

Abstract
Integration of a therapeutic gene into the host cell genome permits stable expression of the gene product in the target cells and its progeny. However, non‐directional integration of any given gene can pose the risk of activating tumor genes or silencing tumor suppressor genes. Therefore, including a safety‐control element into integrating vector systems is an important advance towards safer human gene therapy. Here, we report on a gene expression cassette that can be potentially exploited in integrating vector systems to eliminate post‐therapeutic tumorigenesis. The Herpes simplex virus thymidine kinase (hsvTK) gene under the transcriptional control of the human telomere reverse transcriptase promoter (hTERTp) was incorporated into a self‐inactivating HIV‐based lentiviral vector. The hTERT promoter is silent in normal somatic cells and re‐activated in tumor cells. Therefore, normal gene‐corrected cells should not express hsvTK from the promoter. However, if some gene‐corrected cells subsequently become tumorigenic and the hTERT promoter is re‐activated, application of ganciclovir (GCV), a clinically used antiviral drug, will achieve selective deletion of the cancerous cells. Our experimental data indicated that the hTERTp‐hsvTK cassette in the lentiviral vector was sufficient to differentiate between tumor cells and normal cells, thus eradicating tumor cells selectively in vitro and in vivo. These results proved the principle of using the element in integrating vectors for safer gene delivery. (Cancer Sci 2005; 96: 607 –613)
Abbreviations:
- CMV
cytomegalovirus
- EGFP
enhanced green fluorescent protein
- GCV
ganciclovir
- hMSC
human mesenchymal stem cells
- HSC
hematopoietic stem cells
- hsvTK
herpes simplex virus thymidine kinase
- hTERT
human telomerase reverse transcriptase
- LTR, long terminal repeat; MOI
multiplicity of infection.
Gene medicine involves the use of genes as therapeutics for the treatment of diseases. Integrating viruses, such as Type‐C mammalian retroviruses and lentiviruses, have evolved highly efficient mechanisms to access their host cell genomes. The frequency of integration and the persistency of transgene expression by vectors derived from these viruses are attractive for achieving long‐term therapeutic effects. Recent reports have overturned the previous perception that retroviruses and lentiviruses randomly integrate into the host cell chromosomes. Instead, they tend to insert into active transcriptional sites.( 1 , 2 , 3 , 4 ) Therefore, when related vectors are used in human gene therapy, there is a risk of activating growth promoting genes, such as LMO2, or of inactivating tumor suppressor genes such as BRCA1.( 5 , 6 ) In fact, leukemia developed in patients in clinical trials for X‐linked severe combined immunodeficiency (X‐SCID) because of vector‐associated insertional mutagenesis.( 7 , 8 ) Thus, improving the safety features of integrating vectors is important for safer human gene therapy.
DNA polymerases are unable to replicate linear chromosomes de novo. Continual cell multiplication causes chromosome shortening, loss of genetic information, and cell senescence.( 9 , 10 , 11 , 12 ) Therefore, compensatory repair of chromosomal ends by telomerase or other alternative mechanisms is required for normal replication of cells and tumorigenesis.( 11 , 13 ) High telomerase activity is found in nearly all immortal cell lines and in more than 85–90% of primary tumors.( 14 ) Importantly, regulation of telomerase activity has been reported to be mainly at the transcriptional level,( 15 ) which provides a rationale for using the hTERT promoter to achieve tumor‐selective targeting. We and other groups have generated replication‐competent adenoviruses controlled by the hTERT promoter or replication‐incompetent adenoviruses expressing therapeutic genes controlled by the hTERT promoter.( 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ) Tests of these vectors in varied tumor models have confirmed the excellent tumor selectivity of this promoter. Based on these findings, we hypothesized that incorporation of a conditional suicide gene, such as hsvTK or sodium‐iodine symporter, driven by the hTERT promoter in an integrating gene therapy vector would confer the capacity of self‐examination of vector‐associated oncogenesis and allow the elimination of such an adverse outcome upon GCV administration.
Materials and Methods
Culture of cells and cell lines
HT‐1080, 293T, YAC‐1 and Jurkat T‐cell leukemia cells were cultured in the medium recommended by American Type Culture Collection. SCCLSU‐1, a cell line derived in our laboratory from a squamous cell carcinoma surgically removed from the tongue of an adult male, was grown in Dulbecco's modified Eagle Medium‐F12 (DMEM‐F12) supplemented to contain 10% fetal bovine serum (FBS), 100 IU/mL penicillin, 100 µg/mL streptomycin, and 2 mM L‐glutamine. Primary human fibroblasts were isolated from donated lung tissues. The protocols were approved by the local Institutional Review Board. Human bone marrow mesenchymal stem cells were generously provided by Dr Darwin J. Prockop (Tulane University Health Sciences Center, New Orleans, LA, USA). The cells were cultured in α‐MEM supplemented to 20% FBS, 100 IU/mL penicillin, 100 µg/mL streptomycin, and 2 mM L‐glutamine.( 24 ) All cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C.
Plasmid constructs
The Herpes simplex virus thymidine kinase (TK) gene was prepared by polymerase chain reaction (PCR) using pGT60‐mIP‐10 (InvivoGen, San Diego, CA, USA) as a template with the primers 5′‐TTTGCATCCCAGTCACCGTCCTTGCCATCAT‐3′ and 5′‐TTTGCGGCCGCTCGACTCAATCTAGTCAGTT‐3′ and the Stratagene PfuTurbo Hotstart DNA polymerase as recommended by the manufacturer. The PCR product was cloned into pCR‐Blunt II‐TOPO (Invitrogen, Carlsbad, CA, USA). Next, the gene was excised with XhoI and NotI and linked to the human telomerase reverse transcriptase promoter, generating the phTERTp‐TK plasmid. Then, the hTERTp‐TK cassette was released and inserted into the SmaI site of a self‐inactivating HIV‐based transgene plasmid pHR‐CMV‐EGFP, which positions the hTERTp‐TK cassette at 37 bp downstream of the EGFP gene. The pHR‐CMV‐EGFP plasmid was derived from pHR(–)‐cCMVsolLacZR(+)W(+) by replacing the lacZ with the EGFP gene.( 25 ) The resultant construct, referred to as pHR‐CMV‐EGFP‐hTERTp‐TK, contains dual transgenes: the CMV promoter‐driven EGFP gene and the hTERT promoter‐driven hsvTK gene.
Vector production, cell transduction, and ganciclovir treatment
A three‐plasmid cotransfection approach was used to produce the HIV‐based vectors.( 25 ) Briefly, 293T cells were transfected by calcium phosphate precipitation with the following three plasmids: (i) the packaging plasmid pCMV‐ΔR8.74; (ii) an envelope plasmid (pCI‐VSVG); and (iii) a transgene plasmid (pHR‐CMV‐EGFP or pHR‐CMV‐EGFP‐hTERTp‐TK). The produced vectors were concentrated approximately 150–200‐fold by ultra‐centrifugation. For the transduction of various cell lines, cells were plated at a density of 1–2 × 105 cells per well on a six‐well tissue culture plate in a complete medium appropriate for each cell type. The next day vectors at the MOI indicated were applied. Six to 18 h later the virus was removed and replaced with fresh medium. Two days after virus addition, the cells were passaged to new plates and were either untreated (control group) or treated with 12.5 µg/mL GCV (experimental group) for a week. Then, they were cultured for an additional week. Cells in both groups were equally passaged only as needed as each well reached confluency. To assess the hTERTp‐TK‐conferred killing ability, crystal violet staining was performed by fixing the cells with 4% glutaraldehyde in phosphate‐buffered saline (PBS) for 30 min at room temperature and staining with crystal violet dye as previously described.( 16 ) To isolate single‐cell clones of HT‐1080 cells transduced with either HIV‐CMV‐EGFP or HIV‐CMV‐EGFP‐hTERT‐TK, cells were infected with 0.5 MOI of each vector, respectively. After 10 days, the cells were plated into 96 well plates at approximately one cell per well and cultured for 2–3 weeks. Based on the expression of EGFP, positive colonies were identified under a fluorescent microscope. Multiple clones with different fluorescence intensities were selected and expanded, as this could have indicated different copy numbers of the transgene in the genomes. For YAC‐1 cells, cloning and transduction was performed in suspension with 20 MOI of HIV‐CMV‐EGFP‐hTERTp‐TK vector. Individual EGFP‐fluorescent cells were identified and directly cloned under a fluorescent microscope. The single‐cell clones were expanded in conditioned medium. To isolate a pure population of Jurkat T‐cell leukemia cells transduced with either HIV‐CMV‐EGFP or HIV‐CMV‐EGFP‐hTERTp‐TK, Jurkat cells were infected with the vectors (MOI = 1) in the presence of polybrene (4 µg/mL). After 6–8 h, the cells were pelleted at 500 g for 10 min and resuspended in fresh medium. The cells were cultured for 6 days and sorted using a fluorescence‐activated cell sorter (FACS) for the 0.2–1% fraction of the population that was the most EGFP‐fluorescent. The collected cells were amplified in culture for 1–2 weeks, and re‐sorted as described earlier to obtain a pure population of the EGFP‐expressing cells.
Thymidine kinase‐mediated tumor deletion in nude mice
The studies were approved by the Louisiana State University Health Sciences Center Institutional and Animal Care Use Committee (IACUC). Nude (nu/nu) mice (athymic NCR‐nu, National Cancer Institute) were injected subcutaneously with 1 × 106 HT‐1080‐EGFP cells into the left flanks, and the same mice with HT1080‐EGFP‐TK cells into the right flanks. For long‐term tumor‐killing studies, only HT‐1080‐EGFP‐TK cells were injected into the left flanks. One to 2 weeks were usually needed for the tumors to grow to a 100–200 mm3 volume and then the animals were treated with 1.25 mg of GCV administered intraperitoneally in 1 mL of sterile PBS. A regime of twice‐daily treatment with the drug was continued for 8 days. Tumor size was measured with digital calipers and the total volume was calculated according to the formula: a 2 × b × 0.5, where a is the smaller tumor diameter and b the larger diameter.
Southern blot analysis
Genomic DNA was isolated from either HT‐1080‐EGFP or HT‐1080‐EGFP‐TK cells. Twelve micrograms of the DNA, digested overnight with EcoRI, was resolved by agarose gel electrophoresis, and transferred under alkaline conditions onto HyBond‐N+ (Amersham Biosciences, Canada). Blots were hybridized with a 32P‐labeled probe to HSV‐TK or to exon 13 of the human cystic fibrosis transmembrane conductance regulator (CFTR) in Ultrahyb (Ambion, Austin, TX, USA) solution. Autoradiography was performed with a Bio‐Rad Molecular Imager FX (Bio‐Rad, Hercules, CA, USA).
Results
To test the hypothesis that hsvTK gene expression controlled by the hTERT promoter can be used in integrating vectors as a safety‐checking element, we selected the HIV‐based vector as our integration vector model. The human TERT promoter (the 1720 b.p. region immediately upstream of the translation start site) was obtained by PCR and was ligated to the gene encoding hsvTK.( 16 ) The resultant expression cassette, named hTERTp‐TK, was subcloned into the SmaI site of pHR‐CMV‐EGFP, a self‐inactivating HIV vector plasmid (Fig. 1a). This final construct, designated pHR‐CMV‐EGFP‐hTERTp‐TK, has dual expression cassettes. One is the EGFP gene driven by the CMV immediate early gene promoter, and the other is hTERTp‐TK. To check that both cassettes functioned independently, we transfected 293T cells (a transformed cell line derived from human embryonic kidney cells) with pHR‐CMV‐EGFP‐hTERTp‐TK or the control plasmid pHR‐CMV‐EGFP by calcium phosphate precipitation. Two days later, 12.5 µg/mL GCV was applied to each culture for a week. Six days after GCV treatment, crystal violet staining demonstrated that cells receiving pHR‐CMV‐EGFP‐hTERTp‐TK were completely ablated, whereas the control pHR‐CMV‐EGFP‐transfected cells were not affected (Fig. 1f). Because of the hsvTK‐GCV bystander effect,( 26 , 27 ) all of the cells were completely eliminated, even though not all of the cells were transfected (Fig. 1e). Next, lentiviral vectors were produced by three‐plasmid cotransfection of 293T cells. The titers of the produced vectors on HT‐1080 cells ranged from 105 to 106 TU/mL as determined by EGFP‐positive colony counting (data not shown).
Figure 1.
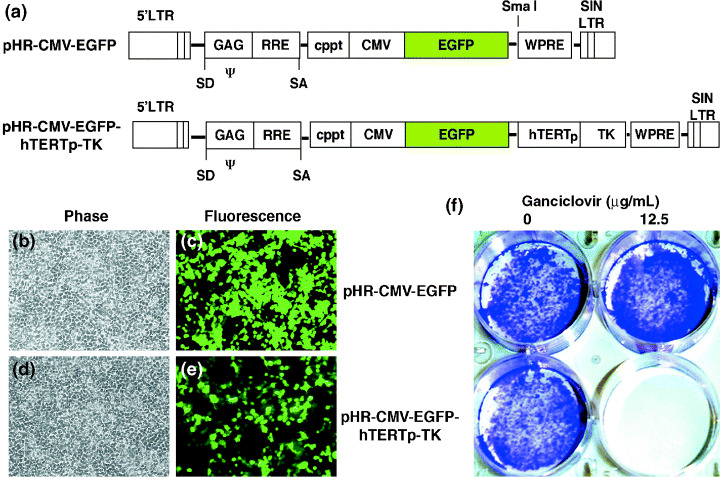
Incorporation of a safety‐control hTERTp‐TK element in a HIV‐based lentiviral vector. (a) Schematic maps of the two HIV‐based vector plasmids: pHR‐CMV‐EGFP and pHR‐CMV‐EGFP‐hTERTp‐TK. Features of the plasmids include a self‐inactivating (SIN) 3′‐LTR, a central polypurine‐rich tract (cppt), a woodchuck postregulatory element (WPRE), and a reporter gene (EGFP) driven by the CMV promoter. The hTERTp‐TK cassette was inserted into the unique SmaI site located 37 bp downstream of the EGFP gene. (b–e) 293T cells were transfected with pHR‐CMV‐EGFP (b,c) or pHR‐CMV‐EGFP‐hTERTp‐TK (d,e). Parts b and d are phase‐contrast micrographs, and c and e are fluorescent micrographs. Green cells are the EGFP‐positive cells. (f) The effect of ganciclovir (GCV) on the cells in b–e. Two days after transfection, the right two wells received 12.5 µg/mL of GCV daily for a week. As a control, the left two wells received no GCV. After drug treatment, the cultures were then fixed with 4% glutaraldehyde and stained with crystal violet dye. Only the cells receiving pHR‐CMV‐EGFP‐hTERTp‐TK were cleared by GCV treatment.
To evaluate whether the hTERTp‐TK element in the lentiviral vector could differentiate between tumor cells versus normal cells, we transduced HT‐1080 (human fibrosarcoma cells), 293T cells, SCCLSU‐1 (human head and neck squamous cell carcinoma cells), primary normal human fibroblasts, and normal hMSC with 10 MOI of the HIV‐CMV‐EGFP‐hTERTp‐TK vector. As controls, the cells were transduced with the identical MOI of the mock vector HIV‐CMV‐EGFP. Two days after transduction, 12.5 µg/mL GCV was applied daily for 6–8 days. Crystal violet staining demonstrated that all the tumor cells (HT‐1080, 293T, and SCCLSU‐1) were eliminated by GCV only when transduced with the hTERTp‐TK‐containing vector (Fig. 2a–c). Further culture for 2 additional weeks after GCV withdrawal produced no cell growth, confirming complete eradication. Notably, SCCLSU‐1 cells were directly isolated from clinically dissected tumors, suggesting that the hTERTp‐TK cassette functions in both established cell lines and primarily isolated tumor cells. Furthermore, we chose two types of leukemia cells to test the potency of hTERTp‐TK. Jurkat cells, derived from a patient with acute lymphoblastic leukemia, were transduced with 10 MOI of HIV‐CMV‐EGFP or HIV‐CMV‐EGFP‐hTERTp‐TK vector. Six days later, the top 1% of the fluorescent cells were obtained by FACS. The collected cells were subsequently amplified, then sorted again to maximize the EGFP‐positive homogeneity. Five thousand cells from the final collections were seeded in medium containing 4 µg/mL GCV. Total cell numbers were counted at various time points and graphed (Fig. 2f). Only the Jurkat cells receiving the hTERTp‐TK‐containing vector were eradicated, whereas the drug treatment did not significantly affect growth of the three controls: (i) HIV‐CMV‐EGFP‐transduced cells receiving no GCV treatment; (ii) HIV‐CMV‐EGFP‐transduced cells treated with GCV; and (iii) HIV‐CMV‐EGFP‐hTERTp‐TK‐transduced cells receiving no GCV treatment. YAC‐1 cells, derived from a mouse T‐cell lymphoma that was induced by murine leukemia virus infection, were also selected for testing whether hTERTp‐TK could differentiate this type of tumor. The cells were transduced with 20 MOI of HIV‐CMV‐EGFP‐hTERTp‐TK vector. Individual EGFP‐positive cells were identified, cloned and expanded. The experimental group received 4 µg/mL GCV treatment daily for a week, and the control group received no treatment. We found that the hTERTp‐TK element permits eradication of the lymphoma cells (Fig. 2g). In contrast to the varied tumor cells, normal primary human fibroblasts and hMSC were not affected by GCV treatment, regardless of transduction of the hTERTp‐TK‐containing vector or the control vector (Fig. 2d,e). Flow cytometric and fluorescent microscopic analyses confirmed that more than 95% of the cells were expressing EGFP (data not shown), suggesting that the resistance to GCV‐mediated killing by these cells is not due to them being refractory to vector transduction. Therefore, we conclude that the hTERTp‐TK cassette in the tested vector model is incapable of ablating normal cells with no or low telomerase activity. MSC in adult bone marrow were shown to have some telomerase activity by using the telomerase repeat amplification assay.( 28 , 29 ) However, our current data proved that the expression of hsvTK by the hTERT promoter in the stem cells is inadequate to elicit GCV‐mediated killing. This finding suggests the possibility of using the safety‐surveillance element in gene delivery to stem cells. In nonexpanding CD34+ HSC, telomerase is undetectable.( 30 ) Direct measurement of telomerase levels in HSC showed an equivalent of 0.06% to 0.4% of the activity of 293 cells.( 31 ) Moreover, telomere shortening occurs in transplanted HSC, suggesting that the telomerase activity is too low to compensate for telomere loss.( 32 , 33 ) Thus, we believe that the hTERTp‐TK element can also be applied in gene delivery to HSC to check and control vector‐insertional oncogenesis.
Figure 2.
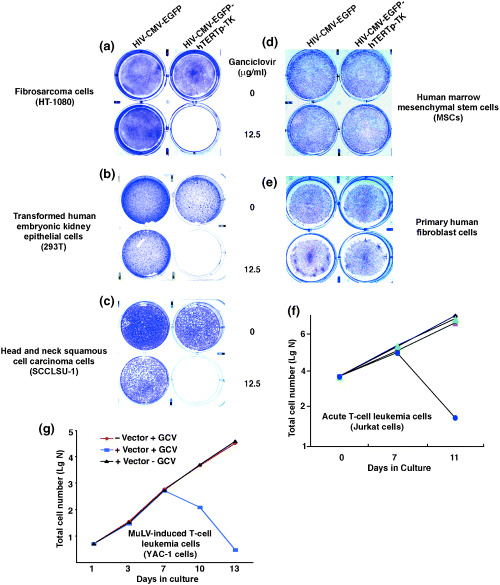
Tumor‐selective elimination by hTERTp‐TK in the lentiviral vector. (a) HT‐1080 cells. (b) 293T cells. (c) SSCLSU‐1 cells (primarily isolated human head and neck squamous cell carcinoma cells). (d) Normal human bone marrow mesenchymal stem cells. (e) Primary human fibroblasts. (a–e) Each group of cells was transduced with 10 MOI of HIV‐CMV‐EGFP‐hTERTp‐TK (right two wells of each panel) or HIV‐CMV‐EGFP (left two wells of each panel). Ganciclovir (GCV) was subsequently applied for a week at the indicated doses, and 6 days after GCV treatment the cells were fixed and stained with crystal violet dye. As noted, tumor cells but not normal cells were eradicated by GCV only when transduced with HIV‐CMV‐EGFP‐hTERTp‐TK, suggesting a tumor‐selective activity of the hTERT promoter. The blue area represents cells grown on the culture surface. (f) GCV effect on Jurkat T cell leukemia cells transduced with HIV‐CMV‐EGFP‐hTERTp‐TK. Jurkat cells were transduced with 10 MOI of either HIV‐CMV‐EGFP‐hTERTp‐TK or HIV‐CMV‐EGFP. The top 1% of the highest EGFP‐expressing population was acquired as described in the experimental protocol. Equal numbers of cells (5000) from each group were cultured in medium containing 4 µg/mL GCV until the end of the experiments. At the indicated times, the cultures were sampled and cell numbers counted microscopically. Blue circles: HIV‐CMV‐EGFP‐hTERTp‐TK transduced cells/+GCV; red rectangles: HIV‐CMV‐EGFP‐hTERTp‐TK transduced cells/‐GCV; green triangles: HIV‐CMV‐EGFP transduced cells/+GCV; black diamonds: HIV‐CMV‐EGFP transduced cells/–GCV. (g) GCV effect on YAC‐1 cells transduced with or without HIV‐CMV‐EGFP‐hTERTp‐TK.
To quantitatively assess the potency of the hTERTp‐TK cassette in tumor cells, we transduced HT‐1080 cells with either the HIV‐CMV‐EGFP‐hTERTp‐TK or the HIV‐CMV‐EGFP vector. Single‐cell clones (HT‐1080‐EGFP‐TK F1, F2, F3, and HT‐1080‐EGFP C1) were obtained by limiting dilution. A GCV dose–response assay was performed in vitro to determine the effective concentration resulting in 50% killing (EC50). The results are summarized in Table 1. From these results, we conclude that the hTERT promoter activity in HT‐1080 allows specific GCV‐associated killing at low micromolar levels.
Table 1.
Concentration of ganciclovir (GCV) required for 50% cell killing (EC50)
| Cell clones | EC50 (µM GCV) |
|---|---|
| HT‐1080‐EGFP‐TK (F1) | 7.2 |
| HT‐1080‐EGFP‐TK (F2) | 3.8 |
| HT‐1080‐EGFP‐TK (F3) | 5.2 |
| HT‐1080‐EGFP (C1) | No effect at 40 |
For gene delivery, an extreme case would be that a single cell was transduced by a single vector, giving rise to a single‐copy vector integration. Therefore, we asked whether a single integrant of hTERTp‐TK is sufficient to eliminate in vivo tumorigenesis. To this end, we performed Southern blot analysis on the HT‐1080‐EGFP‐TK F3 clone with a probe to the hsvTK gene or to exon 13 of the human CFTR gene (Fig. 3b). As a control, a clone from the HIV‐CMV‐EGFP‐transduced cells was also selected. Flow cytometric analyses of EGFP transgene expression were performed (Fig. 3a). We noticed that the progeny of the F3 clone had relatively weak and varied levels of EGFP expression, although they were from a single transduced cell. For establishment of subcutaneous tumors, cells (1 × 106) from each of clones F3 and C1 were injected into the rear flanks of five nu/nu mice, respectively (left flanks: HT‐1080‐EGFP‐TK; right flanks: HT‐1080‐EGFP). Two weeks after the xenograft, solid tumors (100–200 mm3) developed (Fig. 3c, top panel). An 8‐day GCV treatment regime was followed (1.25 mg in 1 mL PBS per dose, i.p., twice a day). Every 3 days, the tumor sizes were measured and tumor volumes were estimated using the formula: (tumor length) × (tumor width)2 × 0.5. As noted, tumors with the integrated hTERTp‐TK element diminished and disappeared (left flanks, bottom panel, Fig. 3c), whereas the control tumors reached the animal welfare limit and the animals were killed at day 16 (right flanks, bottom panel, Fig. 3c). Statistical data are also shown (Fig. 3d). To ensure that every single tumor cell in the tumor mass was eradicated, a separate experiment was performed where only the HT‐1080‐EGFP‐TK cells were injected into one flank of a group of five nu/nu mice. After GCV treatment, the established tumors again regressed and disappeared (Fig. 3e). The animals were housed for more than 2 months after the GCV treatment, and during this time no tumor recurrence was observed in any of the five tested animals. Therefore, a single‐copy integration of the lentivector containing hTERTp‐TK is sufficient to eradicate tumorigenesis in vivo. To confirm these results, we further tested the HT‐1080‐EGFP‐TK F2 clone in vivo. Although F2 tumors had a slower growth rate, taking approximately 3 weeks to develop to a size of 50–100 mm3 in volume, they responded to the GCV treatment in a similar manner as the F3 clone (Fig. 3e).
Figure 3.
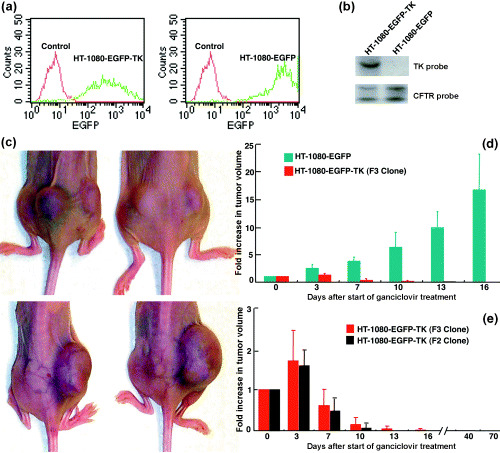
Single integration of the lentivector containing hTERTp‐TK is sufficient for self‐elimination of tumorigenesis in vivo. (a) Flow cytometric analysis of the F3 clone of HT‐1080‐EGFP‐TK cells (left panel) and the HT‐1080‐EGFP cells (right panel). Untransduced cells served as controls. (b) Southern blot analysis of the EcoRI‐digested genomic DNA (15 µg per lane) from the HT‐1080‐EGFP‐TK cells and HT‐1080‐EGFP cells. The EcoRI enzyme has a unique restriction site in the vector that is located outside of the HSV‐TK gene. Upper panel: A blot hybridized with the 32P‐labeled probe specific for the HSV‐TK gene. HT‐1080‐EGFP‐TK cells (left lane) with a single radioactive band indicating a single hTERT‐TK integration. In contrast, HT‐1080‐EGFP cells (right lane) had no radioactive band. Lower panel: An identical blot hybridized with the probe specific for the entire exon 13 of the cystic fibrosis transmembrane conductance regulator (CFTR) gene as a control. EcoRI cuts exon 13 of the CFTR gene into two parts. Therefore, both HT‐1080‐EGFP‐TK and HT‐1080‐EGFP clones gave two radioactive bands as expected (left and right lanes). (c) Photographs of xenografted tumors on two animals, developed from HT‐1080‐EGFP‐TK cells (left rear flank) and HT‐1080‐EGFP cells (right rear flank) on the same two animals before (upper panel) and after GCV treatment (lower panel). The tumors were apparent on both flanks prior to GCV application. Sixteen days after GCV treatment the HT‐1080‐EGFP‐TK tumors had disappeared, whereas the control HT‐1080‐EGFP tumors continued growing. (d) Quantitative analyses of tumor dynamics in five animals after starting GCV treatment. Green bars: HT‐1080‐EGFP control tumors; red bars: HT‐1080‐EGFP‐TK tumors. The y‐axis indicates the increase in tumor volume compared with the initial tumor size when starting the GCV treatment. (e) Long‐term study of tumor elimination by the hTERTp‐TK element. Five mice were inoculated with only the HT‐1080‐EGFP‐TK (F3 clone, red bars) tumor cells so that tumor growth dynamics could be monitored over longer periods of time. The hTERTp‐TK‐GCV effects on tumor formation were similar to those seen in Panel d. Up to 70 days post GCV treatment, the mice remained healthy and tumor‐free. Another HT‐1080‐EGFP‐TK clone (F2, black bars) was also tested in four animals. Tumors were totally eradicated 13 days after GCV treatment.
In order to evaluate the long‐term stability of the hTERTp‐TK cassette in cells receiving gene transfer, the HT‐1080‐EGFP‐TK cells were subjected to two rounds of cloning process and were serially passaged to greater than 60 population doublings in culture. Then, 106 cells were seeded into each nu/nu mouse to form subcutaneous tumors. Subsequently, the tumors were dissected, passed through a 70‐µm sieve and washed. The harvested tumor cells were seeded into another batch of nu/nu mice. After 2 weeks, tumor growth was observed (100 mm3 ± 50, n = 3). Similarly, a GCV regime was followed (1.25 mg in 1 mL PBS per dose, i.p., twice a day). After drug treatment, all the tumors completely regressed. No tumor regrowth occurred within 70 days of regression. Mice implanted with the same cells without GCV treatment grew tumors to a size such that the animals had to be killed. These results suggest that the vector with the hTERTp‐TK element is stable in transduced cells.
Discussion
We have demonstrated that the hTERTp‐TK cassette in the HIV‐based lentiviral vector system can differentiate between tumoral and normal cells, suggesting the possibility of exploiting the cassette for post‐therapeutic elimination of vector‐associated insertional oncogenesis in human gene therapy. Gene therapy trials for SCID have been the only success in the gene therapy field. Unfortunately, three children treated in this way developed T‐cell leukemia, which has bred concern about the risks associated with this therapy. Therefore, improving the safety features of the current integrating vector systems has become imperative. Our findings offer a potential strategy to overcome the problem.
The HIV‐based lentiviral vectors have the advantage of transducing non‐dividing cells. Therefore, this type of vector is a very promising tool for delivering genes to quiescent cells. Previous reports have indicated that lentiviral vectors integrate into a known tumor‐suppressor gene,( 6 ) which raises the possibility of vector insertional oncogenesis. Thus, the addition of a safety‐checking mechanism to the vector will allow self‐deletion of any tumorized cells, if vector insertion leads to oncogenesis and the telomerase promoter is activated. We also believe that the hTERTp‐TK cassette can be incorporated into Moloney murine leukemia viral (MuLV) vectors, the vector system used for the X‐SCID trials, to improve their safety. Therefore, further evaluation of the hTERTp‐TK cassette in the MuLV system is warranted.
Although the current study focused on assessing the hTERTp‐TK cassette as a safety‐guard element in the lentiviral vector, we believe that the vector with this cassette could also be used for direct tumor targeting. In previous publications, we and others reported that the hTERT promoter drives adenoviral E1 gene expression, thus restricting adenovirus replication only in tumor cells.( 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ) The adenovirus‐associated cytolysis mechanism is different from that of suicide gene targeting through the hTERTp‐TK cassette. Using a lentiviral system to deliver the hsv‐TK gene to tumor cells has been studied by several groups.( 34 , 35 , 36 , 37 , 38 , 39 ) However, the promoters used to drive TK gene expression were either ubiquitously active or tissue‐specific. Therefore, exploiting the hTERT promoter could be advantageous in terms of restricting TK‐gene expression to a broad spectrum of tumor cells to increase the vector‐targeting specificity. This proposed research is worthy of being pursued, but is beyond the scope of this study.
Acknowledgments
We thank Donna Bertucci and Anand Viswanathan for expert technical assistance, Olga Sirin for her assistance in vector preparation and titering, and Dr Jakob Reiser for his critical reading of the manuscript and helpful discussions. This work was partially supported by the Louisiana Gene Therapy Research Consortium. F.P. is a research fellow of the National Hemophilia Foundation.
References
- 1. Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV‐1 integration in the human genome favors active genes and local hotspots. Cell 2002; 110: 521–9. [DOI] [PubMed] [Google Scholar]
- 2. Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003; 300: 1749–51. [DOI] [PubMed] [Google Scholar]
- 3. Laufs S, Gentner B, Nagy KZ et al. Retroviral vector integration occurs in preferred genomic targets of human bone marrow‐repopulating cells. Blood 2003; 101: 2191–8. [DOI] [PubMed] [Google Scholar]
- 4. De Palma M, Montini E, Santoni de Sio FR et al. Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood 2005; 105: 2307–15. [DOI] [PubMed] [Google Scholar]
- 5. Hacein‐Bey‐Abina S, Von Kalle C, Schmidt M et al. LMO2‐associated clonal T cell proliferation in two patients after gene therapy for SCID‐X1. Science 2003; 302: 415–9. [DOI] [PubMed] [Google Scholar]
- 6. Woods NB, Muessig A, Schmidt M et al. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood 2003; 101: 1284–9. [DOI] [PubMed] [Google Scholar]
- 7. Cavazzana‐Calvo M, Hacein‐Bey S, De Saint Basile G et al. Gene therapy of human severe combined immunodeficiency (SCID)‐X1 disease. Science 2000; 288: 669–72. [DOI] [PubMed] [Google Scholar]
- 8. Hacein‐Bey‐Abina S, Von Kalle C, Schmidt M et al. A serious adverse event after successful gene therapy for X‐linked severe combined immunodeficiency. N Engl J Med 2003; 348: 255–6. [DOI] [PubMed] [Google Scholar]
- 9. Blackburn EH. Structure and function of telomeres. Nature 1991; 350: 569–73. [DOI] [PubMed] [Google Scholar]
- 10. Yu GL, Bradley JD, Attardi LD, Blackburn EH. In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature 1990; 344: 126–32. [DOI] [PubMed] [Google Scholar]
- 11. Blackburn EH. Telomere states and cell fates. Nature 2000; 408: 53–6. [DOI] [PubMed] [Google Scholar]
- 12. Greider CW. Telomeres and senescence: the history, the experiment, the future. Curr Biol 1998; 8: R178–81. [DOI] [PubMed] [Google Scholar]
- 13. Harley CB, Kim NW. Telomerase and cancer. Important Adv Oncol 1996: 57–67. [PubMed]
- 14. Kim NW, Piatyszek MA, Prowse KR et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266: 2011–5. [DOI] [PubMed] [Google Scholar]
- 15. Horikawa I, Cable PL, Afshari C, Barrett JC. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res 1999; 59: 826–30. [PubMed] [Google Scholar]
- 16. Lanson NA Jr, Friedlander PL, Schwarzenberger P, Kolls JK, Wang G. Replication of an adenoviral vector controlled by the human telomerase reverse transcriptase promoter causes tumor‐selective tumor lysis. Cancer Res 2003; 63: 7936–41. [PubMed] [Google Scholar]
- 17. Huang TG, Savontaus MJ, Shinozaki K, Sauter BV, Woo SL. Telomerase‐dependent oncolytic adenovirus for cancer treatment. Gene Ther 2003; 10: 1241–7. [DOI] [PubMed] [Google Scholar]
- 18. Kim E, Kim JH, Shin HY et al. Ad‐mTERT‐delta19, a conditional replication‐competent adenovirus driven by the human telomerase promoter, selectively replicates in and elicits cytopathic effect in a cancer cell‐specific manner. Hum Gene Ther 2003; 14: 1415–28. [DOI] [PubMed] [Google Scholar]
- 19. Wirth T, Zender L, Schulte B et al. A telomerase‐dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res 2003; 63: 3181–8. [PubMed] [Google Scholar]
- 20. Irving J, Wang Z, Powell S et al. Conditionally replicative adenovirus driven by the human telomerase promoter provides broad‐spectrum antitumor activity without liver toxicity. Cancer Gene Ther 2004; 11: 174–85. [DOI] [PubMed] [Google Scholar]
- 21. Gu J, Fang B. Telomerase promoter‐driven cancer gene therapy. Cancer Biol Ther 2003; 2: S64–70. [PubMed] [Google Scholar]
- 22. Zhang Q, Nie M, Sham J et al. Effective gene‐viral therapy for telomerase‐positive cancers by selective replicative‐competent adenovirus combining with endostatin gene. Cancer Res 2004; 64: 5390–7. [DOI] [PubMed] [Google Scholar]
- 23. Huang Q, Zhang X, Wang H et al. A novel conditionally replicative adenovirus vector targeting telomerase‐positive tumor cells. Clin Cancer Res 2004; 10: 1439–45. [DOI] [PubMed] [Google Scholar]
- 24. Sekiya I, Larson BL, Smith JR, Pochampally R, Cui JG, Prockop DJ. Expansion of human adult stem cells from bone marrow stroma: conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells 2002; 20: 530–41. [DOI] [PubMed] [Google Scholar]
- 25. Park F, Ohashi K, Chiu W, Naldini L, Kay MA. Efficient lentiviral transduction of liver requires cell cycling in vivo. Nat Genet 2000; 24: 49–52. [DOI] [PubMed] [Google Scholar]
- 26. Engelmann C, Heslan JM, Fabre M, Lagarde JP, Klatzmann D, Panis Y. Importance, mechanisms and limitations of the distant bystander effect in cancer gene therapy of experimental liver tumors. Cancer Lett 2002; 179: 59–69. [DOI] [PubMed] [Google Scholar]
- 27. Asklund T, Appelskog IB, Ammerpohl O et al. Gap junction‐mediated bystander effect in primary cultures of human malignant gliomas with recombinant expression of the HSVtk gene. Exp Cell Res 2003; 284: 185–95. [DOI] [PubMed] [Google Scholar]
- 28. Gronthos S, Zannettino AC, Hay SJ et al. Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J Cell Sci 2003; 116: 1827–35. [DOI] [PubMed] [Google Scholar]
- 29. Parsch D, Fellenberg J, Brummendorf TH, Eschlbeck AM, Richter W. Telomere length and telomerase activity during expansion and differentiation of human mesenchymal stem cells and chondrocytes. J Mol Med 2004; 82: 49–55. [DOI] [PubMed] [Google Scholar]
- 30. Engelhardt M, Kumar R, Albanell J, Pettengell R, Han W, Moore MA. Telomerase regulation, cell cycle, and telomere stability in primitive hematopoietic cells. Blood 1997; 90: 182–93. [PubMed] [Google Scholar]
- 31. Yui J, Chiu CP, Lansdorp PM. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood 1998; 91: 3255–62. [PubMed] [Google Scholar]
- 32. Allsopp RC, Cheshier S, Weissman IL. Telomerase activation and rejuvenation of telomere length in stimulated T cells derived from serially transplanted hematopoietic stem cells. J Exp Med 2002; 196: 1427–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Allsopp RC, Morin GB, DePinho R, Harley CB, Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSC during serial transplantation. Blood 2003; 102: 517–20. [DOI] [PubMed] [Google Scholar]
- 34. Marcello A, Giaretta I. Inducible expression of herpes simplex virus thymidine kinase from a bicistronic HIV1 vector. Res Virol 1998; 149: 419–31. [DOI] [PubMed] [Google Scholar]
- 35. Kong B, Wang W, Liu C et al. Efficacy of lentivirus‐mediated and MUC1 antibody‐targeted VP22‐TK/GCV suicide gene therapy for ovarian cancer. In Vivo 2003; 17: 153–6. [PubMed] [Google Scholar]
- 36. Uch R, Gerolami R, Faivre J et al. Hepatoma cell‐specific ganciclovir‐mediated toxicity of a lentivirally transduced HSV‐TkEGFP fusion protein gene placed under the control of rat alpha‐fetoprotein gene regulatory sequences. Cancer Gene Ther 2003; 10: 689–95. [DOI] [PubMed] [Google Scholar]
- 37. Gerolami R, Uch R, Faivre J et al. Herpes simplex virus thymidine kinase‐mediated suicide gene therapy for hepatocellular carcinoma using HIV‐1‐derived lentiviral vectors. J Hepatol 2004; 40: 291–7. [DOI] [PubMed] [Google Scholar]
- 38. Pellinen R, Hakkarainen T, Wahlfors T et al. Cancer cells as targets for lentivirus‐mediated gene transfer and gene therapy. Int J Oncol 2004; 25: 1753–62. [PubMed] [Google Scholar]
- 39. Germain E, Roullin VG, Qiao J, De Campos Lima PO, Caruso M. RD114‐pseudotyped retroviral vectors kill cancer cells by syncytium formation and enhance the cytotoxic effect of the TK/GCV gene therapy strategy. J Gene Med 2005; 7: 389–97. [DOI] [PubMed] [Google Scholar]