

Abstract
This phase I, open‐label study investigated the Toll‐like receptor 9 agonist, PF‐3512676, in combination with carboplatin and paclitaxel in Japanese patients with advanced, non‐small‐cell lung cancer (NSCLC). Patients (n = 12) with treatment‐naive stage IIIB or IV NSCLC received single‐agent PF‐3512676 subcutaneously once during the first 7 days (monotherapy phase) in three escalating dose levels (0.1, 0.2, and 0.4 mg/kg) followed by a combination phase during which patients received 0.1 or 0.2 mg/kg PF‐3512676 subcutaneously on days 8 and 15 of each 3‐week cycle of carboplatin (area under the curve, 6 mg × min/mL) and paclitaxel (200 mg/m2). Safety and pharmacokinetics of PF‐3512676 were assessed during monotherapy and combination therapy phases. PF‐3512676 was tolerable as monotherapy or in combination with chemotherapy in patients with NSCLC. Most common treatment‐related, non‐hematologic adverse events (AEs) throughout the study were injection‐site reactions (n = 12, 100%) and flu‐like symptoms (n = 11, 91.7%) that were each grade 1 or 2 in all but one patient. All patients experienced neutropenia and leukopenia (≥grade 3 in 11 [91.7%] and seven [58.3%] patients, respectively). One patient in dose level 2 had a dose‐limiting toxicity: grade 3 rash and grade 3 increase in γ‐glutamyltransferase during combination therapy. Mean PF‐3512676 half‐life ranged from 4.8 to 21.6 h (longer with higher doses). Four (33%) patients had objective responses (one complete response, three partial responses), and seven (58%) patients achieved stable disease. PF‐3512676 as monotherapy and in combination with chemotherapy had an acceptable safety profile in Japanese patients with treatment‐naive NSCLC. (Cancer Sci 2009)
Worldwide, lung cancer accounts for 1.3 million deaths per year, and cancers of the lung, trachea, and bronchus are the leading cause of cancer‐related death in Japanese men. Non‐small‐cell lung cancer (NSCLC) accounts for approximately 80% of lung cancers,( 1 ) and the vast majority (∼70%) of cases of NSCLC are locally advanced or metastatic at diagnosis.( 2 ) The current standard first‐line treatment for patients with stage IIIB or IV NSCLC and good performance status is doublet chemotherapy with a platinum agent (e.g. carboplatin or cisplatin) in combination with paclitaxel, docetaxel, gemcitabine, or vinorelbine.( 3 , 4 ) This treatment is associated with objective response rates of approximately 20% to 40% and median survival of 8 to 10 months, which is not considered satisfactory to patients.( 4 , 5 ) Therefore, development of more effective treatment regimens is warranted for the unmet medical needs of patients with advanced NSCLC.
Toll‐like receptors (TLRs) are a family of specialized immune receptors that induce protective immune responses upon detection of highly conserved pathogen‐expressed molecules. To date, 10 different TLRs have been identified in humans.( 6 , 7 ) Each TLR binds one or more distinct pathogen‐expressed molecules and can function as an immune system ‘alarm signal,’ leading to initiation of appropriate host immune defenses.( 6 , 8 ) In humans, TLR9 is expressed primarily by plasmacytoid dendritic cells (pDCs) and B cells. It recognizes unmethylated cytosine‐phosphate‐guanine (CpG) dinucleotide sequences commonly found in bacterial and viral DNA.( 9 ) TLR9 can also be stimulated using synthetic oligodeoxynucleotides (ODNs) containing one or more unmethylated CpG dinucleotide motifs. This stimulation leads to activation of type 1 helper T cell (TH1)‐like innate immunity, including upregulated production of interleukin (IL)‐6, IL‐12p40, interferon‐alpha (IFN‐α), and IFN‐inducible chemokines such as interferon‐γ‐inducible protein 10 (IP‐10).( 10 ) Innate immune activation with a TLR9 agonist may enhance tumor antigen presentation and promote an antitumor immune response.
PF‐3512676 (formerly known as CpG 7909) is a TLR9 agonist that has been tested in clinical trials for the treatment of patients with several types of cancer.( 11 ) This synthetic CpG ODN can induce potent innate and adaptive immune TH1 responses, and to a lesser extent, TH2 immune responses in murine models.( 9 , 10 , 11 , 12 ) Preclinical evidence supporting the use of PF‐3512676 in lung cancer was provided by studies with a murine Lewis lung cancer model in which mice treated with PF‐3512676 in combination with paclitaxel had significantly prolonged survival compared to mice treated with either drug given alone (P < 0.0001).( 13 ) This preclinical evidence, combined with the promising clinical activity of PF‐3512676 in other types of advanced cancer, supported investigation in patients with NSCLC. In non‐clinical studies in mice investigating efficacy of PF‐3512676 plus paclitaxel in the metastatic Renca renal cell carcinoma (RCC) models, survival following treatment with PF‐3512676 was longer with regional divided dosing and weekly administration compared with temporal divided dosing using twice‐weekly administration. Furthermore, in early clinical studies, elevations of IP‐10 observed after dosing with PF‐3512676 returned to baseline levels after about 1 week. Therefore, PF‐3512676 was administered weekly with rotation of administration sites in clinical studies. Chemotherapy and PF‐3512676 were not co‐administered because chemotherapy was intended to cause decomposition of tumor cells and release of tumor antigens. PF‐3512676 was administered after chemotherapy so that pDCs activated through the TLR9 pathway might present these antigens, thus increasing the number of antigen‐specific, cytotoxic T cells.
The safety of PF‐3512676 has been studied in more than 800 subjects, including more than 400 cancer patients. The extensive human clinical experience demonstrates that PF‐3512676 is safe and well tolerated. In phase I studies in Western patients, 0.0025 to 0.81 mg/kg PF‐3512676 weekly subcutaneous doses have been evaluated. To date, no organ dysfunction meeting the protocol‐defined dose‐limiting toxicity (DLT) criteria has been reported in any of these studies, and the maximum tolerated dose has not been defined.
In phase I or II studies of single‐agent PF‐3512676, the minimum dose level at which objective response was reported was 0.16 mg/kg weekly in patients with cutaneous T‐cell lymphoma or RCC and 6 mg (approximately 0.10 mg/kg) weekly in patients with advanced melanoma.( 14 , 15 , 16 ) A subsequent, randomized phase II study in Western patients with chemotherapy‐naive NSCLC investigated PF‐3512676 (0.2 mg/kg) in combination with standard taxane/platinum doublet chemotherapy (n = 74) and chemotherapy alone (n = 37).( 17 ) The PF‐3512676 dose of 0.2 mg/kg was selected to be above the minimum dose associated with antitumor activity in phase I and II studies of single‐agent PF‐3512676 and below the dose level (0.24 mg/kg) that, in the same single‐agent studies, had been well tolerated for up to 6 months by the majority of patients. In the randomized phase II NSCLC study, the response rate in the PF‐3512676 plus chemotherapy arm was higher than that in the chemotherapy‐alone arm (30%vs 19% confirmed response rate, respectively). In addition, there was a trend toward improved median overall survival with addition of PF‐3512676 to chemotherapy (12.3 months compared with 6.8 months for chemotherapy alone, P = 0.188). One‐year survival was 50% and 33% for PF‐3512676 plus chemotherapy and chemotherapy alone, respectively. Common adverse events (AEs) considered related to treatment with PF‐3512676 and not to chemotherapy were injection‐site reactions and flu‐like symptoms. Other, less‐common AEs considered related to treatment with PF‐3512676 were febrile neutropenia, anemia, and thrombocytopenia. Overall, a 0.2 mg/kg dose of PF‐3512676 in combination with taxane/platinum doublet chemotherapy appeared to have promising antitumor activity as well as a favorable safety profile and was recommended for further study in patients with advanced NSCLC.( 17 )
The present phase I study was conducted to investigate the safety and pharmacokinetics of PF‐3512676 both as monotherapy and in combination with carboplatin and paclitaxel as first‐line therapy for Japanese patients with advanced NSCLC.
Patients and Methods
Patients. Patients aged 20 to 75 years with histopathologically or cytologically diagnosed, previously untreated stage IIIB or IV NSCLC were eligible. To enroll in the study, patients were required to have a life expectancy ≥3 months, an Eastern Cooperative Oncology Group performance status (ECOG PS) of ≤1, and at least one measurable lesion of ≥20 mm according to Response Evaluation Criteria in Solid Tumors (RECIST).( 18 ) Patients were also required to have adequate renal, liver, and bone marrow function (serum creatinine <1.5 × upper limit of normal [ULN], total bilirubin <1.5 × ULN, aspartate aminotransferase and alanine aminotransferase <2.5 × ULN, absolute neutrophil count ≥2000/mm3, platelets ≥100 000/mm3, and hemoglobin ≥10 g/dL).
Patients were excluded if they had brain or central nervous system metastases that were symptomatic or requiring treatment; any other malignancies within the past 5 years (except non‐melanoma skin cancer or adequately treated in situ cervical cancer, gastric cancer, or colorectal cancer); autoimmune or antibody‐mediated diseases; possible hypersensitivity to ODNs or castor oil; or hepatitis B or C infection. In addition, patients were excluded if they had participated in any other clinical trials; had received other investigational drugs within the previous 3 months; were pregnant or lactating; had uncontrolled infections or hypertension; had certain cardiac abnormalities; or required chronic treatment with therapeutic doses of systemic corticosteroids.
This study was conducted according to the Declaration of Helsinki and its amendments, Japanese Good Clinical Practice guidelines, and in agreement with the Institutional Review Board at the National Cancer Center Hospital (Tokyo, Japan). All patients provided written informed consent prior to study procedures.
Study design and treatments. This was an open‐label phase I study in patients with advanced NSCLC. Patients received single‐agent PF‐3512676 subcutaneously on day 1, followed by 7 days of observation. If safety was confirmed, the patient immediately proceeded to the combination therapy phase. During combination therapy, carboplatin (area under the curve [AUC] 6 mg × min/mL) and paclitaxel (200 mg/m2) were administered by intravenous (i.v.) infusion on day 1 and PF‐3512676 was administered subcutaneously on days 8 and 15 of a 3‐week cycle. Treatments were administered for a maximum of six cycles. Dexamethasone (20 mg) and chlorpheniramine maleate (10 mg) were administered by i.v. infusion 1 h before and ranitidine (50 mg) by i.v. infusion at least 30 min before each administration of paclitaxel.
During the monotherapy phase, patients in dose levels 1 and 2 were to be administered 0.1 mg/kg and 0.2 mg/kg PF‐3512676, respectively. These doses were to be maintained during the combination therapy phase. Patients in dose level 3 were to receive 0.4 mg/kg PF‐3512676 in the monotherapy phase and 0.2 mg/kg during the combination therapy phase. The three treatment arms with a maximum dose of 0.4 mg/kg PF‐3512676 during the monotherapy phase were designed to establish one of the primary endpoints of this study: the pharmacokinetic (PK) profile of PF‐3512676 in Japanese patients. Another study objective was to determine whether the same dose (0.2 mg/kg) of PF‐3512676 that was used in combination with chemotherapy in the phase II and III studies of this agent in Western patients with NSCLC would also be recommended in Japanese patients. Therefore, PF‐3512676 in dose level 3 was reduced from 0.4 mg/kg to 0.2 mg/kg when patients moved from the monotherapy to the combination phase. Patients with no DLT in the monotherapy phase could move immediately into the combination phase. For patients in level 3 only, any DLT observed during the monotherapy phase would have led to extension of the duration of this phase of the study by 1 week; if severity of toxicity decreased to ≤grade 1, patients would then continue into the combination therapy phase. A DLT was defined as any of the following: ≥grade 3 febrile neutropenia accompanied by infection; ≥grade 3 non‐hematologic toxicity; ≥grade 3 injection site reaction; ≥grade 3 thrombocytopenia requiring transfusion; grade 4 flu‐like symptoms; grade 4 neutropenia lasting 7 days; or grade 4 thrombocytopenia. DLT evaluation took place during monotherapy and the first cycle of combination therapy. PF‐3512676 activates the immune system, and commonly associated AEs include flu‐like symptoms and mild neutropenia believed to be the result of transient migration of neutrophils into peripheral tissues. This is distinct from bone‐marrow suppression and may not necessarily be an indication of an increased risk of infection. Therefore, in this study, ≥grade 3 neutropenia was not considered a DLT unless it was accompanied by infection. If, following a DLT, continuation of study was judged to be possible with dose reduction of chemotherapeutic agents, and if study protocol dose‐reduction criteria were satisfied, treatment could be continued. The dose of carboplatin could also be reduced to AUC 4.5 mg × min/mL and/or paclitaxel to 150 mg/m2 if, in the absence of a DLT, patients had specific, predesignated hematologic or non‐hematologic adverse events. These dose modifications were based on those reported for the Four‐Arm Comparative Study.( 19 ) The planned sample size for dose levels 1 and 3 was three patients each. If one DLT was observed in dose level 1 or 3, three additional patients were to be enrolled. The planned number of patients in dose level 2 in this study was predefined to be six patients. If >1 DLT was observed in dose levels 1 or 2, the study would not have progressed to the next level. Dose level 2 in this study was the same dose used in preceding clinical studies in Western patients.
Primary endpoints were evaluation of safety and PK of PF‐3512676 during the monotherapy and combination therapy phases. Secondary endpoints included evaluation of patient immune function and objective tumor response according to RECIST.
Pretreatment assessment and follow‐up studies. History, physical examination (including temperature, blood pressure, heart rate, and weight) ECOG PS, and routine laboratory studies were performed at baseline, before each treatment cycle, and at end of the study. Routine laboratory studies included serum electrolytes, renal and liver function tests, complete blood count and differential white blood cell counts, coagulation studies, and urinalysis. Physical examination and complete blood count were also performed on days 2, 3, and 4 of the monotherapy phase and on days 1, 8, 9, 10, 11, and 15 of the first cycle of combination therapy. After patients completed one cycle of monotherapy and one cycle of combination therapy, these tests were performed on days 1, 8, and 15 of all other cycles of combination therapy. An electrocardiogram was performed at baseline as well as at 3 and 24 h after administration of PF‐3512676 monotherapy. Severity of AEs and other symptoms were evaluated according to Common Terminology Criteria for AEs (CTCAE) version 3.0. Relevant radiologic studies to assess measurable and evaluable disease were repeated after every other cycle, and responses were scored according to RECIST.
Pharmacokinetics. To compare the PK of PF‐3512676 in the monotherapy phase with its PK during the combination therapy phase, blood samples were collected predose and at 1, 2, 3, 5, 7, 10, 24, 48, 72, and 96 h postdose in the monotherapy phase as well as predose and at 1, 2, 3, 5, 7, 10, 24, 48, 72, and 96 h postdose on day 8 of the first cycle of the combination therapy phase. For each sample, 4 mL of whole blood was collected in a tube containing EDTA‐2K dipotassium salt. Collected samples were centrifuged at 1000g for 10 min, and resultant plasma was stored in aliquots at or below −70°C until analysis. Concentrations of PF‐3512676 were determined by Pharmaceutical Product Development (Richmond, VA, USA) using a validated hybridization assay with capture and detection probes complementary to either the 3′ or 5′ portions of the molecule. Pharmacokinetic parameters were calculated and summarized using descriptive statistics.
Pharmacodynamics. To evaluate patient immune function, blood samples were collected to measure the serum concentrations of IP‐10, IL‐6, IFN‐α, IL‐12p40, monocyte chemotactic protein‐1 (MCP‐1), and C‐reactive protein (CRP). Serum samples were collected predose and at 1, 3, 7, 24, 48, 72, 96, and 168 h postdose of PF‐3512676 in the monotherapy phase. During the combination therapy phase, samples were collected on day 8 of the first cycle of combination therapy predose and at 1, 3, 7, 24, 48, 72, 96, and 168 h postdose. For each sample, ≥3 mL of whole blood was collected, stored at room temperature for 30 min, and then centrifuged at 1000g for 10 min. Resultant serum was stored in aliquots at or below −70°C until analysis. Serum levels of IFN‐α, IL‐12p40, and MCP‐1 were determined by the Human Custom Three‐Plex Beads Kit (Invitrogen/Biosource, Carlsbad, CA, USA). Multianalyte profiling was performed on the BioPlex® Suspension Array System, and acquired fluorescence data were analyzed by the BioPlex Manager software versions 4.1 (BioRad Laboratories, Hercules, CA, USA). The levels of CRP, IP‐10, and IL‐6 were determined by ELISA (enzyme‐linked immunosorbent assay). C‐reactive protein was quantified with the C‐reactive Protein (hsCRP) EIA kit (ALPCO Diagnostics, Salem, NH, USA). Interleukin‐6 and IP‐10 were detected using the Quantikine® HS Human IL‐6 Immunoassay kit and Quantikine® Human CXCL10/IP‐10 Immunoassay kit (R&D systems, Minneapolis, MN, USA), respectively. The levels of IFN‐α, MCP‐1, IL12‐p40, and CRP were determined at Mitsubishi Chemical Medicine (Tokyo, Japan). The levels of IP‐10 and IL‐6 were determined at Quest Pharmaceutical Services (Newark, DE, USA).
Results
Patient characteristics. From June 2006 to March 2007, a total of 12 patients were enrolled, and all patients were treated with PF‐3512676 monotherapy and at least one cycle of combination therapy. There were seven male and five female patients in this study, and median age was 60 (range, 41–69) years (Table 1). Most patients had stage IV disease (8/12, 67%) and adenocarcinoma (9/12, 75%). Forty‐two total cycles of combination therapy were administered, and the median number of combination therapy cycles per patient was four (range, 1–6).
Table 1.
Characteristics of patients
| Enrolled patients, n | 12 |
| Age (years), median (range) | 60 (41–69) |
| Gender, n (%) | |
| Men | 7 (58) |
| Women | 5 (42) |
| Baseline ECOG performance status | |
| 0 | 7 |
| 1 | 5 |
| Histologic classification of NSCLC, n (%) | |
| Adenocarcinoma | 9 (75) |
| Squamous cell carcinoma | 2 (17) |
| Other | 1 (8) |
| Clinical stage, n (%) | |
| IIIB | 4 (33) |
| IV | 8 (67) |
ECOG, Eastern Cooperative Oncology Group; NSCLC, non‐small‐cell lung cancer.
Safety. A list of any‐grade AEs with incidence of 30% or more in either the monotherapy phase or the entire study (both monotherapy and combination therapy phases) is presented in Table 2. Many treatment‐related AEs observed during the combination therapy phase were likely to be at least in part related to PF‐3512676, as they also developed in patients during the monotherapy phase. Treatment‐related AEs that occurred in >30% of patients during monotherapy included injection‐site reactions (n = 12, 100%), flu‐like symptoms (n = 11, 91.7%), lymphocytopenia (n = 6, 50.0%), leukopenia (n = 4, 33.3%), and anemia (n = 4, 33.3%). Neutropenia was also observed (n = 3, 25.0%). Through the entire study period the most common treatment‐related AEs of any grade were injection‐site reactions, neutropenia, and leukopenia (n = 12, 100% for each); anemia, flu‐like symptoms, and lymphocytopenia (n = 11, 91.7% each) were also very common.
Table 2.
Treatment‐related adverse events occurring in >30% of patients in either the PF‐3512676 monotherapy phase or entire study (both monotherapy and combination therapy phases)
| Level† (evaluable patients, n) | Patients, n (%) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entire study (monotherapy phase + combination therapy phase) | Monotherapy phase | |||||||||||||||
| Level 1 (n = 3) | Level 2 (n = 6) | Level 3 (n = 3) | All levels (n = 12) | Level 1 (n = 3) | Level 2 (n = 6) | Level 3 (n = 3) | All levels (n = 12) | |||||||||
| All grades | ≥Grade 3 | All grades | ≥Grade 3 | All grades | ≥Grade 3 | All grades | ≥Grade 3 | All grades | ≥Grade 3 | All grades | ≥Grade 3 | All grades | ≥Grade 3 | All grades | ≥Grade 3 | |
| Adverse events, hematologic | ||||||||||||||||
| Leukopenia | 3 | 2 | 6 | 3 | 3 | 2 | 12 (100) | 7 (58.3) | 2 | 0 | 1 | 0 | 1 | 0 | 4 (33.3) | 0 |
| Neutropenia | 3 | 3 | 6 | 5 | 3 | 3 | 12 (100) | 11 (91.7) | 1 | 0 | 1 | 0 | 1 | 0 | 3 (25.0) | 0 |
| Lymphocytopenia | 2 | 2 | 6 | 1 | 3 | 1 | 11 (91.7) | 4 (33.3) | 2 | 0 | 3 | 0 | 1 | 1 | 6 (50.0) | 1 (8.3) |
| Anemia | 3 | 1 | 5 | 1 | 3 | 1 | 11 (91.7) | 3 (25.0) | 1 | 0 | 2 | 0 | 1 | 0 | 4 (33.3) | 0 |
| Thrombocytopenia | 2 | 0 | 2 | 2 | 3 | 0 | 7 (58.3) | 2 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Monocytopenia | 1 | 0 | 1 | 0 | 3 | 0 | 5 (41.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Adverse events, non‐hematologic | ||||||||||||||||
| Injection‐site reactions | 3 | 0 | 6 | 0 | 3 | 1 | 12 (100) | 1 (8.3) | 3 | 0 | 6 | 0 | 3 | 0 | 12 (100) | 0 |
| Flu‐like symptoms | 2 | 0 | 6 | 0 | 3 | 1 | 11 (91.7) | 1 (8.3) | 2 | 0 | 6 | 0 | 3 | 0 | 11 (91.7) | 0 |
| Anorexia | 1 | 1 | 4 | 0 | 2 | 1 | 7 (58.3) | 2 (16.7) | 0 | 0 | 2 | 0 | 0 | 0 | 2 (16.7) | 0 |
| Malaise | 2 | 0 | 3 | 0 | 2 | 0 | 7 (58.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ALT increased | 1 | 0 | 3 | 0 | 2 | 0 | 6 (50.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Constipation | 0 | 0 | 3 | 0 | 2 | 0 | 5 (41.7) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 (8.3) | 0 |
| Diarrhea | 1 | 0 | 4 | 0 | 0 | 0 | 5 (41.7) | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 (16.7) | 0 |
| AST increased | 0 | 0 | 2 | 0 | 2 | 0 | 4 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nausea | 1 | 0 | 3 | 0 | 0 | 0 | 4 (33.3) | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 (8.3) | 0 |
†Level 1: (Mono) PF‐3512676 0.1 mg/kg → (Combo) PF‐3512676 0.1 mg/kg + carboplatin AUC 6 + paclitaxel 200 mg/m2; Level 2: (Mono) PF‐3512676 0.2 mg/kg → (Combo) PF‐3512676 0.2 mg/kg + carboplatin AUC 6 + paclitaxel 200 mg/m2; Level 3: (Mono) PF‐3512676 0.4 mg/kg → (Combo) PF‐3512676 0.2 mg/kg + carboplatin AUC 6 + paclitaxel 200 mg/m2. ALT, alanine‐aminotransferase; AST, aspartate‐aminotransferase; Combo, combination therapy; Mono, monotherapy.
Only injection‐site reactions and flu‐like symptoms occurred with similar frequency in both monotherapy and combination therapy phases, suggesting these AEs were most closely related to treatment with PF‐3512676. Certain AEs such as thrombocytopenia, monocytopenia, and malaise that were observed during the combination therapy phase were not observed at all during monotherapy phase, suggesting they were most closely related to chemotherapy.
Seven patients discontinued study therapy; one patient in dose level 1 discontinued as the result of progressive disease, while the remaining six patients (85.7%) discontinued as a result of AEs or laboratory abnormalities (one patient in dose level 1, three patients in dose level 2, and two patients in dose level 3). All of the discontinuations resulting from AEs or laboratory abnormalities occurred during combination therapy, and the AEs that led to discontinuation varied. The patient in dose level 1 discontinued as a result of grade 2 nausea and grade 2 vomiting that were related to both PF‐3512676 and chemotherapy. One patient in dose level 2 discontinued after having multiple hematologic AEs that were related to both PF‐3512676 and chemotherapy: grade 4 anemia, and grade 2 neutropenia and leukopenia. Another patient in dose level 2 discontinued after having PF‐3512676‐related, grade 2 flu‐like symptoms (this event was considered unrelated to chemotherapy). The third discontinuation in dose level 2 was the result of grade 3 increase in γ‐glutamyltransferase that was considered related to PF‐3512676 and chemotherapy and a grade 3 rash considered related to chemotherapy, but not to PF‐3512676. One discontinuation in dose level 3 was the result of grade 2 peripheral neuropathy that was considered to be related to paclitaxel. The other was a patient who developed PF‐3512676‐related grade 3 anorexia and flu‐like symptoms (these events were considered unrelated to chemotherapy).
Although all patients reported treatment‐related AEs of ≥grade 3, no serious AEs were reported. No DLTs occurred during the monotherapy phase. One patient in level 2 experienced a DLT in the combination therapy phase. This patient developed grade 3 rash and grade 3 increase in γ‐glutamyltransferase on days 9 and 10 of the first cycle of combination therapy, respectively. Both events decreased to grade 2 by day 13 of the same cycle and to grade 1 after completion of the DLT observation period. The patient discontinued study therapy as a result of these AEs. No further DLTs were observed. Therefore, the study progressed to the highest planned dose level.
Efficacy. Of 12 patients treated with PF‐3512676 and chemotherapy, one patient (8%) achieved a complete response (CR) and three patients (25%) had partial responses (PRs). All objective responses were among patients treated in dose levels 1 and 2. In addition, seven patients (58%) had stable disease (SD).
Pharmacokinetics. The plasma concentration profiles of PF‐3512676 were similar in the monotherapy and combination therapy phases (Fig. 1), and overall pharmacokinetic parameters of PF‐3512676 were not different with addition of chemotherapy (Table 3). Median time to highest plasma concentration ranged from 2–3 h and mean peak plasma concentration (C max) of PF‐3512676 appeared to be dose dependent. Furthermore, mean half‐life (t 1/2) of PF‐3512676 varied with dose, ranging from 4.8 to 21.6 h during the monotherapy phase and from 7.9 to 9.5 h in combination therapy phase, with longer t 1/2 for higher doses of PF‐3512676. Based on these PK data, accumulation of PF‐3512676 was not observed in this study.
Figure 1.
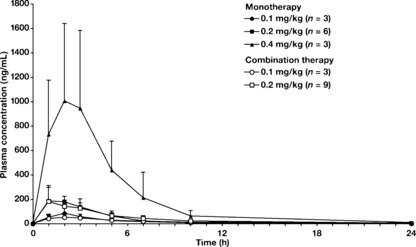
Pharmacokinetics (PK) of PF‐3512676 were similar during monotherapy and combination therapy phases. To compare the PK of PF‐3512676 in the monotherapy phase with the PK of the combination therapy phase, blood samples were collected predose and at 1, 2, 3, 5, 7, 10, 24, 48, 72, and 96 h postdose in the monotherapy phase as well as predose and at 1, 2, 3, 5, 7, 10, 24, 48, 72, and 96 h postdose on day 8 in the first cycle of the combination therapy phase. A custom‐designed hybridization enzyme‐linked immunosorbent assay was used. Mean plasma concentration ± SD of each time point for each group is shown.
Table 3.
Pharmacokinetics of PF‐3512676
| Dose level | n | Mean C max, ng/mL (SD) | Mean AUC(0–∞), ng × h/mL (SD) | Median t max, hours (range) | Mean t 1/2, hours (SD) |
|---|---|---|---|---|---|
| Monotherapy | |||||
| 0.1 mg/kg | 3 | 90 (36) | 376 (73) | 2 (2–3) | 4.8 (3.4) |
| 0.2 mg/kg | 6 | 217 (90) | 856 (127) | 2 (1–3) | 12.8 (14.0) |
| 0.4 mg/kg | 3 | 1010 (633) | 5270 (2450) | 2 (2–2)† | 21.6 (16.4) |
| Combination therapy | |||||
| 0.1 mg/kg | 3 | 55 (19) | 379 (55) | 3 (2–3) | 7.9 (6.2) |
| 0.2 mg/kg | 9 | 226 (124) | 1340 (775) | 2 (1–3) | 9.5 (6.9) |
†All patients had reached maximum concentration of PF‐3512676 by 2 h postdose. AUC, area under the curve; C max, peak plasma concentration; SD, standard deviation; t ½, half‐life; t max, time to maximum plasma concentration.
Pharmacodynamics. IFN‐α, IL‐12p40, IL‐6, IP‐10, CRP, and MCP‐1 were evaluated following treatment with PF‐3512676 during both monotherapy and combination therapy phases for all dose levels. For each assayed cytokine or protein, detected levels began to escalate at approximately 3 h postdose, but time to peak concentration varied from approximately 24 to 96 h (Fig. 2). Levels returned to predose concentrations by ∼168 h postdose. Pharmacodynamic profiles of the cytokines and proteins during the combination therapy phase were similar to their corresponding profiles in the monotherapy phase, although there was a trend toward lower peak cytokine and protein levels in the combination therapy phase. However, it must be noted that there was considerable variation in individual predose and maximum concentrations. Cytokine and protein profiles of patients who achieved objective responses were not different from those of patients without evidence of antitumor activity.
Figure 2.
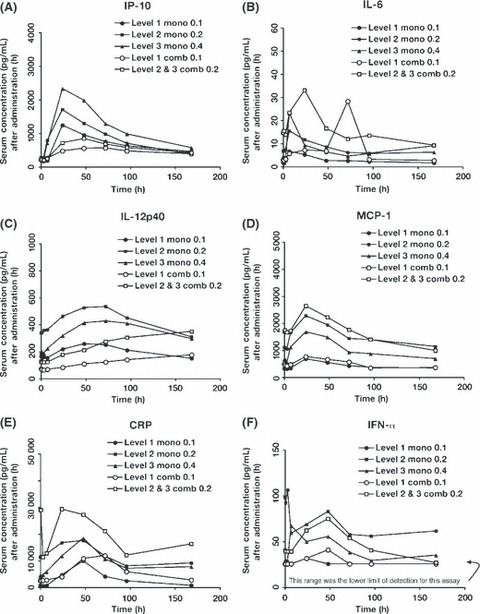
Pharmacodynamics of cytokines, monocyte chemotactic protein‐1 (MCP‐1), and C‐reactive protein (CRP) were similar during monotherapy and combination therapy phases of the study. The levels of (A) interferon‐γ‐inducible protein 10 (IP‐10), (B) interleukin (IL)‐6, (C) IL‐12p40, (D) MCP‐1, (E) CRP, and (F) interferon‐alpha (IFN‐α) were evaluated in patient sera at predose (0 h) and at 1, 3, 7, 24, 48, 72, 96, 168 h postdose of PF‐3512676 during the monotherapy and combination therapy phases. Sample collection for the combination therapy phase occurred during the first cycle of treatment, and predose was on day 8 before treatment. In each case, the level of the assayed protein began to escalate after 3 h. The time at which highest expression was achieved varied but generally returned to baseline by 168 h postdose. comb, combination therapy; h, hour; mono, monotherapy.
Discussion
This phase I study was conducted to examine the safety and PK of PF‐3512676 as a single agent and in combination with carboplatin/paclitaxel therapy in Japanese patients with previously untreated NSCLC. Treatment with carboplatin and paclitaxel is a standard approach for patients with advanced NSCLC in Japan.( 19 ) American Society of Clinical Oncology guidelines for treatment of previously untreated stage IV NSCLC recommend combination chemotherapy, but suggest stopping this treatment if patients do not respond after three or four cycles.( 4 ) Furthermore, in a recent phase III trial in Japanese patients, the median number of cycles of first‐line platinum‐based chemotherapy was three.( 20 ) In this phase I study, patients received a median of four cycles of chemotherapy combined with PF‐3512676. Therefore, SC delivery of PF‐3512676 was considered tolerable either as monotherapy or in combination therapy at the highest doses tested in this study (0.4 mg/kg and 0.2 mg/kg, respectively).
Through the entire study period, the most common treatment‐related AEs of any grade were injection‐site reactions, neutropenia, leukopenia, anemia, flu‐like symptoms, and lymphocytopenia. Injection‐site reactions and flu‐like symptoms were likely related to treatment with PF‐3512676 alone, as they occurred with similar frequency in both the monotherapy and the combination therapy phases. There was no clear dose relationship for these AEs during PF‐3512676 monotherapy. Other AEs (eg, leukopenia, neutropenia, lymphocytopenia, anemia, and anorexia) observed during both phases of the study were probably related to treatment with both PF‐3512676 and chemotherapy, because they occurred more frequently during the combination therapy phase than the monotherapy phase. There was no indication of cumulative toxicity. These safety results are similar to those from a previous phase II study in Western patients.( 17 ) In that study, the most common AEs related to PF‐3512676 and not to chemotherapy were mild to moderate injection‐site reactions and flu‐like symptoms. Other less common AEs considered related to treatment with PF‐3512676 were neutropenia, anemia, and thrombocytopenia.
Across this study, the most frequently occurring AEs of ≥grade 3 were hematologic (e.g. neutropenia, leukopenia, or lymphocytopenia). Hematologic AEs were observed at all dose levels and were qualitatively similar to those reported with carboplatin and paclitaxel doublet chemotherapy.( 21 ) When evaluating safety in studies of doublet chemotherapy, it is important to note that the incidence of ≥grade 3 neutropenia after doublet chemotherapy may be higher in Japanese patients( 19 ) than in Western patients.( 5 , 20 , 22 ) Although the small number of patients included in this study precludes a definitive comparison, 11 patients (91.7%) in the present study had ≥grade 3 neutropenia, and this is similar to the frequency reported (84%) in Japanese patients with NSCLC receiving doublet chemotherapy alone.( 19 )
Because the administration and observation periods were brief in this phase I study, patient blood samples were not analyzed for immunopathological changes that could potentially be indicative of autoimmune events. However, no symptoms suggestive of autoimmune disease were observed. Some patients in other PF‐3512676 clinical trials developed positive tests for anti‐DNA antibodies. The potential significance of these serologic results is not yet clear.
The PK profiles of PF‐3512676 observed during the monotherapy and combination therapy phases were similar. The effect of PF‐3512676 on the PK of carboplatin and paclitaxel was not evaluated in this study. Median time required to achieve maximum plasma concentration (2–3 h) was consistent across all PF‐3512676 doses with or without the addition of chemotherapy. The C max increased with the dose administered and was highest in dose level 3 monotherapy in which patients received 0.4 mg/kg PF‐3512676. The time required to clear drug from the body appeared to be dose dependent; shortest t 1/2 (4.8 h) was found in the 0.1 mg/kg dose level monotherapy phase, and longest t 1/2 (21.6 h) was observed in the 0.4 mg/kg monotherapy phase. However, these data may be confounded by the small number of patients per group and resultant high SD as well as the assay sensitivity level at the lowest dose level. Therefore, it is unclear whether clearance is truly dose dependent. Linearity was also not clearly defined because of the small sample size and the large variation in PK parameters.
The objective response rate (33%) in this study was similar to the rate of confirmed responses (30%) found in the previous phase II study.( 17 )
Treatment with PF‐3512676 alone or in combination with chemotherapy, regardless of dose, modulated several cytokines and other proteins. Immunomodulation was transient, and all increases had dissipated by ∼168 h postdose. The most robust responses observed were increases in the levels of IP‐10 and IL‐6, and this was consistent with the TH1‐like pattern of activation of the innate immune system previously observed in normal human volunteers subcutaneously injected with PF‐3512676.( 10 ) IP‐10 is a potent chemokine for activated T lymphocytes and regulates cell proliferation, apoptosis, and adhesion molecule expression.( 23 ) Its elevation is indicative of TLR9 activation. There appeared to be a trend toward reduced stimulation of cytokine and chemokine production in the combination therapy phases compared with monotherapy. Although the relevance of this finding is unclear, it should be noted that in this study design, patients who received monotherapy were treatment‐naive, while patients who received combination therapy had already received monotherapy with PF‐3512676. Increasing the single‐agent dose to 0.4 mg/kg seemed to result in a similar pattern of cytokine and chemokine production to that observed with lower doses. Cytokine and chemokine profiles from patients who achieved CR or PR were similar to those from patients without evidence of antitumor activity. However, the small sample size in this study may have confounded these results, and further investigation in future, larger studies would be required for confirmation.
In addition to the present study, PF‐3512676 has been investigated in two phase III clinical studies in which combination with platinum‐based doublet chemotherapy was compared with platinum‐based doublet chemotherapy alone in Western patients with previously untreated advanced NSCLC.( 24 , 25 ) In those studies, addition of PF‐3512676 to doublet chemotherapy did not produce an improvement in overall survival and was associated with increased toxicity. After completion of the study described in this manuscript and based on results from these phase III studies, all clinical trials of PF‐3512676 in combination with cytotoxic chemotherapy agents for treatment of NSCLC were discontinued. However, clinical trials in other settings and in combination with targeted or immunotherapeutic agents are ongoing or planned.
In conclusion, PF‐3512676 as a single agent and in combination with carboplatin and paclitaxel had an acceptable safety profile in Japanese patients with treatment‐naive NSCLC, and PF‐3512676 showed evidence of immune activation in the study. It is, therefore, still considered to have potential utility as an anticancer agent.
Disclosure Statement
Financial support for this study was provided by Pfizer, Inc. Junichi Hashimoto and Emiko Ohki are employed by and hold stock in Pfizer Japan. Yuichiro Ohe receives speaker’s bureau honoraria from Pfizer Japan. None of the other authors has a conflict to disclose. Financial support for medical editorial assistance was provided by Pfizer, Inc.
Acknowledgments
The authors thank Fumiaki Koizumi, MD, PhD; Kazuto Nishio, MD, PhD; and Koji Kono, MD, PhD, for their immunologic advice. The authors also thank Tamara Fink, PhD, ProEd Communications Inc.,® for her medical editorial assistance with this manuscript.
References
- 1. Okamoto I, Moriyama E, Fujii S et al. Phase II study of carboplatin‐paclitaxel combination chemotherapy in elderly patients with advanced non‐small cell lung cancer. Jpn J Clin Oncol 2005; 35: 188–94. [DOI] [PubMed] [Google Scholar]
- 2. Molina JR, Adjei AA, Jett JR. Advances in chemotherapy of non‐small cell lung cancer. Chest 2006; 130: 1211–9. [DOI] [PubMed] [Google Scholar]
- 3. Pfister DG, Johnson DH, Azzoli CG et al. American Society of Clinical Oncology treatment of unresectable non‐small‐cell lung cancer guideline: update 2003. J Clin Oncol 2004; 22: 330–53. [DOI] [PubMed] [Google Scholar]
- 4. Grossi F, Aita M, Follador A et al. Sequential, alternating, and maintenance/consolidation chemotherapy in advanced non‐small cell lung cancer: a review of the literature. Oncologist 2007; 12: 451–64. [DOI] [PubMed] [Google Scholar]
- 5. Schiller JH, Harrington D, Belani CP et al. Comparison of four chemotherapy regimens for advanced non‐small‐cell lung cancer. N Engl J Med 2002; 346: 92–8. [DOI] [PubMed] [Google Scholar]
- 6. Iwasaki A, Medzhitov R. Toll‐like receptor control of the adaptive immune responses. Nat Immunol 2004; 5: 987–95. [DOI] [PubMed] [Google Scholar]
- 7. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124: 783–801. [DOI] [PubMed] [Google Scholar]
- 8. Takeda K, Kaisho T, Akira S. Toll‐like receptors. Annu Rev Immunol 2003; 21: 335–76. [DOI] [PubMed] [Google Scholar]
- 9. Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest 2007; 117: 1184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krieg AM, Efler SM, Wittpoth M et al. Induction of systemic TH1‐like innate immunity in normal volunteers following subcutaneous but not intravenous administration of CPG 7909, a synthetic B‐class CpG oligodeoxynucleotide TLR9 agonist. J Immunother 2004; 27: 460–71. [DOI] [PubMed] [Google Scholar]
- 11. Krieg AM. Toll‐like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008; 27: 161–7. [DOI] [PubMed] [Google Scholar]
- 12. Krieg AM. Therapeutic potential of Toll‐like receptor 9 activation. Nat Rev Drug Discov 2006; 5: 471–84. [DOI] [PubMed] [Google Scholar]
- 13. Weeratna RD, Bourne LL, Sullivan SM et al. Combination of a new TLR9 agonist immunomodulator (CpG 7909) and paclitaxel for treatment of metastatic Lewis Lung Carcinoma (LLC). J Clin Oncol 2004; 22 (Suppl): 699. Abstract 7346. 14966094 [Google Scholar]
- 14. Kim Y, Girardi M, McAuley S, Schmalbach T. Cutaneous T‐cell lymphoma (CTCL) responses to a TLR9 agonist CpG immunomodulator (CPG 7909), a phase I study. J Clin Oncol 2004; 22 (14 Suppl): 580. Abstract 6600. 14726506 [Google Scholar]
- 15. Pashenkov M, Goess G, Wagner C et al. Phase II trial of a toll‐like receptor 9‐activating oligonucleotide in patients with metastatic melanoma. J Clin Oncol 2006; 24: 5716–24. [DOI] [PubMed] [Google Scholar]
- 16. Thompson JA, Kuzel T, Bukowski R et al. Phase Ib trial of a targeted TLR9 CpG immunomodulator (CPG 7909) in advanced renal cell carcinoma (RCC). J Clin Oncol 2004; 22 (14 Suppl): 416. Abstract 4644. 14691124 [Google Scholar]
- 17. Manegold C, Gravenor D, Woytowitz D et al. Randomized phase II trial of a toll‐like receptor 9 agonist oligodeoxynucleotide, PF‐3512676, in combination with first‐line taxane plus platinum chemotherapy for advanced‐stage non‐small‐cell lung cancer. J Clin Oncol 2008; 26: 3979–86. [DOI] [PubMed] [Google Scholar]
- 18. Therasse P, Arbuck SG, Eisenhauer EA et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 19. Ohe Y, Ohashi Y, Kubota K et al. Randomized phase III study of cisplatin plus irinotecan versus carboplatin plus paclitaxel, cisplatin plus gemcitabine, and cisplatin plus vinorelbine for advanced non‐small‐cell lung cancer: Four‐Arm Cooperative Study in Japan. Ann Oncol 2007; 18: 317–23. [DOI] [PubMed] [Google Scholar]
- 20. Scagliotti GV, De Marinis F, Rinaldi M et al. Phase III randomized trial comparing three platinum‐based doublets in advanced non‐small‐cell lung cancer. J Clin Oncol 2002; 20: 4285–91. [DOI] [PubMed] [Google Scholar]
- 21. Gandara DR, Ohe Y, Kubota K et al. Japan‐SWOG common arm analysis of paclitaxel/carboplatin in advanced stage non‐small cell lung cancer (NSCLC): a model for prospective comparison of cooperative group trials. J Clin Oncol 2004; 22 (Suppl): 14s. Abstract 7007. [Google Scholar]
- 22. Smit EF, Van Meerbeeck JP, Lianes P et al. Three‐arm randomized study of two cisplatin‐based regimens and paclitaxel plus gemcitabine in advanced non‐small‐cell lung cancer: a phase III trial of the European Organization for Research and Treatment of Cancer Lung Cancer Group – EORTC 08975. J Clin Oncol 2003; 21: 3909–17. [DOI] [PubMed] [Google Scholar]
- 23. Neville LF, Mathiak G, Bagasra O. The immunobiology of interferon‐gamma inducible protein 10 kD (IP‐10): a novel, pleiotropic member of the C‐X‐C chemokine superfamily. Cytokine Growth Factor Rev 1997; 8: 207–19. [DOI] [PubMed] [Google Scholar]
- 24. Hirsh V, Boyer M, Rosell R et al. Randomized phase III trial of paclitaxel/carboplatin with or without PF‐3512676 as first line treatment of advanced non‐small cell lung cancer (NSCLC) [oral presentation]. J Clin Oncol 2008; 26 (Suppl): 428s. Abstract 8016. [Google Scholar]
- 25. Manegold C, Thatcher N, Benner RJ et al. Randomized phase III trial of gemcitabine/cisplatin with or without PF‐3512676 as first line treatment of advanced non‐small cell lung cancer (NSCLC) [oral presentation]. J Clin Oncol 2008; 26 (Suppl): 428s. Abstract 8017. [Google Scholar]