

Abstract
Growth, survival and differentiation of hematopoietic cells are regulated by the interactions between hematopoietic growth factors and their receptors. The defect in these interactions results in a failure of hematopoiesis, while aberrantly elevated and/or sustained activation of these signals cause hematologic malignancies. Among them, constitutively activating mutations of the receptor tyrosine kinases (RTKs), such as c‐Kit, platelet‐derived growth factor receptor (PDGFR) and FLT3, are often involved in the pathogenesis of various types of hematologic malignancies. Constitutive activation of RTKs is provoked by several mechanisms including chromosomal translocations and various mutations involving their regulatory regions. Chromosomal translocations commonly generate chimeric proteins consisting of the cytoplasmic domain of RTKs and the dimerization or multimerization motif of the fusion partner, resulting in the constitutive dimerization of RTKs. On the other hand, missense, insertion or deletion mutations in the regulatory regions, such as juxtamembrane domain, activation loop, and extracellular domain, also cause constitutive activation of RTKs mainly by preventing the auto‐inhibitory regulation. Oncogenic RTKs activate downstream signaling molecules such as Ras/MAPK, PI3‐K/Akt/mTOR, and STATs as well as ligand‐activated wild type RTKs. However, their signals are quantitatively and qualitatively different from wild type RTKs. Based on these findings, several agents that target oncogenic RTKs or their downstream molecules have been developed: imatinib and FLT3 inhibitors for RTKs themselves, farnesyltransferase inhibitors, mTOR inhibitors and MEK inhibitors for the downstream signaling molecules. As promising results have been obtained in several clinical trials using these agents, the establishment of these molecular targeted agents is expected. (Cancer Sci 2008; 99: 479–485)
Growth, differentiation, and survival of hematopoietic cells are regulated by a number of soluble factors such as cytokines and hormones as well as intercellular interactions through the cell surface antigens. Cytokines bind to their cognate cell‐surface receptors, which immediately activate intracellular cascades either through their intrinsic enzymatic activity or their association with other catalytic proteins. Then, the activated intracellular downstream signaling cascades control the transcription of their effector genes directly or indirectly. The loss of function of this signaling leads to the failure of hematopoiesis. Conversely, aberrantly elevated and/or sustained activation of these signals can be a causative event of hematologic malignancies.( 1 , 2 , 3 ) Among them, considerable attention has been paid to oncogenic receptor tyrosine kinases (RTKs). These include receptors for macrophage colony‐stimulating factor (c‐FMS or CSF‐1R), FLT3, stem cell factor (c‐KIT), and platelet‐derived growth factor (PDGF), which are also involved in normal hematopoiesis. Here, we will briefly review the recent findings on the mechanisms of aberrant activation of RTKs and their roles in the development of hematological malignancies. Also, we will focus on several new agents that target these oncogenic RTKs and their downstream molecules for the treatment of hematologic malignancies.
Structures and classification of RTKs
Receptor tyrosine kinases are membrane‐bound enzymes composed of a characteristic extracellular ligand‐binding domain, a transmembrane domain, a highly conserved intracellular kinase domain, and a C‐terminal tail. RTKs include 20 subfamilies: class I including epidermal growth factor receptor; class II including insulin‐like growth factor‐1 (IGF1) receptor; class III including PDGF receptor (PDGFR), c‐FMS, c‐KIT, and FLT3R; and class IV including fibroblast growth factor receptor (FGFR). Among them, type III RTKs, which are characterized by five immunoglobulin‐like extracellular domains and two intracellular kinase domains separated by the kinase insert, are frequently mutated and involved in the pathogenesis of hematopoietic malignancies (Fig. 1).( 2 , 3 )
Figure 1.
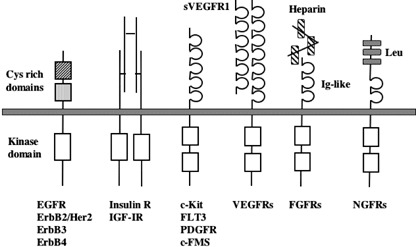
Structures and classification of receptor tyrosine kinases.
Mechanisms of activation of RTKs
Generation of fusion genes by chromosomal translocations. Reciprocal chromosomal translocations involving RTKs are observed in several types of hematologic malignancies. Among RTKs, PDGFR and FGFR1 are the frequent targets of this type of activation (Table 1). For example, TEL(ETV6)‐PDGFRβ is formed in t(5;12)(q31;p12), Huntington interacting protein‐1 (HIP1)‐PDGFRβ in t(5;7)(q33;q11.2), H4/D10S170‐PDGFRβ in t(5;10)(q33;q11.2), Rabaptin‐5‐PDGFRβ in t(5;17)(q33;p13), and CEV14‐PDGFRβ in t(5;14)(q33;q32).( 2 , 3 ) The fusion partners are completely unrelated, but commonly possess the motifs, which are assumed to be utilized for the ligand‐independent homodimerization of each RTK (Fig. 2). Most patients with the PDGFRβ rearrangement reveal common clinical features resembling chronic myelogenous leukemia (CML) or chronic myelomonocytic leukemia (CMML), which are characterized by leukocytosis accompanied by marked eosinophilia, a variable degree of monocytosis, and splenomegaly. Transformation to acute leukemia occurs in only a minority of these patients with a highly variable latent period from 9 months to 12 years. Exceptionally, CEV14‐PDGFRβ caused by t(5;14) (q33;q32) has been reported to be associated with acute myeloid leukemia (AML) relapse.( 4 ) In addition to PDGFRβ, PDGFRα is also involved in chromosomal translocation. FIP1L1‐PDGFRα generated by the interstitial deletion at 4q12 was reported to act as a causative gene for hypereosinophilic syndrome (HES)/chronic eosinophilic leukemia (CEL).( 5 ) Moreover, a recent paper identified novel fusion genes, KIF5B‐PDGFRα from t(4;10)(q12;p11), STRN‐PDGFRα from t(2;4)(p24;q12), and ETV6‐PDGFRα from t(4;12)(q2?3;p1?2) in patients with CEL.( 6 , 7 ) Meanwhile, a rare variant translocation t(4;22)(q12;q11) yielding BCR‐PDGFRα was detected in a patient with CML‐like myeloproliferative disorder (MPD).( 8 ) On the other hand, the rearrangements of FGFR1 generated by chromosomal translocations involving 8p11 cause MPDs with common clinical features characterized by splenomegaly and marked eosinophilia.( 3 , 9 ) So, these MPDs are now recognized as a distinct disease entity referred to as ‘8p11 myeloproliferative syndrome (EMS)’ or ‘stem cell leukemia–lymphoma syndrome (SCLL)’.( 9 ) The most frequent type of chromosomal translocation is t(8;13)(p11;q12), which yields the ZNF198‐FGFR1 fusion product. Other fusion genes are FOP‐FGFR1 caused by t(6;8)(q27;p11), CEP110‐FGFR1 by t(8;9)(p11;q33), and BCR‐FGFR1 by t(8;22)(p11;q22). Among EMS, patients with BCR‐FGFR1 translocation have clinical and morphological characteristics very similar to typical, BCR‐ABL‐positive CML. In addition, t(4;14)(p16.3;q32.3) leading to the overexpression of FGFR3 was identified in approximately 15% of multiple myeloma patients and cell lines.( 10 ) In some cases, the translocated FGFR3 gene contains an activating mutation K650E. FGFR3 is also involved in the t(4;12)(p16;p13)‐associated peripheral T cell lymphoma (PTCL) that progresses into AML, which generates the TEL‐FGFR3 fusion protein with constitutive tyrosine kinase activity.( 11 )
Table 1.
Chromosomal translocations involving receptor tyrosine kinases
| Genetic abnormality | Chromosomal translocation | Disease type |
|---|---|---|
| TEL‐PDGFRβ | t(5;12)(q31;p12) | Atypical CML/CMML |
| HIP1‐PDGFRβ | t(5;7)(q33; q11.2) | |
| H4‐PDGFRβ | t(5;10)(q33; q11.2) | |
| Rabaptin‐5‐PDGFRβ | t(5;17)(q33;p13) | |
| CEV14‐PDGFRβ | t(5;14)(q33;q32) | AML relapse |
| FIP1L1‐PDGFRα | Interstitial deletion at 4q12 | CEL |
| KIF5B‐PDGFRα | t(4;10)(q12;p11) | |
| STRN‐PDGFRα | t(2;4)(p24;q12) | |
| ETV6‐PDGFRα | t(4;12)(q2?3; p1?2) | |
| BCR‐PDGFRα | t(4;22)(q12;q11) | CML‐like MPD |
| ZNF198‐FGFR1 | t(8;13)(p11;q12) | 8p11 myeloproliferative syndrome (EMS)/stem cell leukemia–lymphoma syndrome (SCLL) |
| FOP‐FGFR1 | t(6;8)(q27;p11) | |
| CEP110‐FGFR1 | t(8;9)(p11;q33) | |
| BCR‐FGFR1 | t(8;22)(p11;q22) | |
| FGFR3 overexpression | t(4;14)(p16.3; q32.3) | Multiple myeloma |
| TEL‐FGFR3 | t(4;12)(p16;p13) | PTCL?AML |
CML, chronic myelogenous leukemia; CMML, chronic myelomonocytic leukemia; AML, acute myeloid leukemia; CEL, chronic eosinophilic leukemia; MPD, myeloproliferative disorder; PTCL, peripheral T cell lymphoma.
Figure 2.
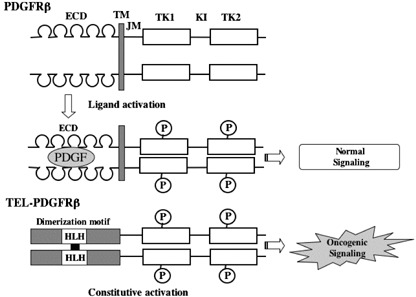
Structures of wild type and chimeric platelet‐derived growth factor receptors (PDGFRs). The representative fusion product of ETV6‐PDGFRβ is shown. Helix‐loop‐helix domain (HLH) in the TEL mediates the dimerization of fused PDGFRβ. ECD, extracellular domain; TM, transmembrane domain; JM, juxtamembrane domain; TK1, tyrosine kinase 1 domain; TK2, tyrosine kinase 2 domain; KI, kinase insert.
Juxtamembrane domain mutation. The juxtamembrane (JM) domain of RTKs interacts with N‐ and C‐terminal kinase lobes and prevents the activation loop from adopting active configuration, thereby serving as a negative regulatory domain. The mutations of this region including a missense point mutation, a deletion, and an insertion have been found in c‐Kit, Flt3 and PDGFRβ genes (Fig. 3). The JM mutations of c‐Kit were first identified in mast cell lines: a point mutation (Val560Gly) in a human mast cell leukemia line, HMC‐1; the deletion of seven amino acids (ΔThr573–His579) in a murine mastocytoma cell line (FMA3).( 12 , 13 ) In addition, JM c‐Kit mutations were detected in canine mast cell tumors, one of the most popular and aggressive neoplasms in dog.( 14 ) Subsequently, similar JM mutations of c‐Kit were found to be present in 50–80% of cases of gastrointestinal stromal tumors (GISTs).( 15 , 16 ) Furthermore, JM domain mutations of PDGFRα including the missense point mutation, insertion, and deletion, were identified in the GISTs without c‐Kit mutations.( 17 )
Figure 3.
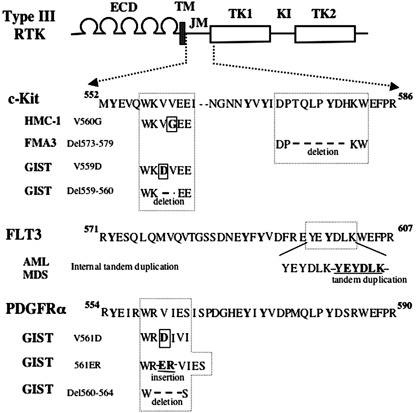
Juxtamembrane domain (JM) mutations of type III receptor tyrosine kinases (RTKs). Representative mutations of type III RTKs, c‐Kit, Flt3 and platelet‐derived growth factor receptor β (PDGFRβ) are shown. Conserved amino acid sequence around the mutated regions are depicted. Conserved tyrosine residues are written in bold. Internal tandem duplications of FLT3 are tandem insertion of various length of amino acid sequences. The mutations of PDGFRα are found in GISTs without c‐Kit mutations. HMC‐1, a human mast cell leukemia cell line; FMA3, a murine mastocytoma cell line; GIST, gastrointestinal stromal tumor; AML, acute myeloid leukemia.
The mutations of the FLT3 genes are in‐frame internal tandem duplications (ITDs) involving the JM and adjacent N‐terminal kinase domain, which result in the duplication of a stretch of several amino acids.( 18 ) ITD is detectable in about 20% of AML cases covering all subtypes in FAB classification and in 10% of myelodysplastic syndrome (MDS) cases, while it is quite rare in acute lymphoblastic leukemia (ALL). In addition, FLT3‐ITD has been shown to be a poor prognostic factor for AML cases in several studies.( 3 , 19 ) To clarify the mechanism of aberrant activation of RTKs caused by the JM domain mutations, Ma and Irusta made alanine‐scanning mutations in the JM domain of c‐Kit and PDGFRβ, respectively.( 20 , 21 ) Consequently, they found that the alanine mutation at the structurally corresponding residues in c‐Kit and PDGFRβ induced the constitutive activation. Based on the structural analysis made by Irusta, the JM domain was supposed to form a short α‐helix followed by a three‐stranded β‐sheet very similar to known structures of WW domains. Furthermore, they found that activation mutations were clustered in the central portions of the first and second β strands and along one face of the β‐sheet. These results suggest that various mutations such as point mutations, deletion, and insertion would invalidate the auto‐inhibitory effect of the JM domain through the disruption of the WW structure. Previous papers including ours showed that both c‐Kit and FLT3 carrying the JM domain mutations constitutively form a homodimer via the extracellular and juxtamembrane domains.( 22 , 23 ) Together, it was speculated that JM mutations would induce the conformational change facilitating the receptor dimerization, which results in the constitutive activation of RTKs.
Tyrosine kinase domain (TKD) point mutations. TKD point mutations of the c‐Kit, FLT3 and PDGFR genes are commonly observed at the Asp residue in the activation loop (Fig. 4). This type of mutation was first identified by us as Asp816Val in a human mast cell leukemia line, HMC‐1, which also carried the JM domain mutation.( 12 ) Subsequently, the mutations of c‐Kit at the same site (Asp816→Val, Tyr, Phe, His) were found in patients with aggressive mastocytosis, MDS, and MPDs associated with mastocytosis.( 24 , 25 , 26 ) Furthermore, the same mutations have been detected in a subset of AML patients,( 27 , 28 ) especially in about 10% of AML cases called CBF AML, in which the AML1(Runx1) complex is deregulated by chromosomal translocations such as t(8;21) and inv(16). Although patients with CBF leukemia commonly have a good prognosis, the concurrent TKD mutation of c‐Kit makes their prognosis poor.( 27 , 28 ) FLT3 also has TKD point mutations at Asp835 to Tyr, Val, or various amino acids, which is found in 7% of AML patients and in some patients with ALL or MDS.( 29 , 30 ) In addition, several novel alterations of FLT3 TKD such as a point mutation at codon 840 or 841, a small deletion yielding Δ835 or Δ836, and a 6‐bp of small insertion between the codon 840 and 841 were identified in AML patients.( 31 , 32 ) Of note, FLT3 TKD mutations are biologically distinct from and have a significantly more favorable prognosis than FLT‐ITD in patients with AML.( 32 ) Furthermore, the activation loop mutations of PDGFRα were found in patients with GIST without c‐Kit mutations.( 17 ) The activation mechanism of the TKD mutations has been extensively analyzed using the c‐Kit mutants. We previously showed that c‐Kit is constitutively activated by the substitution of Asp814 not only into Val and Tyr, but also into a wide variety of amino acids,( 33 ) suggesting that Asp814 would play a crucial role in regulating enzymatic activity of c‐Kit. Although the JM domain mutations induced constitutive dimerization via the extracellular domain, the TKD mutant does not show a similar reaction. By generating c‐Kit mutants lacking the extracellular domain, we demonstrated that TKD mutants can dimerize independently of the extracellular domain.( 34 ) Furthermore, using a series of c‐Kit mutants carrying Tyr→Phe conversion of the intracellular 20 tyrosine residues or C‐terminal deletion, we found that Tyr719Phe mutation and C‐terminal 70 amino acids deletion abolished kinase activity of c‐Kit.( 35 ) Although the C‐terminal deletion disrupted kinase activity of the wild type c‐Kit, Tyr719Phe did not, suggesting that Tyr719 is specifically required for the activation of the c‐Kit TKD mutant. Since Tyr719 is the binding site for the p85 subunit of PI3‐K, it is postulated that the binding between Tyr719 of c‐Kit and the p85 subunit is necessary for the c‐Kit TKD mutant to form an active configuration. Recently, the crystal structure analysis of the RTK revealed that the activation loop in the wild‐type RTK is oscillating between an active and inactive conformation, which is shifted to an active form when tyrosine residues in the loop are phosphorylated. So, it is speculated that activation loop mutations would evoke a conformational change similar to that caused by the tyrosine phosphorylation in the loop, thereby inducing constitutive activation of RTKs.
Figure 4.

Tyrosine kinase domain (TKD) mutations of type III receptor tyrosine kinases. Mutations of activation loop in TKD involved the conserved aspartate residue among c‐Kit, FLT3 and growth factor receptor α (PDGFRα). PDGFRα mutations are found in gastrointestinal stromal tumors without c‐Kit mutations.
Extracellular domain (ED) mutations. ED mutations in c‐Kit, c‐FMS and FGFR3 have been identified in several hematological malignancies. c‐Kit mutations in the ED were located in the exon 8, causing the in‐frame deletion plus insertion with consistent loss of Asp419. Gari et al. reported that this mutation was associated with AML harboring a mutation in either subset of CBF AML; 7/21 cases (33%) with inv(16) and 1/19 cases (5.2%) with t(8;21).( 36 ) In agreement with their findings, Care et al. detected the same mutation in 15/63 cases (23.8%) with inv(16) and 1/22 cases (4.5%) with t(8;21).( 37 ) In addition, Boell et al. found the c‐Kit mutation in the exon 8 in 10/46 cases (22%) with inv(16) and 6/50 cases (12%) with t(8;21).( 38 ) Similarly, the ED mutations of c‐FMS at Leu301 located in the fourth immunoglobulin domain were found in acute myelomonocytic leukemia and MDS.( 39 , 40 ) Although c‐Kit mutants harboring the ED mutations have not been verified to be constitutively active, the ED mutants of c‐FMS was proved to be constitutively active and oncogenic.( 41 ) Since the fourth Ig‐like domain is implicated in the stabilization of ligand‐induced dimerization, these mutations are supposed to induce a conformational shift facilitating dimerization and activation. The ED mutations of FGFR3 involving Arg248 or Tyr373 were identified in a small subset of multiple myeloma. FGFR possesses three Ig‐like domains, and the first Ig‐like domain interacts with the ligand binding domain in the second and third Ig‐like domains.( 42 ) This intramolecular interaction is assumed to prevent the accidental activation of FGFR by the ligand, serving as an autoinhibition mechanism. So, the ED mutation of FGFR was supposed to disturb this autoinhibitory mechanism, resulting in the constitutive activation.
Signal transduction from RTKs
Upon the ligand‐binding to the extracellular domain, RTK dimerizes and induces transphophorylation of tyrosine residues in the cytoplasmic domains, which serve as docking sites for several adaptor molecules harboring SH2 domain or PTB (phosphotyrosine binding domain). These adaptor molecules recruit and activate downstream signaling molecules such as Ras/MAPK, PI3‐Kinase, phospholipase C‐γ, JNK, STATs, NF‐κB pathways through tyrosine‐ or serine/threonine‐phosphorylations (Fig. 5). Among them, Ras/MAPK, PI3‐K and STAT pathways act as the major oncogenic signaling pathways.( 1 , 2 , 3 )
Figure 5.
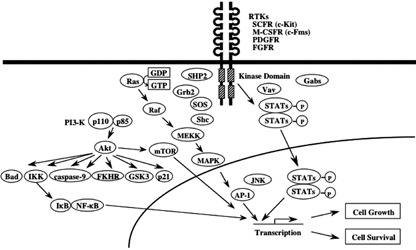
Signal transduction from receptor tyrosine kinases (RTKs). Both ligand activated RTKs and oncogenic RTKs activate a common set of signaling molecules such as Ras/MAPK, PI3‐K/Akt, and STATs. However, their signals are supposed to be quantitatively and/or qualitatively different.
Ras family of proteins belongs to the large superfamily of GTPases that localize to the inner surface of the plasma membrane.( 43 , 44 ) Ras proteins play a pivotal role in a number of signaling pathways mediated by RTKs. Ligand‐activated and autophosphorylated RTKs create phosphotyrosyl‐binding sites for adapter proteins such as Shc and Grb2, which in turn recruit guanine nucleotide exchange factors (GEFs), thereby inducing Ras activation. Once induced, Ras activates Raf serine/threonine kinase, which then phosphorylates MAPK kinases (otherwise known as MEKs) (Fig. 5). In addition to Ras, PKC is a potent activator of Raf‐1/MAPK cascade, whereas its role as a downstream effector of RTKs has not been clearly demonstrated. Activated MAPKs (or extracellular signal‐regulated kinases [ERKs]), move into the nucleus and phosphorylate and activate nuclear transcription factors such as Elk‐1. ERKs can also activate other kinases such as RSKs (also known as MAPK‐activated protein kinases), which regulate cell‐cycle regulation and apoptosis. ERK‐activated RSK kinase catalyzes the proapoptotic Bcl‐2 family protein Bad, thereby inhibiting Bad‐mediated apoptosis. Furthermore, the Ras‐Raf‐MEK‐ERK cascade modulates cellular proliferation by regulating the expression and activity of several proteins, including cell‐cycle regulators (e.g. cyclin D1, p21WAF( 1 ) p27KIP( 1 ) and cdc25A) and transcription factors (e.g. c‐fos, c‐jun, and c‐myc). Aberrant MEK and ERK activity has been demonstrated in AML and CML cells.( 1 , 3 , 43 , 44 ) As for the roles of Ras/MAPK in the growth and survival of hematopoietic cells, we and other groups previously showed that dominant‐negative Ras and a chemical inhibitor of MAPK blocked proliferation and survival of hematopoietic cells mediated by oncogenic RTKs, respectively, indicating the importance of the Ras/MAPK signaling in the RTK‐dependent malignant transformation of hematopoietic cells.( 45 , 46 )
PI3‐K is another important signaling pathway controlling serine/threonine phosphorylation.( 1 , 3 ) PI3‐K consists of two subunits, the p85 regulatory subunit and the p110 catalytic subunit (Fig. 5). The p85 subunit binds to the ligand‐activated and autophosphorylated RTKs. As a result, the p110 subunit and their downstream substrate Akt (also called PKB) are recruited to the membrane. PI3‐K/Akt pathway activates several downstream targets including p70 RSK, forkhead transcription factors (FOXOs), and NF‐κB. The serine/threonine kinase Akt is an important component of the cell survival machinery. PI3‐K‐activated Akt provokes a number of signaling events. For example, Akt phosphorylates an NF‐κB inhibitor, IκB. Upon phosphorylation, IκB is degraded by 26S proteasome and releases NF‐κB, which then moves into the nucleus and induces a number of target genes involved in cell survival such as Bcl‐XL and IAPs. Akt also phosphorylates the proapoptotic protein Bad, which leads to higher levels of free anti‐apoptotic Bcl‐XL, thereby inhibiting the cell‐death protease, caspase‐9. A tumor suppressor, PTEN, is a phosphatase that removes a phosphate from the 3 position of the inositol ring of the PIP3,4,5 phospholipids. PTEN has been shown to act as a negative regulator for Akt through its phosphatase activity. Furthermore, the mammalian target of rapamycin (mTOR) is known to be another important downstream effector of the PI3‐K/Akt signaling pathway, which mediates cell survival and proliferation as a serine/threonine kinase. With regard to the biologic roles of PI3‐K/Akt in hematologic malignancies, we previously showed that tyrosine 719 in c‐Kit, which is utilized for the PI3‐K‐binding, is essential for the transforming activity of the TKD mutant of c‐Kit by exchanging tyrosine residues in the cytoplasmic domain to phenylalanine (as described in the above section).( 35 , 47 ) However, since KITWT–Tyr719Phe knock‐in mouse did not show an apparent abnormality in hematopoiesis, it was speculated that PI3‐K signaling is dispensable for normal hematopoiesis under physiologic conditions.( 48 , 49 )
STATs are coded by six known mammalian genes and include 10 different proteins including different isomers of STAT1, 3, 4, and 5.( 50 , 51 ) Like other transcription factors STATs have a well‐defined structure including a DNA‐binding domain, a conserved NH2‐terminal domain, a COOH‐terminal transactivation domain, and SH2 and SH3 domains. Upon tyrosine phosphorylation by upstream TKs, activated STATs dimerize and translocate into the nucleus, where they activate specific target genes (Fig. 5). A number of previous studies have shown that STATs mediate cytokine‐dependent cell growth and survival by regulating the expression of cyclins, c‐myc, and Bcl‐XL. Besides the normal signaling from ligand‐activated cytokine receptors, the TKD mutant of c‐Kit induces constitutive activation of STAT3 and STAT1 in a ligand‐independent manner.( 52 , 53 ) TEL‐PDGFRβ can activate STAT1 and STAT5 through JAK‐independent pathway in BaF3 cells, while wild‐type PDGFRβ can scarcely activate these STATs.( 54 , 55 ) FLT‐ITD also activates STAT3 and STAT5 constitutively, which is far more effective than ligand‐activated wild type FLT3.( 45 , 46 ) Thus, the aberrant activation of STAT is the common characteristic of constitutive active RTKs. Regarding the biologic functions of STATs as downstream signaling molecules of oncogenic RTKs, it was shown that dominant‐negative STAT3 suppressed the growth and survival mediated by oncogenic c‐Kit.( 52 ) Also, we previously showed that dominant negative STAT5 inhibited the growth of 32D cells transformed by FLT‐ITD as efficiently as dominant negative Ras.( 46 ) These results suggest that STATs would play a crucial role in malignant transformation caused by oncogenic RTKs. In order to identify the target genes activated by FLT3‐ITD, we previously compared the mRNA expression profile between the cells expressing FLT3‐ITD and those expressing wild type FLT3 by a microarray analysis.( 56 ) Consistent with the previous observation that STATs are aberrantly activation by FLT3‐ITD, several known target genes of STATs were specifically induced by FLT‐ITD. Among STAT3/5 target genes, we found that Pim‐2 mRNA was induced by FLT3‐ITD. In addition, the inhibition of Pim‐2 activity by the kinase‐dead Pim‐2 mutant suppressed the transforming activity of FLT3‐ITD. These results suggest that the STAT5/Pim‐2 cascade alone may be sufficient to execute the biologic function of FLT3‐ITD, whereas other molecules can also contribute to this process. As for other STAT3/5 target genes, we found that SOCS2 and SOCS3 were also specifically induced by FLT3‐ITD. Since SOCS family proteins can act as a negative regulator of JAK/STAT pathways, this result may indicate that the negative feedback system still works even in the cells transformed by oncogenic RTKs. However, as we previously found that the SOCS1 gene was silenced by the hypermethylation in its promoter region in about 70% of AML cases, this result may indicate that this SOCS‐mediated anti‐oncogenic system may be disrupted in more advanced stage or in some type of hematologic malignancies.( 57 ) Other interesting findings from the microarray analysis of FLT3‐ITD are the repressed expression of transcription factors, C/EBPα and PU.1, which are critical mediators of myeloid differentiation. Because loss of function mutations of C/EBPα or PU.1 have been found in AML, our results suggest that FLT3‐ITD would work as both a differentiation blocker and an augmenter of proliferation and survival.
New drugs targeting oncogenic RTKs and their downstream molecules
As described above, aberrant activation of RTKs and their downstream molecules are involved in the pathogenesis of various hematologic malignancies, suggesting that these molecules are particularly attractive targets for therapy.( 1 , 2 , 3 )
Imatinib originally synthesized to inhibit activity of BCR‐ABL was found to inhibit that of PDGFR and c‐Kit more efficiently. In fact, imatinib has been shown to be effective on GIST carrying the mutations of c‐Kit or PDGFR, and its clinical use is now admitted. In addition, imatinib has been shows to be effective on CMML patients with t(5;12)(q33;p13) yielding TEL‐PDGFRβ and HES/CEL patients with the interstitial deletion at 4q12 yielding FIP1L1‐PDGFRα.( 5 , 58 )
Also, several FLT3 inhibitors, such as MLN518, PKC412, CEP701, and SU11248, have been shown to be selectively cytotoxic to AML blasts harboring the mutation either FLT‐ITD or FLT‐TKD in vitro.( 59 , 60 ) However, in several clinical trails, the effects of these agents were observed in a minority of patients and were only transient. So, the development of a more potent new drug is required to control deregulated FLT3 activity in AML patients.
Inhibition of Ras signal transduction has been the subject of intense research. Ras activity can be inhibited by preventing its membrane localization.( 43 , 44 ) Farnesyltransferase, which induces the COOH‐terminal prenylation of Ras, is the key enzyme responsible for membrane localization of Ras as well as geranylgeranyltransferase I that regulates geranylgeranylation. So, several farnesyltransferase inhibitors (FTIs) have already been developed, and clinical trials for cancers including hematologic malignancies are now under way.( 43 , 44 ) Among them, two FTIs, R115777 and SCH66336, have been reported to be effective against several hematologic malignancies such as AML, high risk MDS, imatinib‐resistant CML, acute or blastic phase of CML, and multiple myeloma, alone or in combination with other drugs. However, when considering the pharmacological effects of FTIs, it should be kept in mind that FTIs also exert their anti‐oncogenic effects through several Ras‐independent mechanisms.( 61 ) Compounds such as geldanamycin and derivatives of radicicol destabilize Raf protein and interfere with Raf signaling.( 1 , 3 ) Several staurosporine derivatives such as UCN‐O1, CGP41251, and PKC412 can inhibit PKC and MAPK signaling, and have been examined in preclinical and clinical studies.( 62 ) Also, MEK inhibitors such as PD098059, PD184352, and UO126 are able to modulate cellular proliferation, differentiation, and apoptosis. PD184352 is now subjected to Phase I trials.( 63 )
Pharmacological inhibitors of PI3‐K, wortmannin, and LY294002 have shown to be effective in preclinical studies.( 64 ) In addition, rapamycin (sirolimus), the prototypic mTOR inhibitor, exhibits anti‐oncogenic activity on AML cells. Three rapamycin analogs, temsirolimus, everolimus, and AP23573, are in clinical trials for various hematologic malignancies: temsirolimus for mantle cell lymphoma, AP23573 for acute leukemia, and everolimus for lymphoma (Hodgkin and non‐Hodgkin) and multiple myeloma.( 65 ) In addition, perifosine, an inhibitor of Akt that exhibited considerable anti‐oncogenic activity on multiple myeloma, is being examined in relapsed multiple myeloma.
Future perspectives
Over the past decade, extensive efforts have been made to elucidate the role of cellular signaling pathways in cellular growth, differentiation, apoptosis, and malignant transformation, which enabled us to establish molecular targeted therapeutics. However, these drugs still have considerable adverse effects on normal tissues and their effects were not satisfactory at all in terms of curing the malignancies. So, we have to further understand the complex pathophysiology of each disease. Also, based on these findings, we have to devise the agents that specifically target the malignant cells without spoiling normal tissues. These attempts would truly realize the concept of molecular‐targeted therapeutics and shed light on patients with hematologic malignancies.
References
- 1. Ravandi F, Talpaz M, Estrov Z. Modulation of cellular signaling pathways: prospects for targeted therapy in hematological malignancies. Clin Cancer Res 2003; 9: 535–50. [PubMed] [Google Scholar]
- 2. Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med 2005; 353: 172–87. [DOI] [PubMed] [Google Scholar]
- 3. Mizuki M, Ueda S, Matsumura I et al . Oncogenic receptor tyrosine kinase in leukemia. Cell Mol Biol 2003; 49: 907–22. [PubMed] [Google Scholar]
- 4. Abe A, Emi N, Tanimoto M et al . Fusion of the platelet‐derived growth factor receptor beta to a novel gene CEV14 in acute myelogenous leukemia after clonal evolution. Blood 1997; 90: 4271–7. [PubMed] [Google Scholar]
- 5. Cools J, DeAngelo DJ, Gotlib J et al . A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 48: 1201–14. [DOI] [PubMed] [Google Scholar]
- 6. Curtis CE, Grand FH, Musto P et al . Two novel imatinib‐responsive PDGFRA fusion genes in chronic eosinophilic leukaemia. Br J Haematol 2007; 138: 77–81. [DOI] [PubMed] [Google Scholar]
- 7. Score J, Curtis C, Waghorn K et al . Identification of a novel imatinib responsive KIF5B‐PDGFRA fusion gene following screening for PDGFRA overexpression in patients with hypereosinophilia. Leukemia 2006; 20: 827–32. [DOI] [PubMed] [Google Scholar]
- 8. Safley AM, Sebastian S, Collins TS et al . Molecular and cytogenetic characterization of a novel translocation t (4;22) involving the breakpoint cluster region and platelet‐derived growth factor receptor‐alpha genes in a patient with atypical chronic myeloid leukemia. Genes Chromosomes Cancer 2004; 40: 44–50. [DOI] [PubMed] [Google Scholar]
- 9. Cross NC, Reiter A. Tyrosine kinase fusion genes in chronic myeloproliferative diseases. Leukemia 2002; 16: 1207–12. [DOI] [PubMed] [Google Scholar]
- 10. Chesi E, Nardini LA, Brents E et al . Frequent translocation t (4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet 1997; 16: 260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yagasaki D, Wakao Y, Yokoyama Y et al . Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T‐cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation, Cancer Res 2001; 61: 8371–4. [PubMed] [Google Scholar]
- 12. Furitsu T, Tsujimura T, Tono T et al . Identification of mutations in the coding sequence of the proto‐oncogene c‐kit in a human mast cell leukemia cell line causing ligand‐independent activation of c‐kit product. J Clin Invest 1993; 92: 1736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsujimura T, Morimoto M, Hashimoto K et al . Constitutive activation of c‐kit in FMA3 murine mastocytoma cells caused by deletion of seven amino acids at the juxtamembrane domain. Blood 1996; 87: 273–83. [PubMed] [Google Scholar]
- 14. London CA, Galli SJ, Yuuki T. et al . Spontaneous canine mast cell tumors express tandem duplications in the proto‐oncogene c‐kit. Exp Hematol 1999; 27: 689–97. [DOI] [PubMed] [Google Scholar]
- 15. Hirota S, Isozaki K, Moriyama Y et al . Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–80. [DOI] [PubMed] [Google Scholar]
- 16. Nishida T, Hirota S, Taniguchi M. et al . Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998; 19: 323–4. [DOI] [PubMed] [Google Scholar]
- 17. Heinrich MC, Corless CL, Duensing A et al . PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299: 708–10. [DOI] [PubMed] [Google Scholar]
- 18. Nakao M, Yokota S, Iwai T et al . Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996; 10: 1911–8. [PubMed] [Google Scholar]
- 19. Kiyoi H, Naoe T, Nakano Y et al . Prognostic implication of FLT3 and N‐RAS gene mutations in acute myeloid leukemia. Blood 1999; 93: 3074–80. [PubMed] [Google Scholar]
- 20. Irusta PM, Luo Y, Bakht O et al . Definition of an inhibitory juxtamembrane WW‐like domain in the platelet‐derived growth factor beta receptor. J Biol Chem 2002; 277: 38627–34. [DOI] [PubMed] [Google Scholar]
- 21. Ma Y, Cunningham ME, Wang X et al . Inhibition of spontaneous receptor phosphorylation by residues in a putative alpha‐helix in the KIT intracellular juxtamembrane region. J Biol Chem 1999; 274: 13399–402. [DOI] [PubMed] [Google Scholar]
- 22. Kitayama H, Kanakura Y, Furitsu T et al . Constitutively activating mutations of c‐kit receptor tyrosine kinase confer factor‐independent growth and tumorigenicity of factor‐dependent hematopoietic cell lines. Blood 1995; 85: 790–8. [PubMed] [Google Scholar]
- 23. Kiyoi H, Towatari M, Yokota S et al . Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998; 12: 1333–7. [DOI] [PubMed] [Google Scholar]
- 24. Longley BJ Jr, Metcalfe DD, Tharp M et al . Activating and dominant inactivating c‐KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA 1999; 96: 1609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nagata H, Worobec AS, Oh CK. et al . Identification of a point mutation in the catalytic domain of the protooncogene c‐kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA 1995; 92: 10560–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Worobec AS, Semere T, Nagata H et al . Clinical correlates of the presence of the Asp816Val c‐kit mutation in the peripheral blood mononuclear cells of patients with mastocytosis. Cancer 1998; 83: 2120–9. [DOI] [PubMed] [Google Scholar]
- 27. Carre RS, Goodeve A, Abu‐Duhier FM et al . Incidence and prognosis of c‐kit and Flt3 mutations in core binding factor (CBF) acute myeloid leukemia. Blood 2002: 746a. [DOI] [PubMed]
- 28. Zwaan CM, Miller M, Goemans BF et al . Frequency and clinical significance of c‐kit exon 17 mutations in childfood acute myeloid leukemia. Blood 2002: 746a.
- 29. Abu‐Duhier FM, Goodeve AC, Wilson GA et al . Identification of novel FLT‐3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol 2001; 113: 983–8. [DOI] [PubMed] [Google Scholar]
- 30. Yamamoto Y, Kiyoi H, Nakano Y et al . Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001; 97: 2434–9. [DOI] [PubMed] [Google Scholar]
- 31. Spiekermann K, Bagrintseva K, Schoch C et al . A new and recurrent activating length mutation in exon 20 of the FLT3 gene in acute myeloid leukemia. Blood 2002; 100: 3423–5. [DOI] [PubMed] [Google Scholar]
- 32. Mead AJ, Linch DC, Hills RK et al . FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 2007; 110: 1262–70. [DOI] [PubMed] [Google Scholar]
- 33. Moriyama Y, Tsujimura T, Hashimoto K et al . Role of aspartic acid 814 in the function and expression of c‐kit receptor tyrosine kinase. J Biol Chem 1996; 271: 3347–50. [DOI] [PubMed] [Google Scholar]
- 34. Tsujimura T, Hashimoto K, Kitayama H et al . Activating mutation in the catalytic domain of c‐kit elicits hematopoietic transformation by receptor self association not at the ligand‐induced dimerization site. Blood 1999; 93: 1319–29. [PubMed] [Google Scholar]
- 35. Hashimoto K, Matsumura I, Tsujimura T et al . Necessity of tyrosine 719 and phosphatidylinositol 3′‐kinase‐mediated signal pathway in constitutive activation and oncogenic potential of c‐kit receptor tyrosine kinase with the Asp814Val mutation. Blood 2003; 101: 1094–102. [DOI] [PubMed] [Google Scholar]
- 36. Gari M, Goodeve A, Wilson G et al . c‐kit proto‐oncogene exon 8 in‐frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol 1999; 105: 894–900. [DOI] [PubMed] [Google Scholar]
- 37. Care RS, Valk PJ, Goodeve AC et al . Incidence and prognosis of c‐KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br J Haematol 2003; 121: 775–7. [DOI] [PubMed] [Google Scholar]
- 38. Boissel N, Leroy H, Brethon B et al . Incidence and prognostic impact of c‐Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF‐AML). Leukemia 2006; 20: 965–70. [DOI] [PubMed] [Google Scholar]
- 39. Ridge SA, Worwood M, Oscier D et al . FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc Natl Acad Sci USA 1990; 87: 1377–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tobal K, Pagliuca A, Bhatt B et al . Mutation of the human FMS gene (M‐CSF receptor) in myelodysplastic syndromes and acute myeloid leukemia. Leukemia 1990; 4: 486–9. [PubMed] [Google Scholar]
- 41. Roussel MF, Downing JR, Rettenmier CW et al . A point mutation in the extracellular domain of the human CSF‐1 receptor (c‐fms proto‐oncogene product) activates its transforming potential. Cell 1988; 55: 979–88. [DOI] [PubMed] [Google Scholar]
- 42. Plotnikov AN, Schlessinger J, Hubbard SR et al . Structural basis for FGF receptor dimerization and activation. Cell 1999; 98: 641–50. [DOI] [PubMed] [Google Scholar]
- 43. Lancet JE, Karp JE. Farnesyltransferase inhibitors in hematologic malignancies: new horizons in therapy. Blood 2003; 102: 3880–9. [DOI] [PubMed] [Google Scholar]
- 44. Morgan MA, Ganser A, Reuter CW. Therapeutic efficacy of prenylation inhibitors in the treatment of myeloid leukemia. Leukemia 2003; 17: 1482–98. [DOI] [PubMed] [Google Scholar]
- 45. Hayakawa F, Towatari M, Kiyoi H. et al . Tandem‐duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL‐3‐dependent cell lines. Oncogene 2000; 19: 624–31. [DOI] [PubMed] [Google Scholar]
- 46. Mizuki M, Fenski R, Halfter H et al . Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000; 96: 3907–14. [PubMed] [Google Scholar]
- 47. Chian R, Young S, Danilkovitch‐Miagkova A et al . Phosphatidylinositol 3 kinase contributes to the transformation of hematopoietic cells by the D816V c‐Kit mutant. Blood 2001; 98: 1365–73. [DOI] [PubMed] [Google Scholar]
- 48. Blume‐Jensen P, Jiang G, Hyman R et al . Kit/stem cell factor receptor‐induced activation of phosphatidylinositol 3′‐kinase is essential for male fertility. Nat Genet 2000; 24: 157–62. [DOI] [PubMed] [Google Scholar]
- 49. Kissel H, Timokhina I, Hardy MP et al . Point mutation in kit receptor tyrosine kinase reveals essential roles for kit signaling in spermatogenesis and oogenesis without affecting other kit responses. EMBO J 2000; 19: 1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rane SG, Reddy EP. JAKs, STATs and Src kinases in hematopoiesis. Oncogene 2002; 21: 3334–58. [DOI] [PubMed] [Google Scholar]
- 51. Darnell JE Jr. STATs and gene regulation. Science 1997; 277: 1630–5. [DOI] [PubMed] [Google Scholar]
- 52. Ning ZQ, Li J, Arceci RJ. Signal transducer and activator of transcription 3 activation is required for Asp (816) mutant c‐Kit‐mediated cytokine‐independent survival and proliferation in human leukemia cells. Blood 2001; 97: 3559–67. [DOI] [PubMed] [Google Scholar]
- 53. Ning ZQ, Li J, McGuinness M et al . STAT3 activation is required for Asp (816) mutant c‐Kit induced tumorigenicity. Oncogene 2001; 20: 4528–36. [DOI] [PubMed] [Google Scholar]
- 54. Sternberg DW, Tomasson MH, Carroll M et al . The TEL/PDGFbetaR fusion in chronic myelomonocytic leukemia signals through STAT5‐dependent and STAT5‐independent pathways. Blood 2001; 98: 3390–7. [DOI] [PubMed] [Google Scholar]
- 55. Wilbanks AM, Mahajan S, Frank DA et al . TEL/PDGFbetaR fusion protein activates STAT1 and STAT5: a common mechanism for transformation by tyrosine kinase fusion proteins. Exp Hematol 2000; 28: 584–93. [DOI] [PubMed] [Google Scholar]
- 56. Mizuki M, Schwable J, Steur C et al . Suppression of myeloid transcription factors and induction of STAT response genes by AML‐specific Flt3 mutations. Blood 2003; 101: 3164–73. [DOI] [PubMed] [Google Scholar]
- 57. Watanabe D, Ezoe S, Fujimoto M et al . Suppressor of cytokine signalling‐1 gene silencing in acute myeloid leukaemia and human haematopoietic cell lines. Br J Haematol 2004; 126: 726–35. [DOI] [PubMed] [Google Scholar]
- 58. Magnusson MK, Meade KE, Nakamura R et al . Activity of STI571 in chronic myelomonocytic leukemia with a platelet‐ derived growth factor β receptor fusion oncogene. Blood 2002; 100: 1088–91. [DOI] [PubMed] [Google Scholar]
- 59. Levis M, Tse KF, Smith BD et al . FLT3 tyrosine kinase inhibitor is selectively cytotoxic to acute myeloid leukemia blasts harboring FLT3 internal tandem duplication mutations. Blood 2001; 98, 885–7. [DOI] [PubMed] [Google Scholar]
- 60. Tse KF, Novelli E, Civin CI et al . Inhibition of FLT3‐mediated transformation by use of a tyrosine kinase inhibitor. Leukemia 2001; 15: 1001–10. [DOI] [PubMed] [Google Scholar]
- 61. Kurzrock R, Sebti SM, Kantarjian HM et al . Phase I Study of a Farnesyl Transferase Inhibitor. R115777, in Patients with Myelodysplastic Syndrome. Blood 2001; 98: 623a. [Google Scholar]
- 62. Propper DJ, McDonald AC, Man A et al . Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C. J Clin Oncol 2001; 19: 1485–92. [DOI] [PubMed] [Google Scholar]
- 63. Sebolt‐Leopold JS. Development of anticancer drugs targeting the MAP kinase pathway. Oncogene 2000; 19: 6594–9. [DOI] [PubMed] [Google Scholar]
- 64. Witzig TE, Kaufmann SH. Inhibition of the phosphatidylinositol 3‐kinase/mammalian target of rapamycin pathway in hematologic malignancies. Curr Treat Options Oncol 2006; 7: 285–94. [DOI] [PubMed] [Google Scholar]
- 65. Smolewski P. Recent developments in targeting the mammalian target of rapamycin (mTOR) kinase pathway. Anticancer Drugs 2006; 17: 487–94. [DOI] [PubMed] [Google Scholar]