

Abstract
Impacts of genetic polymorphisms of the ATP‐binding cassette (ABC) transporter BCRP/MXR1/ABCP (ABCG2) on drug response have been implicated; however, the hitherto reported data involve some inconsistencies. To re‐evaluate the effect of single nucleotide polymorphisms (SNP) of ABCG2 in vitro, we created a total of seven variant cDNAs (V12M, Q141K, F208S, S248P, F431L, S441N and F489L) by site‐directed mutagenesis and stably expressed each of them in Flp‐In‐293 cells using the Flp recombinase system. Multicolor fluorescence in situ hybridization mapping analysis revealed that one single copy of ABCG2 cDNA was incorporated into the telomeric region of chromosome 12p. It was proven that mRNAs of those integrated ABCG2 variants were expressed evenly in Flp‐In‐293 cells. However, the protein expression levels varied among those variants. In particular, expression of the F208S and S441N variants was markedly low, suggesting the instability of these variant proteins. Drug resistance profiles of Flp‐In‐293 cells expressing two major SNP variants (V12M and Q141K) toward the drug SN‐38 demonstrated that the IC50 value (drug concentrations producing a 50% reduction of cell growth) for Q141K was approximately 50% of that for wild type. The contributions of the minor SNP variants (F208S, S248P, F431L, S441N and F489L) to drug resistance toward SN‐38, mitoxantrone, doxorubicin, daunorubicin or etoposide were significantly lower than wild type. Based on our functional validation, the above‐mentioned non‐synonymous polymorphisms as well as acquired mutants (R482G and R482T) of ABCG2 were classified into four groups. Furthermore, new camptothecin analogs synthesized by our research group had potent effects in circumventing ABCG2‐mediated drug resistance without any influence from major non‐synonymous polymorphisms. (Cancer Sci 2007; 98: 231–239)
Abbreviations:
- ABC
ATP‐binding cassette
- ABCG2
BCRP/MXR1/ABCP
- D‐MEM
high‐glucose Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethyleneglycol‐bis(2‐aminoethyl)‐N,N,N′, N′‐tetracetic acid
- ER
endoplasmic reticulum
- FCS
fetal calf serum
- FISH
fluorescence in situ hybridization
- FRT
Flp recombination target
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HRP
horseradish peroxidase
- IC50
drug concentrations producing a 50% reduction of cell growth
- MTT
3‐(4,5‐dimethyl‐2‐thiazol‐2‐yl)‐2,5‐diphenyl‐2H‐tetrazolium bromide
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- RT‐PCR
reverse transcription–polymerase chain reaction
- SNP
single nucleotide polymorphism
- SSC
300 mM sodium chloride and 30 mM sodium citrate (pH 7.0).
Human ABCG2 ( 1 , 2 , 3 ) is a member of the ABC transporter gene family. The ABCG2 gene is located on chromosome 4q22, spans over 66 kb, and consists of 16 exons ranging from 60 to 532 bp.( 4 ) The ABCG2 protein is a so‐called ‘half ABC transporter’, existing as a homodimer linked through a cysteinyl disulfide bond at Cys603.( 5 , 6 , 7 ) ABCG2 protein is expressed endogenously in placental trophoblast cells, the epithelium of the small intestine and liver canalicular membrane, as well as in ducts and lobules of the breast. Apical localization in the epithelium of the small intestine and colon indicates a possible role for human ABCG2 in regulating the uptake of orally (p.o.) administered drugs as well as xenobiotics.( 8 )
Overexpression of ABCG2 reportedly confers cancer cell resistance to anticancer drugs, such as topotecan, irinotecan (CPT‐11) and mitoxantrone.( 9 , 10 , 11 , 12 ) In the case of drug resistance to irinotecan, SN‐38‐resistant PC‐6/SN2–5H human lung carcinoma cells were shown to overexpress ABCG2 with reduced intracellular accumulation of SN‐38, an active metabolite of CPT‐11, and the SN‐38–glucuronide conjugate.( 12 ) Plasma membrane vesicles prepared from those cancer cells or ABCG2‐transfected cells transported both SN‐38 and SN‐38–glucuronide ATP‐dependently.( 13 , 14 ) It has also been reported that ABCG2‐transfected cells are resistant to photosensitizers, such as hematoporphyrin IX, pheophorbide a, and chlorine e6, suggesting a possible role for ABCG2 in cellular resistance to photodynamic therapy.( 15 ) In this regard, we have most recently demonstrated that ABCG2 transports hematoporphyrin in an ATP‐dependent manner.( 16 )
SNP of ABCG2 have been suggested as a significant factor in a patient's response to medication and the risk of diseases.( 17 , 18 , 19 , 20 , 21 , 22 ) Sequencing of the ABCG2 gene from human samples has revealed over 80 different, naturally occurring sequence variations.( 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 ) However, information is still limited regarding the functional impact of genetic polymorphisms of ABCG2. In addition, it was noticed that some discrepancies existed among reports in terms of the transport function and drug resistance profile of SNP variants of ABCG2.( 25 , 28 , 30 , 31 ) The reason for such discrepancies is not known, but it may be due to differences in their experimental procedures.
In the present study, we aimed to re‐evaluate the impact of the genetic polymorphisms of ABCG2 on drug resistance by functionally validating its SNP in vitro. To analyze quantitatively the effect of non‐synonymous SNP of ABCG2 on the protein expression level and the drug resistance profile, we used the Flp‐In method to integrate one single copy of ABCG2 variant cDNA into FRT‐tagged genomic DNA. By using this method, we could exclude the random integration of ABCG2 cDNA into the chromosomal DNA of host cells. Furthermore, we have examined the pharmacological potency of our new camptothecin analogs( 32 , 33 ) to determine whether they could circumvent ABCG2‐associated drug resistance in human tumor cells without being influenced by the genetic polymorphisms of ABCG2. The relevant experimental data are presented herein, and the potential impact of genetic polymorphisms of ABCG2 on drug resistance is discussed.
Materials and Methods
Chemicals and biological reagents. The following reagents and drugs were purchased from the commercial sources indicated in parentheses: ATP, 5‐bromo‐2′‐deoxyuridine, salmon sperm DNA, E. coli tRNA, bovine serum albumin, etoposide, doxorubicin, and daunorubicin (Sigma‐Aldrich, St Louis, MO, USA); l‐glutamine, KCl, methanol, acetic acid, formamide, mitoxantrone, vincristine, and prazosin (Wako Pure Chemical Industries, Osaka, Japan); creatine kinase, creatine phosphate, EGTA, EDTA, Tris, HEPES, and D‐MEM (Nacalai Tesque, Inc., Kyoto, Japan); FCS (Dainippon Pharmaceuticals, Osaka, Japan); antibiotic‐antimycotic cocktail solution and hygromycin B (Invitrogen, Carlsbad, CA, USA); Hoechst 33258, Nick translation Kit, biotin‐16‐dUTP, digoxigenin‐11‐dUTP (Roche, Mannheim, Germany); Cy3‐labeled streptavidin (GE Healthcare UK, Buckinghamshire, UK); and Multicolor FISH‐Human (Cambio, Cambridge, UK). SN‐38 and its analogs were generously provided by Yakult Honsha Co. (Tokyo, Japan). All other chemicals used were of analytical grade.
SNP data on non‐synonymous polymorphisms of the human ABCG2 gene. SNP data on the polymorphisms of ABCG2 were obtained from the NCBI dbSNP database and recent publications.( 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 )
Preparation of plasmids carrying ABCG2 variant cDNA. WT ABCG2 cDNA inserted into the pcDNA5/FRT plasmid( 7 ) was used as the template, and non‐synonymous SNP variants were generated using the QuikChange Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA), as described previously.( 16 ) The mutations were confirmed by sequencing the inserted cDNA.
Expression of ABCG2 and its variants in Flp‐In‐293 cells. Flp‐In‐293 cells (Invitrogen) were maintained in D‐MEM supplemented with 10% (v/v) heat‐inactivated FCS, 2 mM l‐glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2 in air. The number of viable cells was determined from counts made in a hemocytometer with Trypan Blue dye exclusion.
Flp‐In‐293 cells were transfected with the ABCG2‐pcDNA5/FRT vector, the Flp recombinase expression plasmid pOG44, and LipofectAmine‐2000 (Invitrogen) according to the manufacturer's instructions. Single colonies resistant to hygromycin B were picked and subcultured. Selection of positive colonies was carried out by immunoblotting, as described below. The resulting cells are described as Flp‐In‐293/ABCG2 cells throughout this manuscript. Mock cells (Flp‐In‐293/Mock) were prepared by transfecting Flp‐In‐293 cells with pcDNA5/FRT and pOG44 vectors in the same manner as described above.
Chromosome preparation and FISH. Chromosome preparation, replication R‐banding and FISH mapping were carried out as described in Matsuda and Chapman.( 34 ) Briefly, the cell cultures were synchronized by thymidine blockage for 16 h, treated with 5‐bromo‐2′‐deoxyuridine for 5.5 h after the release of excessive thymidine, and harvested after colcemid treatment for 0.5 h. ABCG2‐pcDNA5/FRT and pcDNA5/FRT plasmid DNAs were labeled with biotin‐16‐dUTP and digoxigenin‐11‐dUTP, respectively, by nick translation for use as probes. After hybridization, the slides were washed in 50% formamide/2× SSC at 37°C and in 1× SSC at room temperature for 20 min each. Detection of the probe signals was carried out with Cy3‐labeled streptavidin and Cy5‐labeled antidigoxigenin for biotin‐labeled and digoxigenin‐labeled probes, respectively. FISH images were captured with the CW4000 FISH application program of Leica Microsystems Imaging Solutions sing a cooled CCD camera mounted on a DMRA2 microscope (Leica Microsystem, Wetzlar, Germany). Multicolor FISH with human chromosome‐specific paints was carried out after hybridization with biotin‐labeled ABCG2‐pcDNA5/FRT according to the manufacturer's instructions for assignment of the chromosomes where the transgenes are located. The multicolor FISH images were captured and merged with the images of the hybridization signals of the ABCG2‐pcDNA5/FRT on the same metaphase spreads using the CW4000 FISH application program.
Detection of mRNA by RT‐PCR. Total RNA was extracted from cultured cells with NucleoSpin RNA II (Macherey‐Nagel, Duren, Germany) according to the manufacturer's protocol. cDNA was prepared from the extracted RNA in a reverse transcription reaction with SuperScript II RT (Invitrogen) and random hexamers, according to the manufacturer's instructions. The mRNA levels of ABCG2 and GAPDH were determined by PCR in an iCycler thermal cycler (Bio‐Rad, Hercules, CA, USA) with the following specific primer sets: ABCG2, 5′‐GATCTCTCACCCTGGGGCTTGTGGA and 5′‐TGTGCAACAGTGTGATGGCAAGGGA; and GAPDH, 5′‐ACTGCCAACGTGTCAGTGGTGGACCTGA and 5′‐GGCTGGTGGTCCAGGGGTCTTACTCCTT. The PCR reaction consisted of hot‐start incubation at 94°C for 2 min and 30 cycles of 94°C for 30 s, 59°C for 30 s, and 72°C for 30 s. After the PCR, products were separated by agarose gel electrophoresis and detected with ethidium bromide under ultraviolet light.
Measurement of mRNA levels by quantitative RT‐PCR. The RNA levels of ABCG2 and GAPDH were determined using the 7500 Fast Real Time‐PCR System (Applied Biosystems, Foster City, CA, USA), TaqMan Fast Universal Master Mix (Applied Biosystems), and TaqMan probes (ABCG2, Hs00184979_m1; GAPDH, Hs99999905_m1) (Applied Biosystems) according to the manufacturer's protocol. The expression levels of ABCG2 were normalized against those of GAPDH.
Immunofluorescence microscopy. ABCG2‐expressing Flp‐In‐293 cells were seeded onto collagen type I‐coated coverslips and incubated under the above‐mentioned culture conditions for 24 h. Cells were fixed with 4% paraformaldehyde in PBS at room temperature for 20 min. Thereafter, cell membranes were permeabilized by incubation with 0.02% Triton X‐100 in PBS at room temperature for 5 min. To block the free aldehyde groups of formaldehyde, cells were treated with glycine (10 mg/mL) in PBS at room temperature for 10 min, which was followed by a further incubation with 0.5% (w/v) bovine serum albumin (BSA) in PBS at room temperature for 1 h. To detect the ABCG2 protein, cells were treated with the BXP‐21 antibody (1:1000 dilution; Signet, Dedham, MA, USA) as the primary antibody and subsequently with the Alexa Fluor 488‐conjugated antimouse IgG antibody (1:1000 dilution; Invitrogen). In the same preparations, nuclear DNA was stained with propidium iodide (4 µg/mL) in PBS containing 0.5% (w/v) BSA. The immunofluorescence of Flp‐In‐293 cells was detected with a confocal laser‐scanning fluorescence microscope IX70/FLUOVIEW (Olympus, Tokyo, Japan).
Gel electrophoresis and detection of ABCG2 protein. The ABCG2 protein expressed in Flp‐In‐293 cells was detected by immunoblotting with BXP‐21, a specific antibody to human ABCG2. Cells were rinsed with ice‐cold PBS and subsequently treated with the lysis buffer containing 50 mM Tris/HCl (pH 7.4), 1% (w/v) Triton X‐100, 1 mM DTT, and a protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). The samples (10 µg of protein) were subjected to sodium dodecylsulfate–polyacrylamide gel electrophoresis in the presence of 2‐mercaptoethanol. Briefly, proteins were separated by electrophoresis on 7.5% polyacrylamide gels and then electroblotted onto Hy‐bond ECL nitrocellulose membranes (GE Healthcare UK, Buckinghamshire, UK). Immunoblotting was carried out using BXP‐21 (1:500 dilution) as the primary antibody and HRP‐conjugated antimouse IgG (1:3000 dilution; Cell Signaling Technology, Beverly, MA, USA) as the secondary antibody. HRP‐dependent luminescence was developed using Western Lighting Chemiluminescent Reagent Plus (PerkinElmer Life Sciences, Boston, MA, USA) and detected in a Lumino Imaging Analyzer FAS‐1000 (Toyobo, Osaka, Japan). To detect GAPDH, used as an internal loading control, immunoblotting was carried out in the same manner as described above, except for the use of mouse monoclonal anti‐GAPDH IgG (1:1000 dilution; American Research Products, Belmont, MA, USA) as the primary antibody.
Drug resistance profiling of ABCG2‐expressing Flp‐In‐293 cells. A growth inhibition assay was carried out by seeding Flp‐In‐293 cells at a density of 2000 cells per well in 96‐well plates containing the culture medium. After 24 h, a test drug (SN‐38, mitoxantrone, doxorubicin, daunorubicin, etoposide or vincristine) was added to the culture medium at different concentrations, and cells were further incubated with the drug in a humidified tissue‐culture chamber (37°C, 5% CO2) for 72 h or 96 h. Surviving cells were detected using the MTT assay.( 7 ) Briefly, 10 µL of MTT solution (5 mg/mL) was added to the culture medium, and cells were incubated for 4 h at 37°C. Thereafter, 100 µL of 10% (w/v) sodium dodecylsulfate in PBS was added to the culture medium, and the mixture was incubated at 37°C overnight. The absorbance of formazan, a metabolite of MTT, in the resulting solution was measured photometrically at a test wavelength of 570 nm and at a reference wavelength of 630 nm in a Multiskan JX system (Dainippon Pharmaceuticals, Osaka, Japan). IC50 values were calculated from dose–response curves (i.e. cell survival vs drug concentration) obtained in multireplicated experiments (n = 6).
ATPase activity measurement. The prazosin‐stimulated ATPase activity of the isolated plasma membrane from ABCG2 variant‐expressing Sf9 cells( 16 ) was determined by measuring inorganic phosphate liberation, as described previously.( 35 )
Results
Flp‐mediated integration of ABCG2 cDNA into FRT‐tagged genomic DNA. To examine the location of cDNA integrated into chromosomal DNA by the Flp‐In method, more than 20 metaphase spreads were examined for both FISH mapping of the ABCG2‐pcDNA5/FRT and pcDNA5/FRT plasmid DNA probes, and multicolor FISH analysis. In the FISH mapping experiment with the ABCG2‐pcDNA5/FRT and pcDNA5/FRT (Fig. 1A), the signals of ABCG2‐pcDNA5/FRT and pcDNA5/FRT were detected on three chromosomes and one chromosome, respectively. Multicolor FISH revealed that the signals of ABCG2‐pcDNA5/FRT were located in the telomeric region of the short arm on one chromosome 12 and in the q22 region on a pair of chromosome 4 (Fig. 1A,B), whereas the hybridization signal of pcDNA5/FRT was detected only in the telomeric region of the short arm on one chromosome 12 (data not shown). Thus, it has been verified that ABCG2 cDNA was incorporated into the telomeric region of chromosome 12p. The number of chromosomes per cell ranged from 57 to 87 in the cell line, and the number of individual chromosomes was varied from one to four among cells. In addition, chromosome rearrangements (e.g. translocations) have occurred frequently in the cell line (Fig. 1B).
Figure 1.
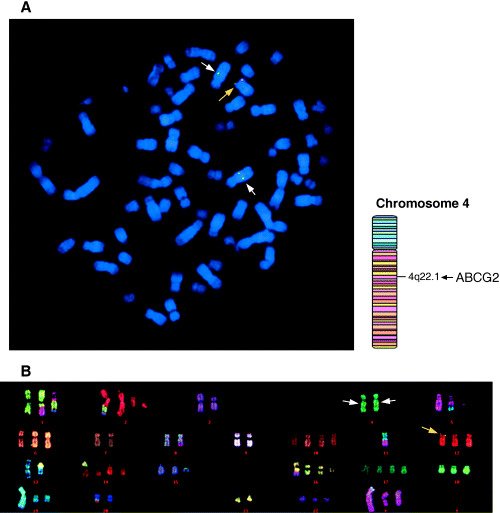
Flp‐mediated integration of the BCRP/MXR1/ABCP (ABCG2) cDNA into Flp recombination target (FRT)‐tagged genomic DNA. (A) Fluorescence in situ hybridization (FISH) analysis of the Flp‐In‐293/ABCG2 cells. (B) Multicolor FISH analysis of the Flp‐In‐293/ABCG2 cells. Biotin‐labeled ABCG2‐pcDNA5/FRT and digoxigenin‐labeled pcDNA5/FRT plasmid DNAs were used for chromosome mapping as probes. Multicolor FISH procedures were carried out using the Multicolor FISH‐Human probe set. In the FISH mapping probed with ABCG2‐pcDNA5/FRT and multicolor FISH‐human probe set the hybridization signal of the transgene was detected in the telomeric region of the short arm of one chromosome 12 (yellow arrows) as overlapped signals of the biotin‐labeled ABCG2‐pcDNA5/FRT and digoxigenin‐labeled pcDNA5/FRT, and two hybridization signals of the biotin‐labeled ABCG2‐pcDNA5/FRT (internal ABCG2 genes, white arrows) were detected in the q22 region on a pair of chromosome 4.
Characterization of WT ABCG2, V12M and Q141K expressed in Flp‐In‐293 cells. As shown in Fig. 2A, mRNA levels of WT ABCG2 as well as its major SNP variants (V12M and Q141K) were represented evenly in Flp‐In cells. The mRNA levels of ABCG2 and GAPDH were measured by quantitative PCR, and the ratios of ABCG2 variants versus GAPDH were plotted. In addition, the ABCG2 and GAPDH proteins were detected by immunoblotting and their expression levels were quantified. There was a linear relationship between the signal intensity of immunoblotting and the logarithmic value of the amount of protein applied to the electrophoresis (data not shown). Based on the linear relationship, the expression levels of ABCG2 or GAPDH in whole‐cell lysate samples were estimated quantitatively. The relative values of protein levels were then normalized to the ratio of WT ABCG2/GAPDH, as described previously.( 16 ) As demonstrated in Fig. 2A, the protein expression level of the Q141K variant was approximately half that of the WT level.
Figure 2.
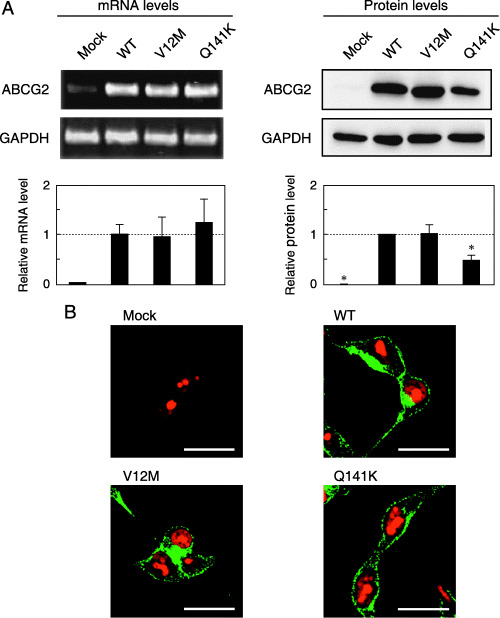
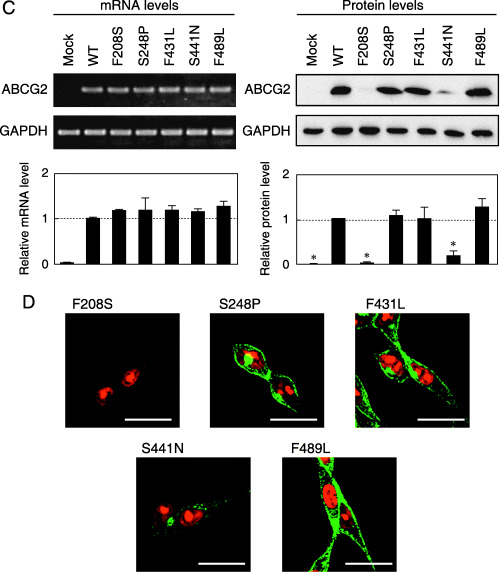
mRNA and protein expression levels as well as immunofluorescence images of wild‐type (WT) BCRP/MXR1/ABCP (ABCG2) and variants expressed in Flp‐In‐293 cells. (A,C) Relative levels of mRNA (A: Major SNP variants; C: Minor SNP variants) were detected by reverse transcription–quantitative polymerase chain reaction with specific primers for ABCG2 and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). Data were calculated as ratios by referring to the GAPDH mRNA levels in Flp‐In‐293 cells and normalized to the ratio of ABCG2/GAPDH. Data are expressed as mean values ± SD (n = 4). Relative levels of ABCG2 protein were detected by immunoblotting. Briefly, ABCG2‐expressing Flp‐In‐293 cells were treated with the lysis buffer containing 50 mM Tris/HCl (pH 7.4), 1% (w/v) Triton X‐100, 1 mM DTT, and protease inhibitors. The sample was homogenized by passing it through a 27‐gauge needle. The resulting homogenated samples (10 µg of protein) were subjected to sodium dodecylsulfate–polyacrylamide gel electrophoresis in the presence of 2‐mercaptoethanol. ABCG2 protein was detected by immunoblotting analysis with BXP‐21 monoclonal antibody. Data were calculated as ratios by referring to the GAPDH protein levels in Flp‐In‐293 cells and normalized to the ratio of ABCG2/GAPDH. Data are expressed as mean values ± SD in triplicate experiments. Statistical significance (*P < 0.05) was evaluated by the Student's t‐test. (B,D) Immunofluorescence (B: Major SNP variants; D: Minor SNP variants) images of WT ABCG2 and variants expressed in Flp‐In‐293 cells. The ABCG2 protein was linked immunologically with Alexa Fluor 488 (green fluorescence), and nuclei were stained with propidium iodide (red fluorescence). Horizontal bars correspond to 20 µm.
Figure 2B depicts the immunofluorescence images of Flp‐In‐293/Mock, Flp‐In‐293/ABCG2 (WT), Flp‐In‐293/ABCG2 (V12M) and Flp‐In‐293/ABCG2 (Q141K) cells. ABCG2 proteins were probed with the BXP‐21 antibody and then labeled with green fluorescent dye (Alexa Fluor 488), whereas DNA in the nuclei was stained with propidium iodide (red fluorescence). In Flp‐In‐293/Mock cells, no green fluorescence was detected, suggesting that endogenous expression of ABCG2 was negligibly low. In contrast, strong green fluorescence was observed at the plasma membrane and within intracellular compartments in Flp‐In‐293 cells expressing WT ABCG2 and the two variants.
Drug resistance profiles of Flp‐In‐293 cells expressing WT ABCG2, V12M and Q141K variants toward camptothecin analogs. To examine the drug resistance profiles, we incubated Flp‐In‐293/ABCG2 (WT), Flp‐In‐293/ABCG2 (V12M) and Flp‐In‐293/ABCG2 (Q141K) cells with SN‐38 and the new camptothecin analogs at different concentrations for 96 h, after which we observed the cell survival rates. Flp‐In‐293/ABCG2 (WT), Flp‐In‐293/ABCG2 (V12M) and Flp‐In‐293/ABCG2 (Q141K) cells exhibited strong resistance to SN‐38, SN‐355 and SN‐398, as represented by large IC50 values (Fig. 3). Interestingly, the V12M variant provided the cells with higher drug resistance to SN‐38, SN‐355 and SN‐398 than did WT ABCG2 (Fig. 3). In contrast, the Q141K variant appeared to render drug resistance at a level only half that of WT. Moderate resistance of those cells was observed to SN‐392. In contrast, none of Flp‐In‐293/ABCG2 (WT), Flp‐In‐293/ABCG2 (V12M) or Flp‐In‐293/ABCG2 (Q141K) cells exhibited resistance toward SN‐22, SN‐343, SN‐348, SN‐349, SN‐351, SN‐352, SN‐353, SN‐364, SN‐397, SN‐443 or SN‐444. These results suggest that camptothecin analogs lacking the hydroxyl or amino group at position 10 or 11 are not substrates for WT ABCG2 or the V12M and Q141K variants.
Figure 3.
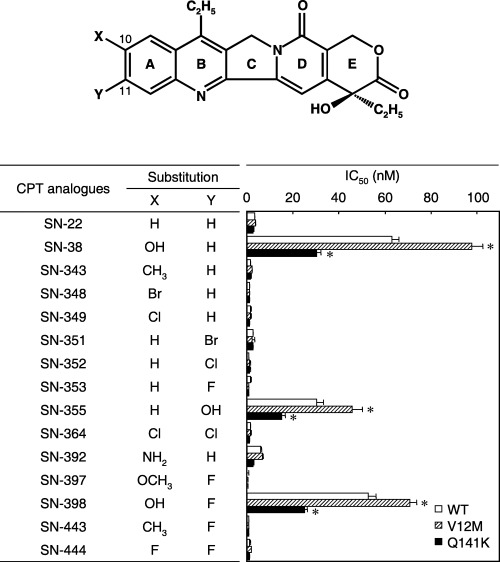
The effect of camptothecin analogs on the growth of Flp‐In‐293/ABCG2 (wild type), Flp‐In‐293/ABCG2 (V12M) or Flp‐In‐293/ABCG2 (Q141K) cells. Camptothecin analogues were synthesized by substituting X and Y at positions 10 and 11, respectively, as indicated in this figure. Drug sensitivity was detected by the MTT assay after a 96‐h drug exposure. IC50 values represent drug concentrations producing a 50% reduction of cell growth. Data are expressed as mean values ± SD in multireplicated experiments (n = 3). The statistical significance was determined according to the two‐sided Student's t‐test (*P < 0.01).
Characterization of the F208S, S248P, F431L, S441N and F489L variants expressed in Flp‐In‐293 cells. Figure 2C demonstrates the mRNA and protein levels of WT ABCG2 and the F208S, S248P, F431L, S441N and F489L variants expressed in Flp‐In‐293 cells. Whereas mRNA levels were almost the same in WT and those variants, the protein levels of the F208S and S441N variants were markedly low. The immunofluorescence images of Flp‐In‐293/ABCG2 (F208S) and Flp‐In‐293/ABCG2 (S441N) cells revealed that those variant proteins were not expressed in the plasma membrane (Fig. 2D). The S441N variant appeared to remain in the intracellular space. The other variants (S248P, F431L and F489L) were expressed in the plasma membrane, as was WT ABCG2.
We examined the drug resistance profiles of Flp‐In‐293 cells transfected with WT ABCG2 or those variants toward SN‐38 and mitoxantrone. Figure 4 shows cell‐survival curves of those cells that were incubated with SN‐38 (Fig. 4A) and mitoxantrone (Fig. 4B) at different concentrations for 72 h. Cells transfected with F 208S or S441N did not exhibit any resistance to SN‐38 ormitoxantrone; their cell survival curves were very similar to that of Flp‐In‐293/Mock cells. On the contrary, the S248P, F431L and F489L variants contributed drug resistance; however, their contribution was not so large as that of WT.
Figure 4.
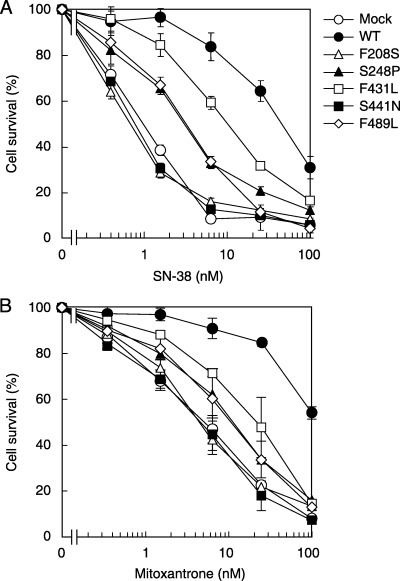
Drug resistance profiles of Flp‐In‐293 cells expressing the wild‐type (WT) BCRP/MXR1/ABCP (ABCG2), F208S, S248P, F431L, S441N or F489L variants toward (A) SN‐38 and (B) mitoxantrone. Cells were incubated in 100 µL of the culture medium containing SN‐38 or mitoxantrone at different concentrations in 96‐well plates in a humidified tissue‐culture chamber (37°C, 5% CO2). After 72 h, surviving cells were detected by the MTT assay. Data are expressed as mean values ± SD in multireplicated experiments (n = 6).
Overview of the drug resistance profiles of Flp‐In‐293 cells expressing WT ABCG2 and SNP variants. Table 1 summarizes the drug resistance profiles of Flp‐In‐293 cells expressing WT ABCG2 and the SNP variants tested in this study. Cells were incubated with SN‐38, mitoxantrone, doxorubicin, daunorubicin, etoposide and vincristine at different concentrations in similar manners as shown in Fig. 4. It is important to note that the IC50 values of Q141K‐expressing cells toward doxorubicin and daunorubicin were greater than that of WT‐expressing cells, whereas Q141K‐expressing cells were more sensitive to SN‐38 and mitoxantron compared with WT‐expressing cells. These results suggest that the substrate specificity of the Q141K variant differs delicately from that of WT ABCG2. Considered overall, the minor SNP variants were less effective in drug resistance than the V12M and Q141K variants.
Table 1.
Resistance profile (IC50) of ABCG2
| Compound | IC50 (nM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mock | Wild type | V12M | Q141K | F208S | S248P | F431L | S441N | F489L | |
| SN‐38 | 0.9 | 40.0 (44.4) | 40.0 (44.4) | 17.0 (18.9) | 0.6 (0.7) | 3.0 (3.3) | 10.0 (11.1) | 0.7 (0.8) | 3.1 (3.4) |
| Mitoxantorone | 5.2 | >100 (>19) | 92.0 (17.7) | 45.0 (8.7) | 4.5 (0.9) | 11.0 (2.1) | 21.0 (4.0) | 4.6 (0.9) | 11.0 (2.1) |
| Doxorubicin | 32.0 | 78.0 (2.4) | 100.0 (3.1) | 110.0 (3.4) | 20.0 (0.6) | 20.0 (0.6) | 40.0 (1.3) | 21.0 (0.7) | 45.0 (1.4) |
| Daunorubicin | 12.0 | 30.0 (2.5) | 50.0 (4.2) | 50.0 (4.2) | 12.0 (1.0) | 21.0 (1.8) | 14.0 (1.2) | 12.0 (1.0) | 19.0 (1.6) |
| Etoposide | 110.0 | 200.0 (1.8) | 220.0 (2.0) | 200.0 (1.8) | 110.0 (1.0) | 120.0 (1.1) | 120.0 (1.1) | 130.0 (1.2) | 170.0 (1.5) |
| Vincristine | 1.4 | 4.0 (2.9) | 5.0 (3.6) | 4.5 (3.2) | 0.6 (0.4) | 4.0 (2.9) | 1.4 (1.0) | 0.8 (0.6) | 2.8 (2.0) |
Relative resistances to mock cells are described in parentheses. IC50, drug concentrations producing a 50% reduction of cell growth.
Prazosin‐stimulated ATPase activity of SNP variants. Because acquired mutants (i.e. R482G and R482T) are known to exhibit prazosin‐stimulated ATPase activity,( 35 ) we examined whether SNP variants carry such ATPase activity. For this purpose, we expressed WT ABCG2, V12M, Q141K, S248P, F431L, F489L, R482G and R482T in Sf9 insect cells and prepared plasma membranes as described previously,( 16 , 35 ) as the plasma membrane of Sf9 cells has lower endogenous background ATPase activity than Flp‐In‐293 cells. None of the SNP variants exhibited prazosin‐stimulated ATPase activity (data not shown), whereas the ATPase activity was markedly stimulated with prazosin in those acquired mutants (R482G and R482T).( 35 )
Discussion
Non‐synonymous polymorphisms of ABCG2 and inconsistencies among previous reports. Honjo et al. first identified non‐synonymous SNP of V12M and Q141K.( 23 ) The V12M polymorphism in exon 2 (34G > A) affects the N‐terminal intracellular region of the protein. Both the WT and the variant amino acids have uncharged, hydrophobic side chains. The V12M polymorphism was found in all ethnic groups tested, with the highest allele frequency in Mexican‐Indians (90% of only five individuals tested), but only 1.7% in a Swedish population.( 20 , 29 ) The Pacific Islanders, South‐east Asians and Hispanics have the Met12 allele at frequencies of 64, 45 and 40%, respectively. Thus, there is a large difference in the allele frequency of the V12M polymorphism among different ethnic groups. This SNP variant, however, is not considered to affect clinical outcomes as no remarkable change was observed in its protein expression level or substrate specificity compared with WT ABCG2 (Table 1).
The Q141K polymorphism located in exon 5 (421C > A) leads to the replacement of the negatively charged glutamic acid residue with a positively charged lysine residue. This polymorphism resides between the Walker A motif (amino acid residues 83–89) and the signature C region (amino acid residues 186–189). The Q141K variant was also detected in all ethnic groups tested, with the allele frequency ranging from 0 to 35% (Africans North of the Sahara, sub‐Saharan Africans and African‐American subjects with low frequencies; Japanese and Chinese populations with high allele frequencies).( 22 , 25 , 27 , 29 )
An investigation of the expression level of ABCG2 in 99 Japanese placenta samples revealed that individuals homozygous for the Q141K variant showed significantly lower expression levels of this transporter protein, whereas the heterozygous samples displayed an intermediate expression level.( 27 ) It has been reported that the SNP Q141K may cause increased plasma concentrations of orally administered diflomotecan in cancer patients who express low levels of ABCG2 (Q141K) protein in the small intestine.( 36 ) In contrast, Zamber et al.,( 29 ) investigating the expression of natural allelic variants of ABCG2 in human intestine, demonstrated no significant differences in mRNA between subjects expressing the Lys141 (K141) allele in heterozygous form compared with the WT Gln141 (Q141) allele.
By using an in‐vitro system, Imai et al. showed that ABCG2 Q141K variant‐transfected PA317 cells had a low‐level of drug resistance associated with decreased protein expression.( 25 ) This finding is apparently in accordance with a report by Kondo et al.( 31 ) However, Morisaki et al.( 30 ) reported that the protein expression level of ABCG2 Q141K varied among clones of HEK293 cells where ABCG2 Q141K cDNA was expressed stably with the conventional pcDNA3.1(+) vector. In contrast, Mizuarai et al.( 28 ) described that approximately equal levels of ABCG2 were expressed in Q141K clones compared to the expression in WT, although they did not present any data in this respect. The reason for these discrepancies is considered to be due to differences in their expression procedure or expression systems. In the conventional vector‐mediated introduction of cDNA into mammalian cells by calcium phosphate coprecipitation or permeation with lipofectamine, integration of cDNA into the host's chromosomal DNA occurs randomly at unpredictable sites and therefore the number of integrated recombinant DNAs is not controllable. Although this results in significant clonal variation and compromises direct comparison of expression among variants, until now no one has carefully investigated these problems.
In the present study, to re‐evaluate the effect of genetic polymorphisms of ABCG2 on the protein expression level and drug resistance profile, we have used the Flp‐In method to integrate cDNA of ABCG2 variants into FRT‐tagged genomic DNA. The Flp‐In method is based on the exchange of an expression cassette within a previously tagged FRT site. The targeting constructs harboring the gene of interest replace the tagged reporter cassette precisely, making use of the Flp recombinase, and therefore the copy number of the recombinant DNA is well controlled, as evidenced by Fig. 1B and Fig. 2A,C. Based on the new method, our present study (Fig. 2A) supports the original report of Imai et al.,( 25 ) suggesting that the protein stability of the Q141K variant is reduced without any detectable changes in its mRNA levels.
As clearly demonstrated in this study, the F208S, S248P, F431L, S441N and F489L variants exhibited greatly altered protein expression levels (Fig. 2C) or drug resistance profiles (Fig. 4 and Table 1). In particular, expression levels of the F208S and S441N variants were markedly low (Fig. 2C). It is likely that such reduced protein levels are associated with the instability or degradation of those variant proteins. In fact, when Flp‐In‐293/ABCG2 (F208S) cells were treated with MG132, a proteasome inhibitor, the protein level recovered up to approximately 50% of the WT level, suggesting the involvement of proteasomes in degradation of the F208S variant (data not shown).
The frequencies of the Asn441 (N441) and Leu489 (L489) alleles were relatively low (less than 1%) compared with those of the Met12 (M12) and Lys141 (K141) alleles.( 19 ) These minor alleles were found only in the Japanese population.( 26 , 27 ) However, the Ser208 (S208) and Pro248 (P248) alleles are registered in the NCBI dbSNP database, but their allele frequencies are not available. The most recent version of NCBI dbSNP does not appear to contain validation for F208S and S248P as bona fide SNP. Thus, the clinical significance of these minor alleles in cancer chemotherapy remains to be elucidated.
Functional classification of non‐synonymous polymorphisms of ABCG2. As one of the specific aims of the present study, we functionally classified the non‐synonymous polymorphisms (V12M, Q141K, F208S, S248P, F431L, S441N and F489L) in terms of their protein expression level, drug resistance profile and prazosin‐stimulated ATPase activity. As summarized in Fig. 5A, the functional properties of these variants were compared with acquired mutations of R482G and R482T. The drug resistance profiles and prazosin‐stimulated ATPase activity of R482G and R482T were reported previously.( 14 , 35 ) Based on the experimental data hitherto obtained, those variants and mutants were classified into four groups (Fig. 5B), where the length of each line reflects the number of differences between the corresponding variant and WT with respect to the items described in Table 1.
Figure 5.
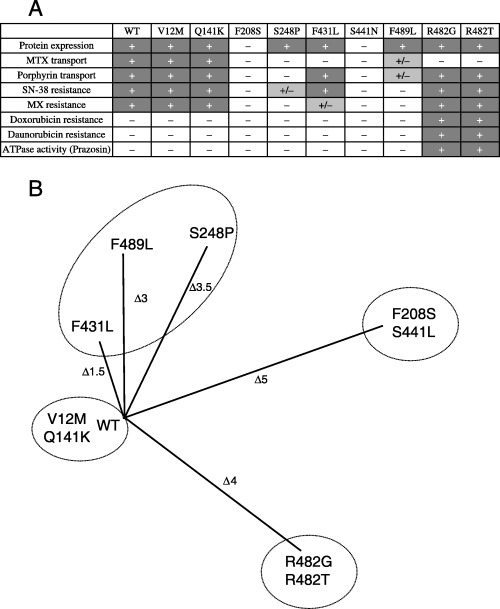
(A) Characterization and (B) classification of wild‐type (WT) BCRP/MXR1/ABCP (ABCG2) and single nucleotide polymorphism (SNP) variants. (A) The properties of WT ABCG2 and SNP variants were characterized as + (positive), – (negative) or +/– (marginal) according to the following indexes: protein expression, transport of methotrexate (MTX) or porphyrin, resistance to SN‐38, mitoxantrone (MX), doxorubicin or daunorubicin, and prazosin‐stimulated ATPase activity. (B) WT ABCG2 and SNP variants are largely classified into four groups, where each line length indicates the difference of a variant from WT according to the characteristic indexes.
As shown in Fig. 5B, it is obvious that WT, V12M and Q141K form one group where protein expression, methotrexate and porphyrin transport, and resistance to SN‐38 and mitoxantrone are positive, but contribution to doxorubicin‐ and daurorubicin‐resistance as well as prazosin‐stimulated ATPase activity are negative. In contrast, the acquired mutants R482G and R482T form another group, which is characterised by a positive contribution to drug resistance toward SN‐38, mitoxantrone, doxorubicin and daurorubicin as well as prazosin‐stimulated ATPase activity. Interestingly, these acquired mutants lack methotrexate transport activity, although they do transport porphyrin.( 16 ) Both F208S and S441N belong to the third group where protein expression levels were extremely low (Fig. 2C). Therefore, their transport activity and contribution to drug resistance are minimal. Finally, S248P, F431L and F489L appear to form the fourth heterogeneous group, where those variant proteins are expressed at normal levels but exhibit different profiles of drug resistance and transport activity.
New camptothecin analogs beyond genetic polymorphisms of ABCG2. In the present study, we tested the pharmacological potency of our new camptothecin analogs to determine whether they could circumvent ABCG2‐associated drug resistance in human tumor cells without the influence of the genetic polymorphisms of AGCG2. We have previously reported that ABCG2 is involved in the active extrusion of SN‐38 and its metabolites from cancer cells.( 13 , 14 ) Based on the structure–activity relationship, we have recently designed novel camptothecin analogs that are not extruded by ABCG2.( 32 , 33 ) As demonstrated in Fig. 3, the new camptothecin analogs that were non‐substrates for ABCG2 circumvented ABCG2‐mediated drug resistance without any influence from major SNP (i.e. V12M and Q141K). Such structure–activity relationship‐based drug molecular design may provide a new approach to develop anticancer drugs that could be used in a large patient population with heterogeneous genetic polymorphisms of the drug‐efflux ABC transporters.
Acknowledgments
We thank Professor Eiry Kobatake and Dr Masayasu Mie (Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology) for their kind help and advice in performing immunofluorescence microscopy experiments. This study was supported by the NEDO International Joint Research Grant program ‘International standardization of functional analysis technology for genetic polymorphisms of drug transporters’.
References
- 1. Doyle LA, Yang W, Abruzzo LV et al. A multidrug resistance transporter from human MCF‐7 breast cancer cells. Proc Natl Acad Sci USA 1998; 95: 15 665 – 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allikmets R, Schriml LM, Hutchinson A, Romano‐Spica V, Dean M. A human placenta‐specific ATP‐binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res 1998; 58: 5337 – 9. [PubMed] [Google Scholar]
- 3. Miyake K, Mickley L, Litman T et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone‐resistant cells: demonstration of homology to ABC transport genes. Cancer Res 1999; 59: 8 – 13. [PubMed] [Google Scholar]
- 4. Bailey‐Dell KJ, Hassel B, Doyle LA, Ross DD. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP‐binding cassette transporter G2) gene. Biochim Biophys Acta 2001; 1520: 234 – 41. [DOI] [PubMed] [Google Scholar]
- 5. Henriksen U, Fog JU, Litman T, Gether U. Identification of intra‐ and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. J Biol Chem 2005; 280: 36 926 – 34. [DOI] [PubMed] [Google Scholar]
- 6. Kage K, Fujita T, Sugimoto Y. Role of Cys‐603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci 2005; 96: 866 – 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wakabayashi K, Nakagawa H, Adachi T et al. Identification of cysteine residues critically involved in homodimer formation and protein expression of human ATP‐binding cassette transporter ABCG2: a new approach using the flp recombinase system. J Exp Ther Oncol 2006; 5: 205 – 22. [PubMed] [Google Scholar]
- 8. Jonker JW, Buitelaar M, Wagenaar E et al. The breast cancer resistance protein protects against a major chlorophyll‐derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA 2002; 99: 15 649 – 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maliepaard M, Van Gastelen MA, De Jong LA et al. Overexpression of the BCRP/MXR/ABCP gene in a topotecan‐selected ovarian tumor cell line. Cancer Res 1999; 59: 4559 – 63. [PubMed] [Google Scholar]
- 10. Ross DD, Yang W, Abruzzo LV et al. Atypical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone‐selected cell lines. J Natl Cancer Inst 1999; 91: 429 – 33. [DOI] [PubMed] [Google Scholar]
- 11. Brangi M, Litman T, Ciotti M et al. Camptothecin resistance: role of the ATP‐binding cassette (ABC), mitoxantrone‐resistance half‐transporter (MXR), and potential for glucuronidation in MXR‐expressing cells. Cancer Res 1999; 59: 5938 – 46. [PubMed] [Google Scholar]
- 12. Kawabata S, Oka M, Shiozawa K et al. Breast cancer resistance protein directly confers SN‐38 resistance of lung cancer cells. Biochem Biophys Res Commun 2001; 280: 1216 – 23. [DOI] [PubMed] [Google Scholar]
- 13. Nakatomi K, Yoshikawa M, Oka M et al. Transport of 7‐ethyl‐10‐hydroxycamptothecin (SN‐38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem Biophys Res Commun 2001; 288: 827 – 32. [DOI] [PubMed] [Google Scholar]
- 14. Yoshikawa M, Ikegami Y, Sano K et al. Transport of SN‐38 by the wild type of human ABC transporter ABCG2 and its inhibition by quercetin, a natural flavonoid. J Exp Ther Oncol 2004; 4: 25 – 35. [PubMed] [Google Scholar]
- 15. Robey RW, Steadman K, Polgar O, Bates SE. ABCG2‐mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther 2005; 4: 187 – 94. [PubMed] [Google Scholar]
- 16. Tamura A, Watanabe M, Saito H et al. Functional validation of the genetic polymorphisms of human ATP‐binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transport. Mol Pharmacol 2006; 70: 287 – 96. [DOI] [PubMed] [Google Scholar]
- 17. Yanase K, Tsukahara S, Mitsuhashi J, Sugimoto Y. Functional SNPs of the breast cancer resistance protein: therapeutic effects and inhibitor development. Cancer Lett 2006; 234: 73 – 80. [DOI] [PubMed] [Google Scholar]
- 18. De Jong FA, De Jonge MJ, Verweij J, Mathijssen RH. Role of pharmacogenetics in irinotecan therapy. Cancer Lett 2006; 234: 90 – 106. [DOI] [PubMed] [Google Scholar]
- 19. Ishikawa T, Tamura A, Saito H, Wakabayashi K, Nakagawa H. Pharmacogenomics of the human ABC transporter ABCG2: from functional evaluation to drug molecular design. Naturwissenschaften 2005; 92: 451 – 63. [DOI] [PubMed] [Google Scholar]
- 20. Backstrom G, Taipalensuu J, Melhus H et al. Genetic variation in the ATP‐binding cassette transporter gene ABCG2 (BCRP) in a Swedish population. Eur J Pharm Sci 2003; 18: 359 – 64. [DOI] [PubMed] [Google Scholar]
- 21. Bosch TM, Kjellberg LM, Bouwers A et al. Detection of single nucleotide polymorphisms in the ABCG2 gene in a Dutch population. Am J Pharmacogenomics 2005; 5: 123 – 31. [DOI] [PubMed] [Google Scholar]
- 22. De Jong FA, Marsh S, Mathijssen RH et al. ABCG2 pharmacogenetics: ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin Cancer Res 2004; 10: 5889 – 94. [DOI] [PubMed] [Google Scholar]
- 23. Honjo Y, Morisaki K, Huff LM et al. Single‐nucleotide polymorphism (SNP) analysis in the ABC half‐transporter ABCG2 (MXR/BCRP/ABCP1). Cancer Biol Ther 2002; 1: 696 – 702. [DOI] [PubMed] [Google Scholar]
- 24. Iida A, Saito S, Sekine A et al. Catalog of 605 single‐nucleotide polymorphisms (SNPs) among 13 genes encoding human ATP‐binding cassette transporters: ABCA4, ABCA7, ABCA8, ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8 . J Hum Genet 2002; 47: 285 – 310. [DOI] [PubMed] [Google Scholar]
- 25. Imai Y, Nakane M, Kage K et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low‐level drug resistance. Mol Cancer Ther 2002; 1: 611 – 16. [PubMed] [Google Scholar]
- 26. Itoda M, Saito Y, Shirao K et al. Eight novel single nucleotide polymorphisms in ABCG2/BCRP in Japanese cancer patients administered irinotacan. Drug Metab Pharmacokinet 2003; 18: 212 – 17. [DOI] [PubMed] [Google Scholar]
- 27. Kobayashi D, Ieiri I, Hirota T et al. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos 2005; 33: 94 – 101. [DOI] [PubMed] [Google Scholar]
- 28. Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced ATPase activity in multidrug transporter ABCG2. Int J Cancer 2004; 109: 238 – 46. [DOI] [PubMed] [Google Scholar]
- 29. Zamber CP, Lamba JK, Yasuda K et al. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics 2003; 13: 19 – 28. [DOI] [PubMed] [Google Scholar]
- 30. Morisaki K, Robey RW, Ozvegy‐Laczka C et al. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol 2005; 56: 161 – 72. [DOI] [PubMed] [Google Scholar]
- 31. Kondo C, Suzuki H, Itoda M et al. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res 2004; 21: 1895 – 903. [DOI] [PubMed] [Google Scholar]
- 32. Yoshikawa M, Ikegami Y, Hayasaka S et al. Novel camptothecin analogues that circumvent ABCG2‐associated drug resistance in human tumor cells. Int J Cancer 2004; 110: 921 – 7. [DOI] [PubMed] [Google Scholar]
- 33. Nakagawa H, Saito H, Ikegami Y, Aida‐Hyugaji S, Sawada S, Ishikawa T. Molecular modeling of new camptothecin analogues to circumvent ABCG2‐mediated drug resistance in cancer. Cancer Lett 2006; 234: 81 – 9. [DOI] [PubMed] [Google Scholar]
- 34. Matsuda Y, Chapman VM. Application of fluorescence in situ hybridization in genome analysis of the mouse. Electrophoresis 1995; 16: 261 – 72. [DOI] [PubMed] [Google Scholar]
- 35. Ishikawa T, Kasamatsu S, Hagiwara Y, Mitomo H, Kato R, Sumino Y. Expression and functional characterization of human ABC transporter ABCG2 variants in insect cells. Drug Metab Pharmacokinet 2003; 18: 194 – 202. [DOI] [PubMed] [Google Scholar]
- 36. Sparreboom A, Gelderblom H, Marsh S et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C > A genotype. Clin Pharmacol Ther 2004; 76: 38 – 44. [DOI] [PubMed] [Google Scholar]