

Abstract
The NADPH oxidase (Nox) family of enzymes generates reactive oxygen species (ROS). At low ROS concentration, intracellular signaling is initiated, whereas at high ROS concentration, oxidative stress is induced. The extensive studies over the years have shed light on the mediating roles of the Nox enzymes in a variety of normal physiological processes ranging from bactericidal activity to remodeling of the extracellular matrix. Consequently, imbalance of Nox activities could be the potential cause of acute or chronic diseases. With regard to functional relationships between Nox isoforms and pathogenesis, it is of particular interest to study whether they are involved in carcinogenesis, because overproduction of ROS has long been implicated as a risk factor in cancer development. We see one remarkable example of the causal relationship between Nox1 and cancer in Ras oncogene‐induced cell transformation. Other studies also indicate that the Nox family of genes appears to be required for survival and growth of a subset of human cancer cells. Thus, the Nox family will be a focus of attention in cancer biology and etiology over the next couple years. (Cancer Sci 2009)
In general, ROS have been considered to be accidentally generated by oxidative enzymes such as mitochondrial respiratory chains and damage to cells by non‐specifically oxidizing DNA, proteins, and lipids. In contrast, phagocytic Nox, whose catalytic subunit consists of a flavoprotein (gp91phox), represents one specifically regulated ROS generation system and its bactericidal activity plays a critical part in innate immunity.( 1 ) Studies over the last decade revealed the gene family of gp91phox homologs – so‐called Nox (Nox1–5 and Duox1 and 2)( 1 )– that produce ROS in various cells in response to stimuli including growth factors, cytokines, and calcium. Nox‐produced superoxide is rapidly converted to hydrogen peroxide, which potentially regulates the target molecules through reversible or irreversible oxidation of redox‐sensitive cysteine residues, depending on the heavy metal catalysts. This allows us to assume that Nox‐generated ROS, at least in part, behave as second messenger‐like molecules at low concentrations and elicit cellular signals as well as biochemical reactions. Aberrant levels of Nox‐derived ROS, whether excess or less, can perturb the balance of cellular homeostasis, ultimately resulting in pathological states. In fact, dysregulation of Nox‐dependent ROS generation is potentially associated with chronic diseases including atherosclerosis, hypertension, inflammation, cancer, and others.( 2 ) With regard to the relationships between ROS and carcinogenesis, it is far from obvious what molecular mechanism governs the action of Nox enzymes mediating the process of cellular transformation. Accordingly, this review focuses on some of the progresses made in studying the functional roles of Nox‐based oxidases in cancer development where overproduction of intracellular ROS is thought to increase the risk of cancer. More complete reviews about other aspects of Nox‐related diseases and Nox structure, function, and regulation are readily available.( 2 , 3 , 4 )
Nox enzymes and their regulation
In terms of structural basis, the Nox enzymes are classified into three distinct groups: Nox1–4, Nox5, and Duox. Nox1–4 contain the well‐conserved catalytic domain consisting of NADPH and FAD‐binding motifs in the cytoplasmic tail and the heme moiety that transfers electrons from NADPH to molecular oxygen in the extracellular or intraphagosome region( 4 ) (Fig. 1). In addition, these Nox proteins require membrane‐associated p22phox as their catalytic partners, whereas Nox5 and Duox do not.( 4 ) Superoxide generated by the Nox enzymes is rapidly converted to H2O2, which in turn diffuses into the cytosol. Nox5 possesses a calmodulin‐like domain with a high affinity for calcium in addition to the basic structure of the first group of Nox isozymes.( 5 ) Duox, referred to as ‘dual oxidases’, have a domain structure similar to that of Nox5, but possess an additional extracellular peroxidase homology domain in the amino terminus.( 6 , 7 )
Figure 1.
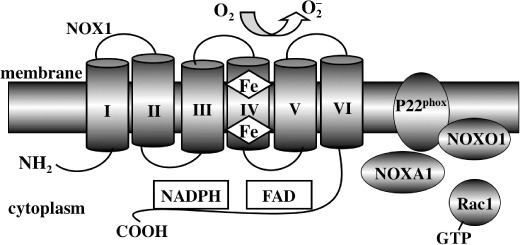
Schematic structure of NADPH oxidase (Nox) 1. Nox1 possesses an NH2‐terminal portion consisting of six transmembrane α‐helices with conserved histidine residues in helices III and V that provide binding sites for two hemes. The COOH‐terminal region of the molecule, facing the cytoplasm, contains the catalytic domain with the binding sites for co‐enzymes FAD and NADPH. Nox1 catalyzes the NADPH‐dependent reduction of oxygen to form superoxide, whereby the hemes transfer an electron from these coenzymes to molecular oxygen. p22phox is required as a catalytic partner for Nox1 and NOXO1 and NOXA1 function as critical regulators. Rac1 is involved in Nox1 activation through interaction with NOXA1. Other Nox isoforms have conserved transmembrane and catalytic domains similar to those of Nox1, while displaying differences in terms of their NH2‐terminal structure, regulatory components, and p22phox dependency.
Regulation of Nox2 involves the cytosolic subunits p47phox, p67phox, and p40phox as well as the small GTPase Rac1 (for macrophages) and Rac2 (for neutrophils).( 4 , 8 ) Activation of phagocytic cells by microorganisms or inflammatory stimuli converts the Rac1 • GDP to Rac1 • GTP, which in turn induces membrane translocation of the cytosolic components through the interaction of Rac1 with p67phox, forming the functional oxidase complexes. On the other hand, Nox1 is regulated by NOXO1 (a homolog of p47phox), NOXA1 (a homolog of p67phox), and Rac1 (Fig. 1).( 9 , 10 , 11 ) The actions of these cytosolic regulators are less clear in Nox3 regulation( 11 ) and are not involved in the activation of Nox4,( 12 ) Nox5,( 13 ) or Duox.( 14 )
Nox1 is abundantly expressed in colon tissues( 15 ) and is detected in intestinal cells stimulated by an inflammatory mediator, interferone (IFN)γ,( 16 ) and a TLR5 ligand, flagellin.( 17 ) Thus, Nox1 is postulated to play a role of mucosal host defense, analogous to phagocytic Nox2. On the other hand, Nox1 appears to be involved in mitogenic cell signaling by growth factors such as platelet‐derived growth factor (PDGF)( 15 ) and EGF( 18 ) in fibroblast cells. Increased expression of Nox1 in human colon cancer cells( 16 ) and actively growing normal intestinal crypt cells( 19 ) (M. Thanuja and T. Kamata, unpublished observation, 2009) also implicates Nox1 in cell growth control. The activity of Nox2, mainly expressed in phagocytic cells, is of critical importance in innate immunity such as microbicidal activity. Dysfunction of this phagocytic oxidase system results in chronic granulomatous disease, an immunodeficiency caused by an inability of phagocytes to kill microorganisms.( 20 )
Nox3 is expressed abundantly in the inner ear( 21 ) and plays an indispensable role in morphogenesis of ocotonia that are required for the perception of motion and gravity.( 22 ) High levels of Nox4 expression are found in distal tubules in the renal cortex.( 23 ) Nox4 participates in multiple signaling processes, including insulin receptor‐mediated glucose transport in adipocytes.( 24 ) Nox5 is expressed in human T and B lymphocytes of spleen and lymph nodes,( 5 ) but its biological role, to date, remains elusive. Duox2 mediates both lactoperoxidase‐dependent microbicidal activity in the airway epithelium( 25 ) and biosynthesis of thyroid hormone, which is supported by the finding that mutations in Duox2 lead to congenital hypothyroidism.( 26 ) Although we have barely scratched the surface of recently accumulated findings, we are already beginning to see the diverse array of structures and functions of the Nox isozymes. What role, if any, Nox oxidases exert in carcinogenesis will be discussed in the sections that follow.
Requirement of Nox1 for oncogenic Ras transformation
The Ras oncogene can transform various mammalian cells and its activation mutation is associated with a variety of malignant human cancers (approximately 30% of all human tumors). The mammalian Ras protein family comprises three isoforms (H‐Ras, K‐Ras, and N‐Ras) that are able to recycle between an active GTP‐bound form and an inactive GDP‐bound conformation. Although protooncogenic Ras proteins tightly regulate normal cell proliferation and differentiation through three major effector pathways such as Raf, phosphatidylinositol 3‐kinase, and RalGDS, oncogenic Ras proteins perturb these effector pathways, resulting in transformation phenotypes.( 27 ) Emerging evidence suggests that generation of ROS appears to be one alternative rate‐limiting factor for the full effect of Ras. Namely, constitutive production of superoxide was induced upon Ras transformation of NIH3T3 cells, and depletion of its dismutated metabolite H2O2 suppressed the unchecked growth of Ras‐transformed cells.( 28 ) Although the nature of the oxidase involved in this process has remained unclear for some time, the discovery of the Nox family opened a new avenue for exploration of the ROS‐mediated mechanism underlying Ras transformation. Mitsushita et al. found that transformation of NRK cells by K‐RasVal12 upregulated transcription of Nox1, and that EGF stimulation of cells also induced Nox1 mRNA transcription, suggesting that Nox1 expression is growth associated.( 18 ) Because a MEK inhibitor or its dominant negative mutant blocked induction of Nox1 expression by K‐RasVal12 or EGF, this indicates that oncogenic Ras and EGF signals enhance the gene expression of Nox1 through the Ras–Raf–MEK–ERK pathway (Fig. 2).
Figure 2.
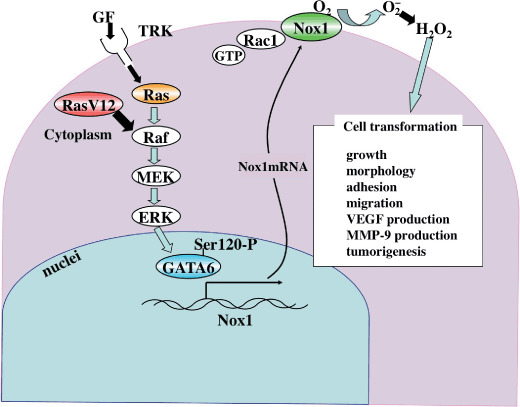
A model for NADPH oxidase (Nox) 1 function in Ras transformation. Growth factor (GF) stimulation of its receptor tyrosine kinase (TRK) induces Nox1 expression via the Ras–Raf–MEK–ERK–GATA‐6 pathway. Nox1, whose activity is regulated by Rac1, generates H2O2 as a signaling molecule in normal physiological processes. In contrast, oncogenic Ras (RasV12) constitutively activates Nox1 expression and increased reactive oxygen species generation perturbs cellular activities including growth, morphology, and migration. MMP, matrix metalloproteinase; VEGF, vascular endothelial growth factor.
What is the functional consequence of Nox1 upregulation in Ras‐transformed cells? Remarkably, introduction of Nox1 siRNA into K‐RasVal12‐transformed NRK (K‐Ras/NRK) cells blocked their anchorage‐independent growth and induced morphological reversion.( 18 ) Furthermore, implantation of K‐Ras/NRK cells carrying Nox1 siRNA into nude mice significantly reduced the rate of tumor growth. These data strongly suggest that an increase in Nox1‐derived ROS is functionally required for maintenance of phenotypes associated with Ras oncogene transformation (Fig. 2). It should be noted that Nox1 per se does not possess the transforming potential, because overexpression of Nox1 alone did not transform NIH3T3 cells( 1 ) (J. Mitsushita and T. Kamata, unpublished data, 2004). With this hierarchy in mind, the molecular mechanism underlying Nox1‐mediated Ras transformation will be described in more detail in subsequent sections.
Transcriptional regulation of Nox1 by Ras oncogene
Besides the control of Nox1 activity by regulatory components, the regulation of Nox1 gene expression is critical for its activity. Nox1 has been known to be preferentially upregulated in colon adenocarcinoma Caco‐2 cells.( 16 , 17 ) Importantly, the amount of Ras • GTP was elevated in Caco‐2 cells, and the increased expression of Nox1 mRNA seemingly correlated with accumulation of active Ras • GTP complexes.( 29 ) Caco‐2 cells do not harbor oncogenic mutations in K‐Ras.( 30 ) However, Ras can be activated by augmented upstream signaling events such as overexpression of EGF receptor and its homolog ErB2,( 31 ) which are frequently detectable in colon tumors. Nox1 transcription in Caco‐2 cells was upregulated by ectopic expression of the activated RasVal12 mutant and driven by a Ras–MEK–ERK responsive element containing the two GATA site sequences TTATCT (–161 to –136 bp and –125 to –100 bp).( 29 ) In fact, GATA‐6 was identified to specifically bind to GATA elements in the Nox1 promoter. The results are consistent with the previous observation that Caco‐2 cells express much higher levels of GATA‐6 mRNA than those of GATA‐4 and GATA‐5.( 32 ) Of note, GATA‐6 was directly phosphorylated at serine residues by ERK in response to Ras activation.( 29 ) The site‐directed mutation of the consensus ERK phosphorylation site Ser‐120 in the GATA‐6 activation domain abolished its transactivation activity and suppressed proliferation of Caco‐2 cells. Similar regulation by ERK‐dependent phosphorylation has been seen in other GATA family members including GATA‐4 (in cardiac myocytes( 33 )). Together, these observations provide compelling evidence that ERK connects the Ras signaling cascade to the GATA‐6 transcription factor, and that the phosphorylation status and hence the activity of GATA‐6 is crucial in the transcriptional activation of Nox1 (Fig. 2). Our recent results indicate that phosphorylation of GATA‐6 dramatically enhances its affinity for regulatory domains of the Nox1 promoter (Y. Adachi and T. Kamata, unpublished data, 2008). Presumably, phosphorylation of GATA‐6 induces conformational changes that favor DNA binding. Brewer et al. also suggested the regulation of Nox1 transcription by a GATA‐binding transcription factor.( 34 )
It has been suggested that GATA‐6 is related to the cancer phenotype because of its increased expression in growing progenitor cells of intestinal crypts, whereas GATA‐4 and GATA‐5 might display tumor suppressor‐like properties because of their upregulation in terminally differentiated intestinal epithelium.( 32 ) Thus, it is tempting to speculate that GATA‐6 promotes colorectal cancer growth by mediating activated Ras‐induced upregulation of Nox1. Intriguingly, among genomic alterations including K‐Ras amplification, GATA‐6 was found to be amplified in pancreatobiliary cancer.( 35 ) Knockdown of GATA‐6 in pancreatic cancer cell lines reduced anchorage‐independent growth,( 35 ) implicating GATA‐6 as a novel candidate for lineage‐specific oncogene in pancreatobiliary cancer. Given that GATA‐6 expression is not restricted to the development process of pancreatic cells, GATA‐6 might well have an oncogenic role in other cell lineages, including colon epithelium. Although Nox1 expression is also induced by various kinds of physiological cues including IFNγ,( 16 ) 25‐dihydroxy vitamin D3,( 16 ) and TLR ligands,( 17 ) it is not clear how such molecules are related to Ras‐mediated Nox1 upregulation.
Regulation of oncogenic Ras‐induced cell morphological changes and cell adhesions by Nox1
Ras oncogene‐transformed cells display morphological alterations, impaired cell adhesions, and augmented cell invasion, as seen in most malignant cancer cells. ROS are implicated in cell migration and adhesion that reflect dynamic actin cytoskeletal organization,( 36 ) and Nox1 seems to provide a cellular source for these oxygen radicals.( 37 ) Strong support for this view comes from the observation that the Rho activity in K‐Ras/NRK cells was suppressed via Rac1, a positive regulator of Nox1, but restored upon abrogation of Nox1‐generated ROS by Nox1 siRNA.( 37 ) How does Nox1 transmit a negative controlling signal to Rho? Using alkylation of redox‐sensitive cysteine thiols,( 38 ) Shinohara et al. have shown that LMW‐PTP is most likely an immediate downstream target of Nox1 involved in this process.( 37 ) In general, H2O2 or ROS can modify the thiols of cysteine residues with different oxidation states, ranging from the disulfide and sulfenyl forms (milder oxidation) to the sulfonyl acid form (much higher oxidation). Only a milder oxidation is believed to exert a possible redox function. LMW‐PTP possesses redox‐sensitive adjacent cysteines (Cys‐12 and Cys‐17) in its catalytic pocket and it has been suggested that oxidation of Cys‐12 and Cys‐17 by H2O2 forms an intramolecular disulfide bond, causing inhibition of its activity.( 39 ) Likewise, Nox1‐generated ROS oxidized LMW‐PTP and impaired its activity, which seemed to involve the formation of a S–S bridge between these cysteine residues.( 37 ) This led to an increase in tyrosine phosphorylation of p190RhoGAP, a GTPase‐activating protein for Rho. p190RhoGAP is thought to be activated following tyrosine phosphorylation by tyrosine kinases such as Src,( 40 ) resulting in inhibition of the Rho activity. Because LMW‐PTP deactivates p190RhoGAP by dephosphorylating its phosphotyrosine residues,( 41 ) oxidative inactivation of LMW‐PTP by Nox1 suppresses the Rho activity by maintaining p190RhoGAP in an active state (Fig. 3).
Figure 3.
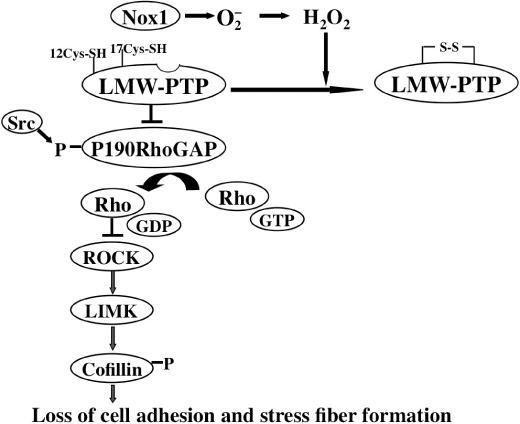
A model for NADPH oxidase (Nox) 1‐mediated suppression of actin stress fiber formation and cell adhesion. Nox1‐generated reactive oxygen species inactivate low molecular weight tyrosine phosphatase (LMW‐PTP) by oxidizing cysteine thiols, resulting in accumulation of active p190RhoGAP possibly tyrosine‐phosphorylated by Src family proteins. This causes downregulation of Rho activity and thereby depresses ROCK–LIMK–coffilin signaling, leading to disruption of actin stress fiber and focal adhesion formation.
Disassembly of actin stress fibers is triggered by downregulation of Rho, which plays an essential regulatory role in the formation of stress fibers and focal contacts.( 42 ) When the Nox1 activity was inhibited by Nox1 siRNA, stress fiber formation mediated by the ROCK–LIM kinase pathway( 43 ) was reactivated, suggesting that Nox1 mediates disruption of actin stress fibers through inactivation of Rho.
As such, Nox1 participates in the control of cell adhesion to the extracellular matrix.( 37 ) Although vinculin, a marker for formation of focal adhesions, accumulated in NRK cells, no vinculin accumulation was detected in K‐Ras/NRK cells. In contrast, silencing of Nox1 expression induced focal adhesions similar to those seen in NRK cells, implying that Nox1 inhibits the formation of focal adhesions by decreasing the Rho activity in K‐Ras/NRK cells. The dominant negative LMW‐PTP (CS) mutant blocked Nox1 siRNA‐induced restoration of stress fibers and focal adhesions, which ensures involvement of the Nox1–LMW‐PTP axis in these biological events.( 37 ) Interestingly, Rho inhibition by LMW‐PTP is also detected upon ligation of integrin to fibronectin, which involves activation of p190RhoGAP and possibly a Nox enzyme.( 44 )
Role of Nox1 in oncogenic Ras‐induced tumor angiogenesis
Angiogenesis, which is characterized by sprouting endothelial cells from preexisting capillaries, is involved in many physiological processes including wound healing, inflammation, and tumor formation.( 45 ) It is well documented that production of VEGF, one of the most potent angiogenesis stimulators, is elevated in tumor cells expressing activated Ras.( 46 ) This appears to be mediated by several transcription factors, including HIF‐1α( 47 ) and Sp1,( 48 ) through the classic Ras–Raf–MEK–ERK pathway. A link between VEGF and Nox1 was initially suggested by the observation that Nox1 was capable of inducing VEGF production in prostate cancer cells,( 49 ) but the detailed mechanism was not described. Komatsu et al. went on further by demonstrating that Nox1 exerted a critical mediating role for Ras‐induced upregulation of VEGF and tumor angiogenesis.( 50 ) Ablation of Nox1 by Nox1 siRNA decreased the tyrosine‐phosphorylated active ERK and subsequently inhibited both ERK‐dependent phosphorylation of Sp1 and Sp1 binding to the VEGF promoter, leading to transcriptional downregulation of VEGF.( 50 ) Surprisingly, the best‐known VEGF‐regulating transcription factor, HIF‐1α, was not affected by Nox1 redox signaling. In xenograft tumor models, tumors derived from K‐Ras/NRK cells carrying Nox1 siRNA exhibited markedly decreased neovascularization, whereas those derived from K‐Ras/NRK cells displayed progressive vascularity.( 50 ) Based on these observations, Nox1 was proposed to mediate oncogenic Ras‐induced upregulation of VEGF and angiogenesis by activating Sp1 through Ras–ERK‐dependent phosphorylation. The activated Ras‐induced Nox1 possibly sustains the activated state of ERK by positive feedback regulation. In this setting, one attractive speculation is that Nox1‐derived H2O2 deactivates mitogen activated protein kinase (MAPK) phosphatases( 51 ) through oxidation of redox‐sensitive cysteines and thereby maintains the oncogenic Ras–MEK‐induced activation of ERK.
Nox1 activity was also required for VEGF production in cells derived from human colorectal adenocarcinoma, assuming a mediating role of Nox1 in angiogenic processes associated with some types of human tumors.( 51 )
Nox1 and human cancer
Nox1 expression in colon cancer has been detected to reach a maximum level in well‐differentiated adenocarcinoma and to decrease in poorly differentiated adenocarcinoma, implying that Nox1 expression does not correlate with the extent of malignancy( 16 , 17 ) (Y. Zhang and T. Kamata, unpublished data, 2007). By the same token, a recent study on patient samples concluded that Nox1 expression is not statistically higher in colon cancer than in normal colon tissues,( 52 ) arguing that Nox1 contributes to colon epithelial differentiation rather than tumorigenesis. By re‐evaluating the statistical analysis, Laurent et al.,( 53 ) however, obtained the contrasting results that increased Nox1 mRNA expression correlated with activating mutations in Gly‐12 and Gly‐13 of K‐Ras, and that the colon tumor phenotype was closely related to overexpression of Nox1. Transgenic mice expressing K‐Ras Gly12Val in the intestinal epithelium also elevated Nox1 expression in the intestine,( 53 ) which fits in very well with oncogenic Ras‐induced upregulation of Nox1 transcription through an ERK–GATA‐6 cascade in CaCO‐2 cells.( 29 ) Because the number of patient samples surveyed was relatively small, more clinical and experimental data would be needed to make any definitive conclusion. Nevertheless, the study has significant implications in understanding a causal relationship between K‐Ras activation and Nox1 expression in colorectal cancer with a high frequency (~45%) of K‐Ras activation mutation. If Nox1 exerts cancer‐promoting effects, it is most likely at an early stage, as Nox1 expression is diminished at a more advanced tumor stage.( 16 ) Another point is that while Nox1 expression predominates, persistent activation of NF‐κB also occurs in well‐differentiated adenocarcinoma.( 54 ) This supports the scenario where the enhanced production of Nox1‐derived ROS activates NF‐κB signaling, which in turn contributes to tumor promotion by inducing inflammatory cytokines or providing potent anti‐apoptotic survival signals to the enterocytes.( 55 )
Additionally, Nox1 may be associated with gastric cancer. Earlier studies indicated that lipopolysaccharide from Helicobacter pylori induced Nox1‐derived ROS through TLR4 in guinea pig gastric pit cells, suggesting that activation of Nox1 is one of the initial innate immune responses against H. pylori.( 56 ) Given that infection with H. pylori is associated with development of gastric adenocarcinoma,( 57 ) one might expect activation of Nox1 to promote gastric carcinogenesis by enhancing inflammation or oxygen radical activity. However, it remains to be determined whether Nox1 is engaged in the pathology of H. pylori in human stomach, as Nox1 and NOXO1 were absent in normal or chronic atrophic gastritis cells.( 58 ) In contrast, significant levels of Nox1 and NOXO1 were detectable in gastric cancer cells (intestinal, diffuse, or signet‐ring cell type) but not normal gastric cells, whereas NOXA1 was constitutively expressed in both normal and tumor tissues.( 59 ) This points to an intriguing possibility that gastric cancer undergoes aberrant control of Nox1 and NOXO1 expression. In addition to gastrointestinal neoplasms, some correlation between ERK activation and increased Nox1 expression was seen in papiloma virus E6/E7‐transformed human keratinocytes.( 60 )
Other Nox enzymes and cancer
ROS production is increased in pancreatic cancer cells( 61 ) and melanoma cells( 62 ) and overexpression of superoxide dismutase attenuates their growth.( 63 ) Yet little is known about the genesis of ROS in these cells. Depletion of Nox4 by Nox4 siRNA results in apoptosis,( 64 , 65 ) possibly through AKT–ASK1 cell survival signaling.( 65 ) Nox4‐generated ROS therefore support resistance to the programmed cell death of pancreatic cancer cells, which contrasts with the long‐believed proapoptotic activity of ROS as in the case of oxidative stress.( 65 ) Inhibition of Nox4 also suppresses growth of melanoma cells,( 66 , 67 ) but this growth suppression is a consequence of G2–M cell cycle arrest, which is accompanied by tyrosine phosphorylation of cdk1 (a hallmark of the G2–M check point) and sequestration of the cell division controlling (cdc)25c phosphatase (a negative regulator of cdk1) to the cytoplasm via complex formation with 14‐3‐3 proteins.( 67 ) The finding is significant because it reveals an alternative deregulation of melanoma cell growth that is dependent on aberrant control of the G2–M check point rather than evasion of the G1–S check directed by cyclin D and CDK4.( 68 ) In another aspect, Nox4‐generated ROS appear to induce the expression of HIF‐2α in renal cell carcinoma where the von Hippel–Lindou tumor suppressor (an E3 ubiquitin ligase of HIF‐2α) is lost,( 69 ) implicating Nox4 as a positive regulator of HIF‐2α independent of von Hippel–Lindou.
A potential role for Nox5 in Barrett's esophageal adenocarcinoma has been suggested. Acid treatment of Barrett's esophageal cells activates expression of Nox5‐S (an EF‐hand motif‐lacking isoform of Nox5) through an increase in intracellular Ca2+ and cAMP‐responsive element‐binding protein,( 70 ) leading to increased cell survival. This may explain why acid reflux accelerates progression from metaplasia to adenocarcinoma in Barrett's esophageal patients. Thus, as illustrated with Nox4 and Nox5, one function of Nox may be to confer anti‐apoptotic activity to tumor cells.
Although numerous studies depict upregulation of Nox enzymes in cancer cells, one unexpected discovery was that the expression of Duox1 and Duox2 was remarkably silenced through hypermethylation of CpG‐rich regions of their promoters in lung cancer cells.( 71 ) Duox may be involved in tissue repair, and silencing of Duox could promote lung carcinogenesis by accumulating tissue injury caused by proinflammatory responses.( 71 ) Table 1 summarizes cancers involving the Nox enzymes described in this review.
Table 1.
Cancers involving NADPH oxidase (Nox) enzymes
| Nox/Duox | Function | Type of cancer |
|---|---|---|
| Nox1 | Cell growth, adhesion, shape, angiogenesis | K‐Ras‐transformed |
| NRK cells | ||
| Cell growth, VEGF production | Colon cancer | |
| Unknown | Gastric cancer (Helicobacter pylori) | |
| Cell growth, unknown | E6/E7‐transformed keratinocytes | |
| Angiogenesis | Prostate cancer | |
| Nox2 | Unknown | Unknown |
| Nox3 | Unknown | Unknown |
| Nox4 | Cell survival | Pancreatic cancer |
| Cell cycle control | Melanoma | |
| HIF2α‐expression | Renal cell carcinoma | |
| Nox5 | Cell survival | Esophageal adenocarcinoma |
| Duox1, Duox2 | Tissue repair, | Lung cancer |
| cell migration |
Duox, dual oxidase; HIF, hypoxia‐inducible factor; NRK, normal rat kidney; VEGF, vascular endothelial growth factor.
Concluding remarks
The available data suggest the potential role of Nox family‐generated superoxide and derived oxidants in carcinogenesis. An important new concept to emerge from these studies is that the effect of Nox family‐derived ROS depends on their signaling inputs rather than their sporadic, genotoxic stresses, which were traditionally believed to be a cause of cancer. However, many issues remain unanswered. Although the referred studies describe that suppression of Nox activity inhibits the transformation phenotype of in vitro cultured cells, direct in vivo evidence is lacking. This concern is raised especially when we consider the complex in vivo processes of tumor progression that involve not only oncogenes and tumor suppressors but also stromal cells constituting tumor microenvironments. To further establish the involvement of Nox in initiation and promotion of tumors, animal model experiments such as knockout and knockin mice, where the expression and activity of Nox isoforms are genetically altered, are critical. Given the close association of cancer progression with chronic inflammation in the epithelium, which is often accompanied by overproduction of ROS, exploration of the underlying mechanism for Nox enzyme‐dependent inflammation is also an area of future focus. Almost all cancers described here are associated with overproduction of ROS due to transcriptional activation of Nox genes. It is also possible that ROS generation is augmented by gain‐of‐function mutation in Nox genes. However, no mutations of this type have been reported thus far, whereas loss‐of‐function mutations in Nox genes were found to be responsible for some inherited diseases (e.g. chronic granulomatous disease,( 20 ) defective gravity sensing,( 22 ) and hypothyroidism( 26 )). It would be of particular interest to explore the possibility of activation mutations in Nox enzymes in certain types of cancers. Further characterization of the biological function of the Nox family of proteins with a focus on the above issues will lead to a better understanding of how these oxidase genes contribute to carcinogenesis.
Abbreviations
| cdk | cyclin dependent kinase |
| Duox | dual oxidase |
| EGF | epidermal growth factor |
| ERK | extracellular‐regulated kinase |
| HIF | hypoxia‐inducible factor |
| IFN | interferone |
| LMW‐PTP | low molecular weight tyrosine phosphatase |
| MEK | mitogen activated kinase/ERK kinase |
| NF‐κB | nuclear factor kappa immunoglobulin enhancer‐binding protein |
| Nox | NADPH oxidase |
| NRK | normal rat kidney |
| Rac | ras‐related C3 botulinum toxin substrate |
| ROS | reactive oxygen species |
| TLR | toll‐like receptor |
| VEGF | vascular endothelial growth factor |
Acknowledgments
I am grateful to F. Ushiyama for assistance in manuscript preparation. This work was supported by a Grant on Cancer Research in Applied Areas from the Ministry of Science and Culture of Japan (T. Kamata) and Grant‐in‐Aid for Scientific Research for Japan Society for Promotion of Science (T. Kamata).
References
- 1. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004; 4: 181–9. [DOI] [PubMed] [Google Scholar]
- 2. Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 2007; 43: 332–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007; 87: 245–313. [DOI] [PubMed] [Google Scholar]
- 4. Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med 2007; 43: 319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banfi B, Molnar G, Maturana A et al . A Ca2+‐activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem 2001; 276: 37 594–601. [DOI] [PubMed] [Google Scholar]
- 6. Edens WA, Sharling L, Cheng G et al . Tyrosine cross‐linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J Cell Biol 2001; 154: 879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Deken X, Wang D, Many MC et al . Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem 2000; 275: 23 227–33. [DOI] [PubMed] [Google Scholar]
- 8. Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun 2005; 338: 677–86. [DOI] [PubMed] [Google Scholar]
- 9. Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 2003; 278: 3510–13. [DOI] [PubMed] [Google Scholar]
- 10. Geiszt M, Lekstrom K, Witta J, Leto TL. Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J Biol Chem 2003; 278: 20 006–12. [DOI] [PubMed] [Google Scholar]
- 11. Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox‐dependent manner: its regulation by oxidase organizers and activators. J Biol Chem 2005; 280: 23 328–39. [DOI] [PubMed] [Google Scholar]
- 12. Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem 2004; 279: 45 935–41. [DOI] [PubMed] [Google Scholar]
- 13. Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline‐rich region of p22phox are dominant inhibitors of Nox1‐ and Nox2‐dependent reactive oxygen generation. J Biol Chem 2005; 280: 31 859–69. [DOI] [PubMed] [Google Scholar]
- 14. Fortemaison N, Miot F, Dumont JE, Dremier S. Regulation of H2O2 generation in thyroid cells does not involve Rac1 activation. Eur J Endocrinol 2005; 152: 127–33. [DOI] [PubMed] [Google Scholar]
- 15. Suh YA, Arnold RS, Lassegue B et al . Cell transformation by the superoxide‐generating oxidase Mox1. Nature 1999; 401: 79–82. [DOI] [PubMed] [Google Scholar]
- 16. Geiszt M, Lekstrom K, Brenner S et al . NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J Immunol 2003; 171: 299–306. [DOI] [PubMed] [Google Scholar]
- 17. Kawahara T, Kuwano Y, Teshima‐Kondo S et al . Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to Toll‐like receptor 5 signaling in large intestinal epithelial cells. J Immunol 2004; 172: 3051–8. [DOI] [PubMed] [Google Scholar]
- 18. Mitsushita J, Lambeth JD, Kamata T. The superoxide‐generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res 2004; 64: 3580–5. [DOI] [PubMed] [Google Scholar]
- 19. Kikuchi H, Hikage M, Miyashita H, Fukumoto M. NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cells. Gene 2000; 254: 237–43. [DOI] [PubMed] [Google Scholar]
- 20. Roos D. X‐CGDbase. a database of X‐CGD‐causing mutations. Immunol Today 1996; 17: 517–21. [DOI] [PubMed] [Google Scholar]
- 21. Banfi B, Malgrange B, Knisz J, Steger K, Dubois‐Dauphin M, Krause KH. NOX3, a superoxide‐generating NADPH oxidase of the inner ear. J Biol Chem 2004; 279: 46 065–72. [DOI] [PubMed] [Google Scholar]
- 22. Paffenholz R, Bergstrom RA, Pasutto F et al . Vestibular defects in head‐tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev 2004; 18: 486–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA 2000; 97: 8010–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahadev K, Motoshima H, Wu X et al . The NAD(P)H oxidase homolog Nox4 modulates insulin‐stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol 2004; 24: 1844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 2003; 17: 1502–4. [DOI] [PubMed] [Google Scholar]
- 26. Moreno JC, Bikker H, Kempers MJ et al . Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med 2002; 347: 95–102. [DOI] [PubMed] [Google Scholar]
- 27. Downward J. Control of ras activation. Cancer Surv 1996; 27: 87–100. [PubMed] [Google Scholar]
- 28. Irani K, Xia Y, Zweier JL et al . Mitogenic signaling mediated by oxidants in Ras‐transformed fibroblasts. Science 1997; 275: 1649–52. [DOI] [PubMed] [Google Scholar]
- 29. Adachi Y, Shibai Y, Mitsushita J, Shang WH, Hirose K, Kamata T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK–ERK‐dependent phosphorylation of GATA‐6. Oncogene 2008; 27: 4921–32. [DOI] [PubMed] [Google Scholar]
- 30. Lorentz O, Cadoret A, Duluc I et al . Downregulation of the colon tumour‐suppressor homeobox gene Cdx‐2 by oncogenic ras. Oncogene 1999; 18: 87–92. [DOI] [PubMed] [Google Scholar]
- 31. Maurer CA, Friess H, Kretschmann B et al . Increased expression of erbB3 in colorectal cancer is associated with concomitant increase in the level of erbB2. Hum Pathol 1998; 29: 771–7. [DOI] [PubMed] [Google Scholar]
- 32. Gao X, Sedgwick T, Shi YB, Evans T. Distinct functions are implicated for the GATA‐4, ‐5, and ‐6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol Cell Biol 1998; 18: 2901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tenhunen O, Sarman B, Kerkela R et al . Mitogen‐activated protein kinases p38 and ERK 1/2 mediate the wall stress‐induced activation of GATA‐4 binding in adult heart. J Biol Chem 2004; 279: 24 852–60. [DOI] [PubMed] [Google Scholar]
- 34. Brewer AC, Sparks EC, Shah AM. Transcriptional regulation of the NADPH oxidase isoform, Nox1, in colon epithelial cells: role of GATA‐binding factor(s). Free Radic Biol Med 2006; 40: 260–74. [DOI] [PubMed] [Google Scholar]
- 35. Kwei KA, Bashyam MD, Kao J et al . Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer. PLoS Genet 2008; 4: e1000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van Wetering S, Van Buul JD, Quik S et al . Reactive oxygen species mediate Rac‐induced loss of cell–cell adhesion in primary human endothelial cells. J Cell Sci 2002; 115: 1837–46. [DOI] [PubMed] [Google Scholar]
- 37. Shinohara M, Shang WH, Kubodera M et al . Nox1 redox signaling mediates oncogenic Ras‐induced disruption of stress fibers and focal adhesions by down‐regulating Rho. J Biol Chem 2007; 282: 17 640–8. [DOI] [PubMed] [Google Scholar]
- 38. Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol 2003; 14: S211–15. [DOI] [PubMed] [Google Scholar]
- 39. Caselli A, Marzocchini R, Camici G et al . The inactivation mechanism of low molecular weight phosphotyrosine‐protein phosphatase by H2O2 . J Biol Chem 1998; 273: 32 554–60. [DOI] [PubMed] [Google Scholar]
- 40. Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol 2001; 3: 361–7. [DOI] [PubMed] [Google Scholar]
- 41. Chiarugi P, Fiaschi T, Taddei ML et al . Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet‐derived growth factor receptor stimulation. J Biol Chem 2001; 276: 33 478–87. [DOI] [PubMed] [Google Scholar]
- 42. Hall A. Rho GTPases and the actin cytoskeleton. Science 1998; 279: 509–14. [DOI] [PubMed] [Google Scholar]
- 43. Maekawa M, Ishizaki T, Boku S et al . Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM‐kinase. Science 1999; 285: 895–8. [DOI] [PubMed] [Google Scholar]
- 44. Nimnual AS, Taylor LJ, Bar‐Sagi D. Redox‐dependent downregulation of Rho by Rac. Nat Cell Biol 2003; 5: 236–41. [DOI] [PubMed] [Google Scholar]
- 45. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1: 27–31. [DOI] [PubMed] [Google Scholar]
- 46. Kranenburg O, Gebbink MF, Voest EE. Stimulation of angiogenesis by Ras proteins. Biochim Biophys Acta 2004; 1654: 23–37. [DOI] [PubMed] [Google Scholar]
- 47. Forsythe JA, Jiang BH, Iyer NV et al . Activation of vascular endothelial growth factor gene transcription by hypoxia‐inducible factor 1. Mol Cell Biol 1996; 16: 4604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gille J, Swerlick RA, Caughman SW. Transforming growth factor‐alpha‐induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP‐2‐dependent DNA binding and transactivation. EMBO J 1997; 16: 750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arbiser JL, Petros J, Klafter R et al . Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci USA 2002; 99: 715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Komatsu D, Kato M, Nakayama J, Miyagawa S, Kamata T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras‐induced vascular endothelial growth factor expression. Oncogene 2008; 27: 4724–32. [DOI] [PubMed] [Google Scholar]
- 51. Kondoh K, Nishida E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim Biophys Acta 2007; 1773: 1227–37. [DOI] [PubMed] [Google Scholar]
- 52. Szanto I, Rubbia‐Brandt L, Kiss P et al . Expression of NOX1, a superoxide‐generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol 2005; 207: 164–76. [DOI] [PubMed] [Google Scholar]
- 53. Laurent E, McCoy JW 3rd, Macina RA et al . Nox1 is over‐expressed in human colon cancers and correlates with activating mutations in K‐Ras. Int J Cancer 2008; 123: 100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fantini MC, Pallone F. Cytokines: from gut inflammation to colorectal cancer. Curr Drug Targets 2008; 9: 375–80. [DOI] [PubMed] [Google Scholar]
- 55. Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKβ is required for prevention of apoptosis mediated by cell‐bound but not by circulating TNFα. Immunity 2003; 19: 725–37. [DOI] [PubMed] [Google Scholar]
- 56. Kawahara T, Kohjima M, Kuwano Y et al . Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol Cell Physiol 2005; 288: C450–7. [DOI] [PubMed] [Google Scholar]
- 57. Hatakeyama M, Higashi H. Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis. Cancer Sci 2005; 96: 835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salles N, Szanto I, Herrmann F et al . Expression of mRNA for ROS‐generating NADPH oxidases in the aging stomach. Exp Gerontol 2005; 40: 353–7. [DOI] [PubMed] [Google Scholar]
- 59. Tominaga K, Kawahara T, Sano T et al . Evidence for cancer‐associated expression of NADPH oxidase 1 (Nox1)‐based oxidase system in the human stomach. Free Radic Biol Med 2007; 43: 1627–38. [DOI] [PubMed] [Google Scholar]
- 60. Chamulitrat W, Schmidt R, Tomakidi P et al . Association of gp91phox homolog Nox1 with anchorage‐independent growth and MAP kinase‐activation of transformed human keratinocytes. Oncogene 2003; 22: 6045–53. [DOI] [PubMed] [Google Scholar]
- 61. Farrow B, Evers BM. Inflammation and the development of pancreatic cancer. Surg Oncol 2002; 10: 153–69. [DOI] [PubMed] [Google Scholar]
- 62. Sander CS, Hamm F, Elsner P, Thiele JJ. Oxidative stress in malignant melanoma and non‐melanoma skin cancer. Br J Dermatol 2003; 148: 913–22. [DOI] [PubMed] [Google Scholar]
- 63. Church SL, Grant JW, Ridnour LA et al . Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc Natl Acad Sci USA 1993; 90: 3113–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem 2004; 279: 34 643–54. [DOI] [PubMed] [Google Scholar]
- 65. Mochizuki T, Furuta S, Mitsushita J et al . Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal‐regulating kinase 1 pathway in pancreatic cancer PANC‐1 cells. Oncogene 2006; 25: 3699–707. [DOI] [PubMed] [Google Scholar]
- 66. Brar SS, Kennedy TP, Sturrock AB et al . An NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am J Physiol Cell Physiol 2002; 282: C1212–24. [DOI] [PubMed] [Google Scholar]
- 67. Yamaura M et al . NADPH oxidase (Nox) 4 contributes to transformation phenotype of melanoma cells by regulating G2–M cell cycle progression. Cancer Res 2009; 69: 2647–54. [DOI] [PubMed] [Google Scholar]
- 68. Kozar K, Sicinski P. Cell cycle progression without cyclin D‐CDK4 and cyclin D‐CDK6 complexes. Cell Cycle 2005; 4: 388–91. [DOI] [PubMed] [Google Scholar]
- 69. Maranchie JK, Zhan Y. Nox4 is critical for hypoxia‐inducible factor 2‐alpha transcriptional activity in von Hippel‐Lindau‐deficient renal cell carcinoma. Cancer Res 2005; 65: 9190–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fu X, Beer DG, Behar J, Wands J, Lambeth D, Cao W. cAMP‐response element‐binding protein mediates acid‐induced NADPH oxidase NOX5‐S expression in Barrett esophageal adenocarcinoma cells. J Biol Chem 2006; 281: 20 368–82. [DOI] [PubMed] [Google Scholar]
- 71. Luxen S, Belinsky SA, Knaus UG. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res 2008; 68: 1037–45. [DOI] [PubMed] [Google Scholar]