility is that p210BCR/ABL‐induced genetic alterations might have occurred in the other regions regulating H2AX transcription, such as the enhancer, which led to the loss of mRNA expression. Alternatively, p210BCR/ABL might have impaired the transcriptional machinery for H2AX mRNA in these tumors by an unknown mechanism. Taken together, our findings demonstrated that p210BCR/ABL induces loss of protein expression through several different mechanisms, including genomic instability and transcriptional inhibition.
It is to be noted that four BA tg/– Bcl11b +/– and two BA tg/– H2AX +/– leukemic mice retained Bcl11b and H2AX protein expression (no. 1, 2, 4, and 7 in the middle panel of Fig. 4a and no. 1 and 4 in the middle panel of Fig. 4b). Thus, the mechanism of how haploinsufficiency of these genes caused disease evolution is to be clarified. Although no obvious phenotypic abnormalities were found in Bcl11b +/– or H2AX +/– mice, previous studies demonstrated that both types of heterozygotes exhibit enhanced susceptibility to hematological malignancies on p53 +/– and p53 −/– backgrounds.( 14 , 15 , 23 ) These results indicated that both genes function as a dosage‐dependent tumor suppressor and their haploinsufficiency predisposes to cancer development in certain genetic backgrounds. Thus, it is possible that haploinsufficiency of Bcl11b and H2AX exerted its oncogenic potential in cooperation with p210BCR/ABL, conferred a growth advantage to p210BCR/ABL‐expressing hematopoietic cells, and consequently developed CML BC. An alternative possibility is that because p210BCR/ABL is known to promote genetic instability,( 3 , 5 ) altered expression of unknown genes synergized with haploinsufficient Bcl11b or H2AX in p210BCR/ABL‐expressing hematopoietic cells, accelerated progression of CML, and eventually caused CML BC.
We finally examined the possible chromosomal abnormalities in the leukemic cells of BA tg/– H2AX +/– mice, as previous reports demonstrated that haploinsufficiency or deficiency of H2AX induced various chromosomal aberrations, especially on a p53 −/– genetic background.( 14 , 15 ) The results showed that two of four tumors exhibited chromosomal abnormalities, which were the presence of an additional chromosome, deletion in part of the long and short arms, and breakage in the body of several chromosomes (Fig. 5). Interestingly, BA tg/– H2AX +/– mice with these chromosomal abnormalities exhibited very high white blood cell (WBC) counts (>1 × 105/µL, see the right top panel of Fig. 2 and Table 2), suggesting that these events conferred a marked proliferative ability to p210BCR/ABL‐expressing hematopoietic cells and exhibited a very aggressive phenotype. We also examined the possible contribution of dysfunction of genes involved in error‐prone non‐homologous end joining, such as DNA ligase IV and XRCC4, by crossing BA tg/– with DNA ligase IV heterozygous mice and XRCC4 heterozygous mice. However, we did not observe disease acceleration or CML BC in BA tg/– DNA ligase IV +/– or BA t g/– XRCC4 +/– double transgenic mice (data not shown), suggesting the possibility that among DNA repair‐associated genes, H2AX might play a unique role in the disease evolution of CML.
The CML BC observed in BA tg/– Bcl11b +/– and BA tg/– H2AX +/– mice were of T‐cell phenotype. Although T‐cell BC is frequently observed in mouse models for CML,( 5 , 24 ) it is rarely detected in human clinical samples. The reason for this discrepancy is not clear but one possibility is that human CML originates from the acquisition of p210BCR/ABL‐transformed hematopoietic stem cells and the T‐cell lineage is rarely involved probably due to its prolonged life span, whereas every cell in transgenic (or knockout) mice inherently contains (or lacks) the target gene and T cells might be more susceptible to the target gene‐induced oncogenic transformation than other types of hematopoietic cells.
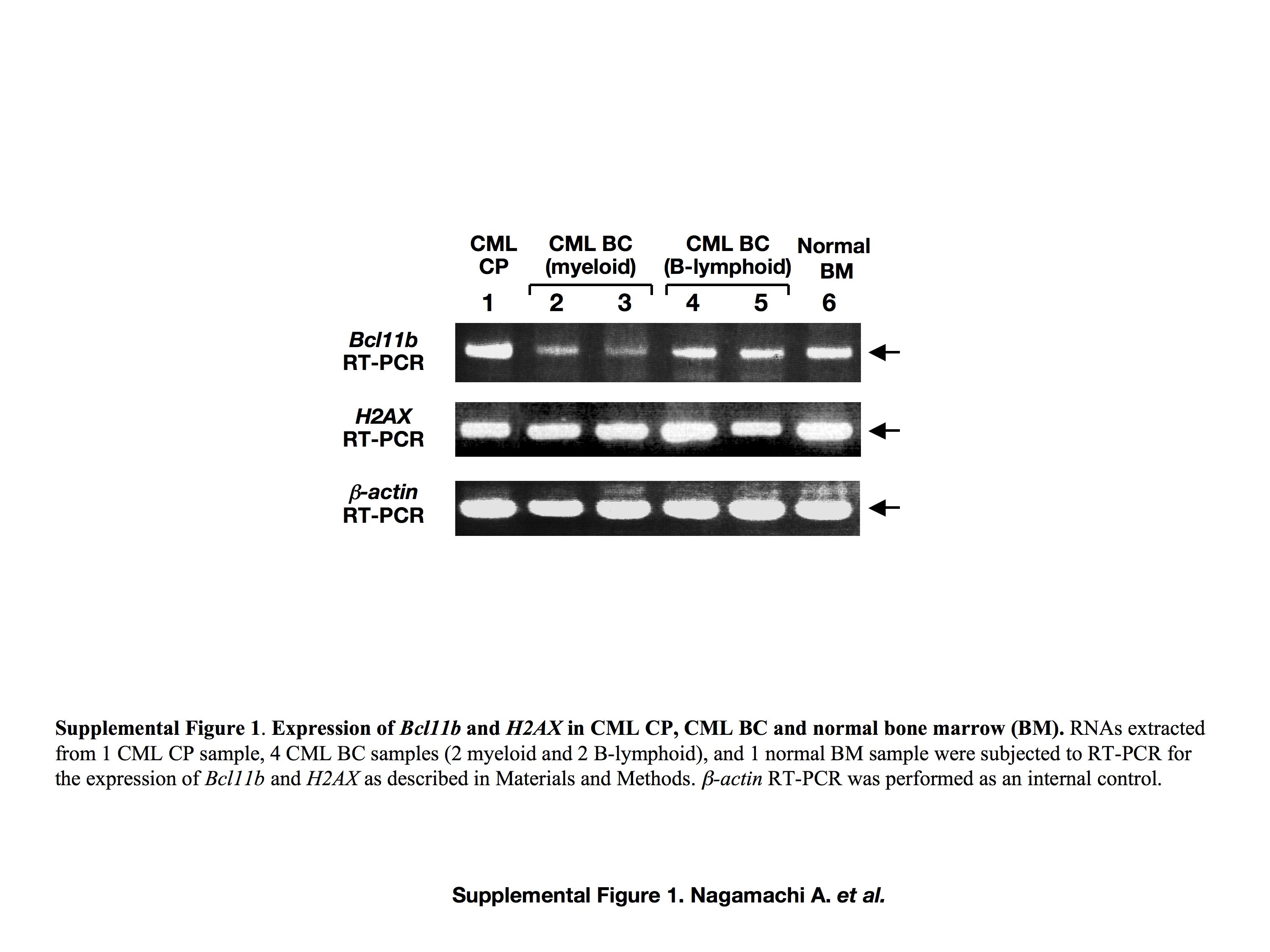
It is intriguing to examine whether acquired expressional loss of Bcl11b and H2AX contributes to human CML BC. We examined Bcl11b and H2AX expression in several CML BC samples by RT‐PCR but did not detect the absence of mRNA expression in either gene (Supporting Information Fig. S1), probably due to the limited number of samples available and a lack of T‐cell crisis cases. Thus, an expanded study is required to clarify the clinical significance of dysfunction of these genes in the development of CML BC.
In the present report, we demonstrated that haploinsufficiency and acquired loss of protein expression of Bcl11b and H2AX cooperate with p210BCR/ABL and induce CML BC. Our findings demonstrated that altered expression of genes involved in cell differentiation or chromosomal integrity contributes to the development of CML BC, which provides insights into the molecular mechanisms underlying the disease evolution of CML.
Supporting information
Fig. S1. Expression of Bcl11b and H2AX in chronic myelogenous leukemia (CML) chronic phase (CP), CML blast crisis (BC), and normal bone marrow (BM). RNA extracted from one CML CP sample, four CML BC samples (two myeloid and two B‐lymphoid), and one normal BM sample were subjected to RT‐PCR for the expression of Bcl11b and H2AX. β‐Actin RT‐PCR was carried out as an internal control.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
{kind=link}
Acknowledgments
This work was supported by a Grant‐in‐Aid from the Ministry of Education, Science, and Culture of Japan, a Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labour, and Welfare of Japan (13‐2), Research Grant of the Princess Takamatsu Cancer Research Fund, Astellas Research Foundation, YASUDA Medical Research Foundation, a Grant‐in‐Aid of The Japan Medical Association, and Japan Leukaemia Research Fund.
References
- 1. Calabretta B, Perrotti D. The biology of CML blast crisis. Blood 2004; 103: 4010–22. [DOI] [PubMed] [Google Scholar]
- 2. Ren R. Mechanisms of BCR‐ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005; 5: 172–83. [DOI] [PubMed] [Google Scholar]
- 3. Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 2007; 7: 441–53. [DOI] [PubMed] [Google Scholar]
- 4. Honda H, Oda H, Suzuki T et al . Development of acute lymphoblastic leukemia and myeloproliferative disorder in transgenic mice expressing p210bcr/abl: a novel transgenic model for human Ph1‐positive leukemias. Blood 1998; 91: 2067–75. [PubMed] [Google Scholar]
- 5. Honda H, Ushijima T, Wakazono K et al . Acquired loss of p53 induces blastic transformation in p210bcr/abl‐expressing hematopoietic cells: a transgenic study for blast crisis of human CML. Blood 2000; 95: 1144–50. [PubMed] [Google Scholar]
- 6. Niki M, Cristofano DA, Zhao M et al . Role of Dok‐1 and Dok‐2 in leukemia suppression. J Exp Med 2004; 200: 1689–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizuno T, Yamasaki N, Miyazaki K et al . Overexpression/enhanced kinase activity of BCR/ABL and altered expression of Notch1 induced acute leukemia in p210BCR/ABL transgenic mice. Oncogene 2008; 29: 3465–74. [DOI] [PubMed] [Google Scholar]
- 8. Wakabayashi Y, Watanabe H, Inoue J et al . Bcl11b is required for differentiation and survival of αβ T lymphocytes. Nat Immunol 2003; 4: 533–9. [DOI] [PubMed] [Google Scholar]
- 9. Bonner WM, Redon CE, Dickey JS et al . γH2AX and cancer. Nat Rev Cancer 2008; 8: 957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bassing CH, Chua KF, Sekiguchi J et al . Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA 2002; 99: 8173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyazaki K, Kawamoto T, Tanimoto K, Nishiyama M, Honda H, Kato Y. Identification of functional hypoxia response elements in the promoter region of the DEC1 and DEC2 genes. J Biol Chem 2002; 277: 47 014–21. [DOI] [PubMed] [Google Scholar]
- 12. Honda H, Ohno S, Takahashi T, Takatoku M, Yazaki Y, Hirai H. Establishment, characterization, and chromosomal analysis of new leukemic cell lines derived from MT/p210bcr/abl transgenic mice. Exp Hematol 1998; 26: 188–97. [PubMed] [Google Scholar]
- 13. Harada H, Harada Y, Tanaka H, Kimura A, Inaba T. Implications of somatic mutations in the AML1 gene in radiation‐associated and therapy‐related myelodysplastic syndrome/acute myeloid leukemia. Blood 2003; 101: 673–80. [DOI] [PubMed] [Google Scholar]
- 14. Bassing CH, Suh H, Ferguson DO et al . Histone H2AX: a dosage‐dependent suppressor of oncogenic translocations and tumors. Cell 2003; 114: 359–70. [DOI] [PubMed] [Google Scholar]
- 15. Celeste A, Difilippantonio S, Difilippantonio MJ et al . H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003; 114: 371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Satterwhite E, Sonoki T, Willis TG et al . The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood 2001; 98: 3413–20. [DOI] [PubMed] [Google Scholar]
- 17. Wakabayashi Y, Inoue J, Takahashi Y et al . Homozygous deletions and point mutations of the Rit1/Bcl11b gene in gamma‐ray induced mouse thymic lymphomas. Biochem Biophys Res Commun 2003; 301: 598–603. [DOI] [PubMed] [Google Scholar]
- 18. Albu DI, Feng D, Bhattacharya D et al . BCL11B is required for positive selection and survival of double‐positive thymocytes. J Exp Med 2008; 204: 3003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Przybylski GK, Dik WA, Wanzeck J et al . Disruption of the BCL11B gene through inv (14) (q11.2q32.31) results in the expression of BCL11B‐TRDC fusion transcripts and is associated with the absence of wild‐type BCL11B transcripts in T‐ALL. Leukemia 2005; 19: 201–8. [DOI] [PubMed] [Google Scholar]
- 20. Riches LC, Lynch AM, Gooderham NJ. Early events in the mammalian response to DNA double‐strand breaks. Mutagenesis 2008; 23: 331–9. [DOI] [PubMed] [Google Scholar]
- 21. Kinner A, Wu W, Staudt C, Iliakis G. c‐H2AX in recognition and signaling of DNA double‐strand breaks in the context of chromatin. Nucl Acids Res 2008; 36: 5678–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Novik KL, Spinelli JJ, Macarthur AC et al . Genetic variation in H2AFX contributes to risk of non‐Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007; 16: 1098–106. [DOI] [PubMed] [Google Scholar]
- 23. Kamimura K, Ohi H, Kubota T et al . Haploinsufficiency of Bcl11b for suppression of lymphomagenesis and thymocyte development. Biochem Biophys Res Commun 2007; 355: 538–42. [DOI] [PubMed] [Google Scholar]
- 24. Gishizky ML, Johnson‐White J, Witte ON. Efficient transplantation of BCR‐ABL‐induced chronic myelogenous leukemia‐like syndrome in mice. Proc Natl Acad Sci USA 1993; 90: 3755–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression of Bcl11b and H2AX in chronic myelogenous leukemia (CML) chronic phase (CP), CML blast crisis (BC), and normal bone marrow (BM). RNA extracted from one CML CP sample, four CML BC samples (two myeloid and two B‐lymphoid), and one normal BM sample were subjected to RT‐PCR for the expression of Bcl11b and H2AX. β‐Actin RT‐PCR was carried out as an internal control.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item