

Abstract
Obesity and related metabolic abnormalities are risk factors for colorectal cancer. A state of chronic inflammation and adipocytokine imbalance may play a role in colorectal carcinogenesis. Statins, which are commonly used for the treatment of hyperlipidemia, are known to possess anti‐inflammatory effects. Statins also exert chemopreventive properties against various cancers. The present study examined the effects of pitavastatin, a recently developed lipophilic statin, on the development of azoxymethane (AOM)‐initiated colonic premalignant lesions in C57BL/KsJ‐db/db (db/db) obese mice. Male db/db mice were administrated weekly subcutaneous injections of AOM (15 mg/kg body weight) for 4 weeks and then were subsequently fed a diet containing 1 ppm or 10 ppm pitavastatin for 8 weeks. Feeding with either dose of pitavastatin significantly reduced the number of colonic premalignant lesions, β‐catenin accumulated crypts, by inhibiting proliferation and the surrounding inflammation. Pitavastatin increased the serum levels of adiponectin while conversely decreasing the serum levels of total cholesterol, tumor necrosis factor‐α (TNF‐α), interleukin (IL)‐6, IL‐18, and leptin. Pitavastatin also caused a significant increase in the expression of phosphorylated form of the AMP‐activated kinase (AMPK) protein on the colonic mucosa of AOM‐treated mice. In addition, the expression levels of TNF‐α, IL‐6, IL‐18, and COX‐2 mRNAs on the colonic mucosa of AOM‐treated mice were decreased by treatment with this agent. These findings suggest that pitavastatin attenuates chronic inflammation and improves the imbalance of adipocytokines, both of which are caused by the presence of excess adipose tissues, thereby preventing the development of colonic premalignancies in an obesity‐related colon cancer model. Therefore, some types of statins, including pitavastatin, may be a useful chemoprevention modality for colon cancer in obese individuals. (Cancer Sci 2010)
Colorectal cancer (CRC) is a serious healthcare problem worldwide due to its substantial morbidity and mortality rates in patients. Recent evidence has indicated that obesity and its related metabolic abnormalities are associated with an increased risk for CRC.( 1 , 2 , 3 ) Several hypotheses have emerged to explain the influence of obesity on the development of CRC, including insulin resistance, alterations in the insulin‐like growth factor (IGF)/IGF‐1 receptor axis and adipocytokine imbalance, such as increased leptin levels and decreased adiponectin levels.( 1 , 2 , 3 ) A state of chronic inflammation, which is induced by excessive production of storage lipids and high circulating glucose levels, also plays a critical role in obesity‐related colorectal carcinogenesis.( 4 ) Adipose tissue constitutively expresses the pro‐inflammatory cytokine tumor necrosis factor‐α (TNF‐α).( 5 , 6 ) Because TNF‐α, a central mediator in chronic inflammatory diseases, stimulates tumor promotion and progression of carcinogenesis, the inhibition of this key pro‐inflammatory molecule may therefore be a novel treatment and prevention strategy for several malignancies, including CRC.( 7 , 8 )
Statins, 3‐hydroxy‐3‐methyl‐glutaryl coenzyme A (HMG‐CoA) reductase inhibitors, are widely used for the treatment of hyperlipidemia and have been shown to exert beneficial effects on both cardiovascular and cerebrovascular diseases.( 9 , 10 ) Statins prevent the conversion of HMG‐CoA to mevalonate and thus decrease serum lipid levels, especially those of low‐density lipoprotein cholesterol and triglyceride. In addition to their lipid‐lowering effect, statins have demonstrated anticancer properties.( 11 , 12 ) Statins suppress inflammation, induce apoptosis, and modulate angiogenesis, thus inhibiting the growth of a wide variety of cancer cells.( 11 , 12 ) Statins induce apoptosis in human colon cancer cells and attenuate inflammation‐related colon carcinogenesis in mice.( 13 , 14 ) Epidemiologic studies have also demonstrated the chemopreventive properties of statins for various types of cancer, including CRC.( 11 , 12 , 15 ) These reports suggest that statin administration might therefore be an effective strategy for preventing the development of CRC. However, no detailed studies on whether statins can prevent the development of obesity‐related CRC have so far been conducted.
In order to develop an effective method to prevent obesity‐related colorectal carcinogenesis, we established a useful preclinical animal model using the colonic carcinogen azoxymethane (AOM) and C57BL/KsJ‐db/db (db/db) mice, which are obese and exhibit hyperlipidemia.( 16 , 17 , 18 , 19 ) In the present study, we investigated the effects of pitavastatin, a recently developed lipophilic statin,( 20 ) on the development of β‐catenin accumulated crypts (BCAC) and aberrant crypt foci (ACF), both of which are putative precursor lesions for colonic adenocarcinoma( 21 , 22 ) and which are utilized as biomarkers to evaluate a number of agents for their potential chemopreventive properties,( 23 , 24 ) in db/db mice treated with AOM.
Materials and Methods
Animals, chemicals, and diets. Four‐week‐old male homozygous db/db mice were obtained from Japan SLC (Shizuoka, Japan). All mice were maintained at the Gifu University Life Science Research Center in accordance with the Institutional Animal Care Guidelines. Azoxymethane (AOM) was purchased from Sigma Chemical (St. Louis, MO, USA). Pitavastatin was obtained from Kowa Pharmaceutical (Tokyo, Japan).
Experimental procedure. The animal experiment, as described previously,( 18 , 19 ) was approved by the Committee of the Institutional Animal Experiments of Gifu University. A total of 34 male db/db mice were divided into five groups. At 5 weeks of age, the mice in Groups 1 (five mice) and 2 (five mice) were given four weekly subcutaneous injections of saline. The mice in Groups 3 (eight mice), 4 (eight mice), and 5 (eight mice) were subcutaneously injected with AOM (15 mg/kg body weight) once a week for 4 weeks. Groups 1 and 3 were fed the basal diet CRF‐1 (Oriental Yeast, Tokyo, Japan), throughout the experiment (for 12 weeks). Group 2 was fed the diet containing 10 ppm pitavastatin throughout the experiment. Groups 4 and 5 were given the diets containing 1 and 10 ppm pitavastatin, respectively, for 8 weeks, starting 1 week after the last injection of AOM. At the termination of the study (17 weeks of age), all mice were sacrificed by CO2 asphyxiation for analyzing histopathology, the number of colonic BCAC and ACF, and clinical chemistry.
Counting the number of BCAC and ACF. The frequencies of BCAC and ACF were determined according to the standard procedures described previously.( 17 , 18 , 19 ) After the resected colons on filter papers were fixed in 10% buffered formalin for 24 h, the mucosal surfaces were stained with methylene blue (0.5% in distilled water) and the number of ACF was counted under a light microscope. After counting the ACF, the distal parts (1 cm from anus) of the colon were cut to count the number of BCAC. To identify BCAC intramucosal lesions, the distal part of the colon (mean area, 0.7 cm2/colon) was embedded in paraffin and a total of 20 serial sections (4‐μm thick each) per mouse were created by an en face preparation.( 17 , 18 , 19 ) For each case, two serial sections were used to analyze BCAC.
Histopathology and immunohistochemical analyses for β‐catenin and PCNA. Three serial sections were cut from paraffin‐embedded tissue blocks. Two sections were subjected to hematoxylin–eosin (H&E) staining for histopathology and β‐catenin immunohistochemistry to count the number of BCAC. The other section was used for immunohistochemical staining of proliferating cell nuclear antigen (PCNA), a G1‐to‐S phase marker, to estimate the proliferative activity of the BCAC. Immunohistochemical staining of β‐catenin and PCNA were performed using the labeled streptavidin–biotin method (LSAB kit; Dako, Glostrup, Denmark) as previously described.( 17 , 18 , 19 ) The primary antibodies included an anti‐β‐catenin antibody (1:1000 final dilution) from BD Transduction Laboratories (Cat. No. 610154; San Jose, CA, USA) and an anti‐PCNA antibody (1:100 final dilution) from Santa Cruz Biotechnology (sc‐7907; Santa Cruz, CA, USA). The negative control sections were immunostained without any primary antibodies. On the PCNA‐immunostained sections, the cells with intensively reacted nuclei were considered to be positive for PCNA, and the indices (%) were calculated in randomly selected 10 BCAC from Group 3, five BCAC from Group 4, and five BCAC from Group 5. The PCNA‐labeling indices for both ACF and surrounding crypts were also determined by representative lesions (n = 8), respectively.
RNA extraction and quantitative real‐time RT‐PCR. The expression levels of the TNF‐α, interleukin (IL)‐6, IL‐18, and cyclooxygenase (COX)‐2 genes were determined in the colonic mucosa of mice from Groups 3 through 5. A quantitative real‐time RT‐PCR analysis was performed as described previously.( 25 ) Total RNA was isolated from the scraped colonic mucosa of the experimental mice using the RNAqueous‐4PCR kit (Ambion Applied Biosystems, Austin, TX, USA). The cDNA was synthesized from 0.2 μg of total RNA using the SuperScript III First‐Strand Synthesis System (Invitrogen, San Diego, CA, USA). The specific primers used for the amplification of TNF‐α, IL‐6, IL‐18, COX‐2, and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) genes are as previously described.( 8 ) Real‐time RT‐PCR was performed in a LightCycler (Roche Diagnostics, Indianapolis, IN, USA) with the SYBR Premix Ex Taq (Takara Bio, Shiga, Japan). The expression levels of the TNF‐α, IL‐6, IL‐18, and COX‐2 genes were normalized to the GAPDH gene expression levels.
Protein extraction and western blot analysis. Total proteins were extracted from the scraped mucosa from the remaining colon of the AOM‐treated mice (Groups 3 through 5) and equivalent amounts of proteins (20 μg/lane) were examined by western blot analysis.( 18 , 19 ) The primary antibodies for AMP‐activated kinase (AMPK) and the phosphorylated form of AMPK (p‐AMPK) were obtained from Cell Signaling Technology (Danvers, MA, USA). An antibody against GAPDH (Chemicon International, Temecula, CA, USA) served as a loading control.
Clinical chemistry. After 6 h of fasting, blood samples were collected at the time of sacrifice to measure the serum concentrations of total cholesterol, triglyceride, adiponectin, leptin, TNF‐α, IL‐6, and IL‐18. The serum total cholesterol and triglyceride levels were assayed as described previously.( 17 , 18 , 19 ) The serum adiponectin (Otsuka, Tokyo, Japan), leptin (R&D Systems, Minneapolis, MN, USA), TNF‐α (Shibayagi, Gunma, Japan), IL‐6 (IBL, Gunma, Japan), and IL‐18 (MBL, Aichi, Japan) levels were determined by an enzyme immunoassay (EIA) according to the manufacturer’s protocol.
Statistical analyses. The results are presented as the mean ± SD and were analyzed using the GraphPad Instat software program, version 3.05 (GraphPad Software, San Diego, CA, USA) for Macintosh. The differences between groups were analyzed by one‐way anova or, as required, by two‐way anova. When anova showed a statistically significant effect (P < 0.05), comparisons of each experimental group with the control group were performed using the Tukey–Kramer multiple comparisons test. The differences were considered statistically significant when the two‐tailed P‐value was <0.05.
Results
General observations. As shown in Table 1, the average body weights of the AOM‐injected groups (Groups 3 through 5) at the termination of the experiment were significantly lower than that of the saline‐injected group (Group 1, P < 0.05, respectively). This might be caused by the toxicity of AOM as observed in previous experiments.( 18 , 19 ) No significant differences were observed in the mean weights of the liver and kidney among the groups. A histopathological examination revealed the absence of toxicity of pitavastatin in these tissues of the mice that received pitavastatin (Groups 2, 4, and 5). As listed in Table 2, the average colon lengths in Groups 3 through 5 were shorter than that of Group 1 (P < 0.05, respectively). However, treatment with pitavastatin did not affect the colon length.
Table 1.
Body, liver, and kidney weights of the experimental mice
| Group no. | Treatment | No. of mice | Final body wt (g) | Relative organ weight (g/100 g body wt) | |
|---|---|---|---|---|---|
| Liver | Kidney | ||||
| 1 | Saline | 5 | 51.2 ± 8.4† | 6.23 ± 1.55 | 0.85 ± 0.25 |
| 2 | Saline + 10 ppm pitavastatin | 5 | 49.2 ± 7.7 | 6.05 ± 0.88 | 0.98 ± 0.23 |
| 3 | AOM alone | 8 | 40.9 ± 6.5* | 4.69 ± 1.10 | 0.95 ± 0.11 |
| 4 | AOM + 1 ppm pitavastatin | 8 | 41.1 ± 8.5* | 5.28 ± 0.79 | 1.05 ± 0.17 |
| 5 | AOM + 10 ppm pitavastatin | 8 | 39.7 ± 6.9* | 5.14 ± 1.02 | 1.10 ± 0.25 |
*Significantly different from Group 1 (P < 0.05). †Mean ± SD. AOM, azoxymethane.
Table 2.
Effects of pitavastatin on AOM‐induced ACF and BCAC formation in the experimental mice
| Group no. | Treatment | No. of mice | Length of colon (cm) | Total no. of ACFs/colon | Total no. of BCACs/cm2 |
|---|---|---|---|---|---|
| 1 | Saline | 5 | 12.2 ± 0.8† | 0 | 0 |
| 2 | Saline + 10 ppm pitavastatin | 5 | 11.7 ± 1.0 | 0 | 0 |
| 3 | AOM alone | 8 | 11.1 ± 0.7* | 80.4 ± 13.4 | 13.6 ± 9.3 |
| 4 | AOM + 1 ppm pitavastatin | 8 | 11.7 ± 0.7* | 73.5 ± 11.4 | 5.4 ± 1.8** |
| 5 | AOM + 10 ppm pitavastatin | 8 | 11.0 ± 1.2* | 72.8 ± 15.2 | 3.4 ± 2.6*** |
*Significantly different from Group 1 (P < 0.05). **Significantly different from Group 3 (P < 0.05). ***Significantly different from Group 3 (P < 0.01). †Mean ± SD. ACF, aberrant crypt foci; AOM, azoxymethane; BCAC, β‐catenin accumulated crypts.
Effects of pitavastatin on AOM‐induced BCAC and ACF formations in the db/db mice. Table 2 summarizes the total numbers of BCAC and ACF in each group. Aberrant crypt foci (ACF) (Fig. 1a,b) and BCAC (Fig. 1c–f) developed in the colons of all mice that received AOM (Groups 3 through 5), but not in those treated without AOM (Groups 1 and 2). The sizes of BCAC and ACF that developed in Group 3 (Fig. 1a,c) tended to be larger compared to those of Groups 4 and 5 (Fig. 1b,d). Interestingly, the inflammatory cell infiltration surrounding BCAC that developed in Group 3 (Fig. 1c) also tended to be more severe compared to that in Groups 4 and 5 (Fig. 1d). The total number of ACF in Groups 4 and 5 was smaller than that of Group 3, but the differences were not statistically significant. As for the total number of BCAC, the values of Groups 4 (P < 0.05) and 5 (P < 0.01) were significantly lower than that of Group 3: the inhibition rates were 60% in Group 4 and 75% in Group 5, respectively (Table 2).
Figure 1.
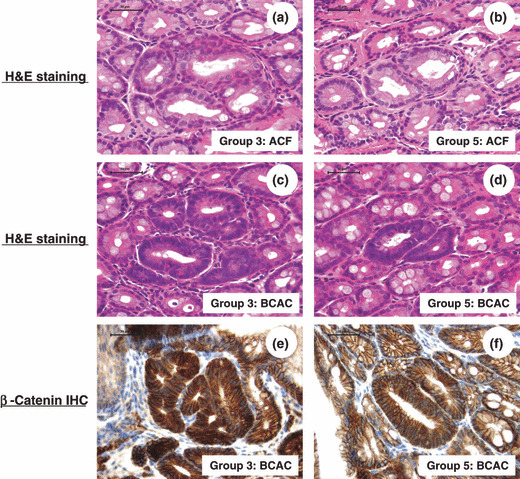
The representative histopathology and β‐catenin‐immunohistochemistry of aberrant crypt foci (ACF) and β‐catenin accumulated crypts (BCAC). (a,b) Aberrant crypt foci (ACF) on the H&E‐stained sections from Groups 3 and 5. (c,d) β‐Catenin accumulated crypts (BCAC) on the H&E‐stained sections from Groups 3 and 5. (e,f) β‐Catenin‐immunohistochemistry of BCAC from Groups 3 and 5. Note: The sizes of ACF and BCAC developed in Group 5 are smaller than those of Group 3. The localization of the accumulated β‐catenin protein is apparent in the cytoplasm and nucleus of atypical cryptal cells in BCAC. IHC, immunohistochemistry. Scale bar, 50 μm.
Effects of pitavastatin on the proliferation activity in BCAC of the AOM‐injected db/db mice. The PCNA‐labeling index of BCAC developed in the AOM‐treated db/db mice was determined by the PCNA‐immunohistochemical sections (Fig. 2a–c). As illustrated in Figure 2(d), the mean PCNA‐labeling index in the 1 ppm (78.7 ± 6.6%) and 10 ppm (71.1 ± 13.4%) pitavastatin‐treated mice was significantly smaller than that in the mice which received AOM alone (95.3 ± 3.0%; P < 0.01 for each comparison). Conversely, neither the PCNA‐labeling indices of ACF (40.6 ± 5.9%) nor the surrounding crypts (23.1 ± 9.5%) were affected by the treatment with 1 ppm (38.6 ± 7.0% or 24.6 ± 5.2%) or 10 ppm pitavastatin (34.9 ± 8.5% or 22.4 ± 8.8%), respectively. These findings indicate that pitavastatin significantly suppresses BCAC in comparison to ACF, at least in part, by reducing cell proliferation selectively in the lesion.
Figure 2.
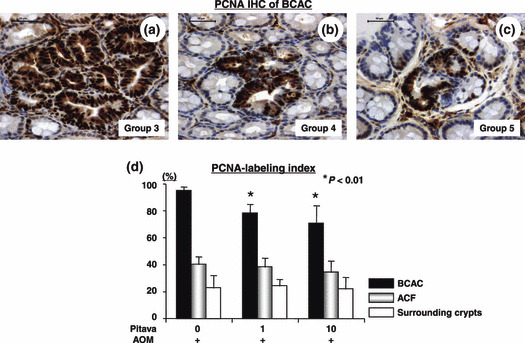
The effect of pitavastatin on cell proliferation in β‐catenin accumulated crypts (BCAC) and aberrant crypt foci (ACF) induced by azoxymethane (AOM) in male db/db mice. (a–c) Representative proliferating cell nuclear antigen (PCNA)‐immunohistochemistry of the BCAC dev‐eloped in the colon of Groups 3, 4, and 5. (d) The mean PCNA‐labeling indices of the BCAC, ACF, and surrounding crypts in the colon of Groups 3 through 5. IHC, immunohistochemistry. Scale bar, 50 μm. *P < 0.01 vs Group 3.
Effects of pitavastatin on serum levels of total cholesterol, triglyceride, adiponectin, and leptin in the db/db mice. The serum concentrations of the total cholesterol, triglyceride, adiponectin, and leptin levels are listed in Table 3. Although there were no significant differences in the serum triglyceride levels, the serum levels of total cholesterol in the pitavastatin‐treated groups (Groups 2, 4, and 5) were significantly lower than those in Groups 1 and 3, regardless of AOM‐injection (P < 0.05 and P < 0.01, respectively). The mice treated with both doses of pitavastatin showed a significant increase in the serum levels of adiponectin when compared to the mice treated without pitavastatin, regardless of AOM‐injection (P < 0.05 and P < 0.05, respectively). In addition, the serum leptin levels of 1 ppm and 10 ppm pitavastatin‐treated mice were significantly lower than those in untreated control mice (P < 0.01 for each comparison).
Table 3.
Serum levels of total cholesterol, triglyceride, adiponectin, and leptin in the experimental mice
| Group no. | Treatment | No. of mice | Total cholesterol (mg/dL) | Triglyceride (mg/dL) | Adiponectin (μg/mL) | Leptin (ng/dL) |
|---|---|---|---|---|---|---|
| 1 | Saline | 5 | 283 ± 44† | 168 ± 42 | 11.2 ± 1.7 | 87.0 ± 14.4 |
| 2 | Saline + 10 ppm pitavastatin | 5 | 193 ± 58* | 174 ± 29 | 15.7 ± 1.7* | 72.6 ± 15.1 |
| 3 | AOM alone | 8 | 170 ± 13** | 132 ± 24 | 13.1 ± 1.3 | 93.6 ± 4.2 |
| 4 | AOM + 1 ppm pitavastatin | 8 | 143 ± 17*** | 138 ± 24 | 18.0 ± 3.2**** | 80.6 ± 6.5*** |
| 5 | AOM + 10 ppm pitavastatin | 8 | 133 ± 22*** | 133 ± 25 | 17.7 ± 3.0**** | 70.7 ± 19.9*** |
*Significantly different from Group 1 (P < 0.05). **Significantly different from Group 1 (P < 0.01). ***Significantly different from Group 3 (P < 0.01). ****Significantly different from Group 3 (P < 0.05). †Mean ± SD. AOM, azoxymethane.
Effects of pitavastatin on the serum level of TNF‐α, IL‐6, and IL‐18 in the db/db mice. The effects of pitavastatin on the serum levels of TNF‐α, IL‐6, and IL‐18 were examined using the EIA method (Fig. 3). In the AOM‐treated mice, not only a high (10 ppm), but also a low (1 ppm) dose of pitavastatin caused a significant decrease in the serum levels of TNF‐α (Fig. 3a) and IL‐18 (Fig. 3c) in comparison to the control mice (P < 0.05, respectively). The serum levels of IL‐6 (Fig. 3b) were also decreased after treatment with pitavastatin, and this difference was significant after treatment with a high dose of this agent (P < 0.05).
Figure 3.
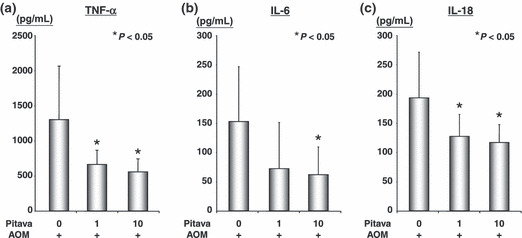
The effect of pitavastatin on the serum levels of tumor necrosis factor‐α (TNF‐α), interleukin (IL)‐6, and IL‐18 in the azoxymethane (AOM)‐treated db/db mice (Groups 3 through 5). The serum concentrations of TNF‐α (a), IL‐6 (b), and IL‐18 (c) were measured by an enzyme immunoassay. Values are the mean ± SD (n = 8). *P < 0.05 vs Group 3.
Effects of pitavastatin on the activation of AMPK and on the expression levels of TNF‐α, IL‐6, IL‐18, and COX‐2 mRNAs in the colonic mucosa of AOM‐injected db/db mice. As shown in Figure 4(a), a western blot analysis demonstrated that treatment with both concentrations of pitavastatin markedly increased the levels of the phosphorylated (i.e. activated) form of AMPK protein in the colonic mucosa of AOM‐treated mice. Real‐time RT‐PCR analyses also revealed that both doses of pitavastatin caused a significant decrease in the expression levels of pro‐inflammatory cytokine TNF‐α (P < 0.05, Fig. 4b), IL‐6 (P < 0.05, Fig. 4c), and IL‐18 (P < 0.01, Fig. 4d) mRNAs in the colonic mucosa of AOM‐injected mice when compared to those of the untreated control mice. In addition, the mRNA expression levels of COX‐2, which is one of the main mediators in the inflammatory pathway and is certainly involved in CRC development,( 26 ) was markedly inhibited by treatment with pitavastatin in comparison to the control mice (P < 0.01, Fig. 4e).
Figure 4.
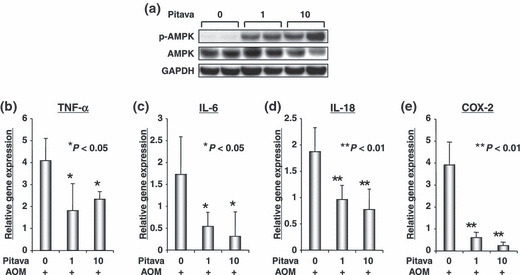
The effect of pitavastatin on the activation of AMP‐activated kinase (AMPK) and on the levels of tumor necrosis factor‐α (TNF‐α), interleukin (IL)‐6, IL‐18, and cyclooxygenase (COX)‐2 mRNAs in the colonic mucosa of AOM‐treated db/db mice (Groups 3 through 5). Total proteins were extracted from the scraped colonic mucosa and equivalent amounts of proteins were examined by a Western blot analysis using p‐AMPK and AMPK specific antibodies (a). An antibody to GAPDH served as a loading control (a). cDNA was synthesized from the colonic mucosa and real‐time RT‐PCR was performed using TNF‐α (b), IL‐6 (c), IL‐18 (d), and COX‐2 (e) specific primers. The expression levels of these genes were normalized to the level of the GAPDH gene (b–e). Each experiment was done in triplicate and the average was subsequently calculated. *P < 0.05 and **P < 0.01 vs Group 3, respectively.
Discussion
The results of the present study clearly indicated that pitavastatin effectively inhibits the development of BCAC (Fig. 1c–f), which are putative precursor lesions for CRC,( 21 , 23 ) in male db/db obese mice (Table 2). This inhibition was most likely associated with the decrease of pro‐inflammatory cytokines, such as TNF‐α, IL‐6, and IL‐18, in serum (Fig. 3a–c) as well as in the colonic mucosa (Fig. 4b–d), and the inhibition of proliferation in BCAC (Fig. 2). Pitavastatin also caused a decrease in the expression levels of COX‐2 mRNA (Fig. 4e), which is a critical target for CRC chemoprevention( 26 ) in the colonic mucosa of AOM‐treated db/db mice. These findings are consistent with previous reports that both pitavastatin and simvastatin significantly suppress inflammation‐related mouse colon carcinogenesis induced by AOM plus dextran sodium sulfate.( 13 , 14 ) Chronic inflammation, which is caused by obesity, plays a crucial role in the pathogenesis of many chronic diseases, including atherogenesis and carcinogenesis. Furthermore, the beneficial effects of statins on cardiovascular disease have also been linked to their anti‐inflammatory properties as well as their lipid‐lowering effects.( 27 ) Therefore, in addition to reducing the risk for cardiovascular disease, pitavastatin is considered to be effective in preventing cancer development in the colon of obese subjects.
The finding that serum TNF‐α levels were decreased by pitavastatin (Fig. 3a) is significant because TNF‐α is an important tumor promoter in inflammation‐related carcinogenesis.( 7 ) In addition, recent studies have revealed that TNF‐α lies at the core of the association between obesity and insulin resistance,( 5 , 28 ) which is a key factor for the development of obesity‐related CRC, and thus may be a critical target for the prevention of this malignancy.( 1 , 2 , 3 , 18 , 19 ) Therefore, it was expected that pitavastatin might be able to improve insulin resistance in the present study. However, contrary to our expectations, there was no clear evidence indicating an improvement in insulin resistance by pitavastatin (data not shown). This result might be explained by the lipid solubility of this statin because lipophilic statins may exacerbate insulin resistance, although hydrophilic statins may improve insulin resistance and reduce the risk of diabetes mellitus onset.( 29 , 30 , 31 ) Therefore, it might be speculated that hydrophilic statins are more effective in suppressing the development of obesity‐related CRC by attenuating inflammation and, likely, improving insulin resistance.
Adiponectin, which is secreted by adipose tissue, is regarded as an anti‐inflammatory adipocytokine because of its ability to down‐regulate the production of TNF‐α and IL‐6.( 32 ) Circulating levels of adiponectin are decreased in obese individuals, and this phenomenon might be linked to obesity‐related carcinogenesis because low adiponectin levels are associated with a higher risk of CRC and an adiponectin‐related gene defect has been shown to be involved in this malignancy.( 33 , 34 , 35 ) Adiponectin suppresses colonic epithelial proliferation in mice that are fed a high‐fat diet.( 36 ) In addition, a higher level of serum leptin, which increases TNF‐α production,( 37 ) exerts tumor‐promoting effects in obesity‐ and inflammation‐related CRC.( 18 , 19 , 38 , 39 ) These reports suggest that abnormalities in the levels of both adiponectin and leptin might be critical targets for the suppression of obesity‐related CRC. Therefore, our observations that pitavastatin increases the adiponectin levels and decreases the leptin levels in the serum of AOM‐treated db/db mice (Table 3) might play a critical role in suppressing obesity‐related colorectal carcinogenesis. The effect of pitavastatin on decreasing the serum TNF‐α levels (Fig. 3a) might be associated with these phenomena because TNF‐α has been shown to decrease per se the expression of adiponectin, while increasing the expression of leptin in adipocytes.( 40 , 41 )
The present study also showed the first evidence demonstrating that treatment with pitavastatin enhances AMPK activation in the colonic mucosa (Fig. 4a). This might be explained by the elevation of serum adiponectin levels (Table 3) because activation of AMPK, which is a critical monitor of cellular energy status, is involved in the signaling cascade of adinopectin receptors.( 42 ) In addition, a recent study has revealed that AMPK controls processes relevant to tumor development and therefore may be a promising target for cancer chemoprevention.( 43 ) Pharmacological AMPK activators including metformin, which is used to treat type 2 diabetes mellitus, can inhibit tumor growth of colon cancer xenografts by inducing apoptosis.( 44 ) AMP‐activated kinase (AMPK) activation is significantly associated with inhibition of COX‐2 expression in CRC cells.( 45 ) Moreover, adiponectin inhibits human CRC cell growth by activating AMPK.( 46 , 47 ) Therefore, pitavastatin may exert chemopreventive properties on obesity‐related colorectal carcinogenesis through the elevation of adiponectin (Table 3) and activation of AMPK (Fig. 4a).
Although both BCAC and ACF are regarded as precancerous lesions in the colon, there are clear biological differences between these lesions.( 23 , 24 ) As a result, both histological abnormalities (Fig. 1) and cell proliferative activity (Fig. 2) are significantly increased in BCAC in comparison to ACF.( 48 ) Compared to ACF, BCAC are frequently accompanied by Paneth cells,( 48 ) which are often present in colon tumors and produce a number of inflammation‐related factors including TNF‐α.( 49 ) In the AOM‐induced mouse colon carcinogenesis model, the incidence and location of BCAC are essentially identical to those of colon tumors, while such a coincidence was not observed for ACF.( 50 ) In addition, the administration of cholic acid, a tumor promoter for colon cancer, in the diet significantly enhanced both the multiplicity and size of AOM‐induced BCAC in the rat, while conversely those of ACF were inhibited by the administration of this agent.( 51 ) These findings might provide sufficient evidence that BCAC are more likely to give rise to carcinomas in the colon. Therefore, our findings, namely that pitavastatin markedly inhibited the development of BCAC, appear to be significant when considering the chemoprevention of CRC itself, although this agent did not significantly suppress the development of ACF (Table 2). These findings may be explained by the biological characteristics of BCAC because these lesions show significant cell proliferative ability and inflammatory responses in comparison to ACF, and can thus be considered a sensitive marker for chemopreventive agents.( 23 , 24 ) Indeed, dietary supplementation with sulindac, a nonsteroidal anti‐inflammatory drug, increased the apoptotic index only in BCAC, but not in ACF.( 52 )
In summary, the prevention of CRC by targeting chronic inflammation and adipocytokine imbalance, which is caused by the dysregulation of energy homeostasis, might be a promising strategy for obese people who are at an increased risk for developing CRC. Some types of statins, including pitavastatin, therefore appear to be potentially effective candidates for this purpose because statins can attenuate inflammation while also improving the imbalance of adipocytokines.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Science, Sports and Culture of Japan (no. 18790457 to M.S. and no. 17015016 to H.M.).
References
- 1. Giovannucci E, Michaud D. The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology 2007; 132: 2208–25. [DOI] [PubMed] [Google Scholar]
- 2. Frezza EE, Wachtel MS, Chiriva‐Internati M. Influence of obesity on the risk of developing colon cancer. Gut 2006; 55: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang CK, Ulrich CM. Hyperinsulinaemia and hyperglycaemia: possible risk factors of colorectal cancer among diabetic patients. Diabetologia 2003; 46: 595–607. [DOI] [PubMed] [Google Scholar]
- 4. Gunter MJ, Leitzmann MF. Obesity and colorectal cancer: epidemiology, mechanisms and candidate genes. J Nutr Biochem 2006; 17: 145–56. [DOI] [PubMed] [Google Scholar]
- 5. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor‐alpha: direct role in obesity‐linked insulin resistance. Science 1993; 259: 87–91. [DOI] [PubMed] [Google Scholar]
- 6. Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin‐6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 2001; 280: E745–51. [DOI] [PubMed] [Google Scholar]
- 7. Szlosarek P, Charles KA, Balkwill FR. Tumour necrosis factor‐alpha as a tumour promoter. Eur J Cancer 2006; 42: 745–50. [DOI] [PubMed] [Google Scholar]
- 8. Shirakami Y, Shimizu M, Tsurumi H, Hara Y, Tanaka T, Moriwaki H. EGCG and Polyphenon E attenuate inflammation‐related mouse colon carcinogenesis induced by AOM and DSS. Mol Med Rep 2008; 1: 355–61. [PubMed] [Google Scholar]
- 9. Hebert PR, Gaziano JM, Chan KS, Hennekens CH. Cholesterol lowering with statin drugs, risk of stroke, and total mortality. An overview of randomized trials. JAMA 1997; 278: 313–21. [PubMed] [Google Scholar]
- 10. Baigent C, Keech A, Kearney PM et al. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366: 1267–78. [DOI] [PubMed] [Google Scholar]
- 11. Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer 2005; 5: 930–42. [DOI] [PubMed] [Google Scholar]
- 12. Gauthaman K, Fong CY, Bongso A. Statins, stem cells, and cancer. J Cell Biochem 2009; 106: 975–83. [DOI] [PubMed] [Google Scholar]
- 13. Yasui Y, Suzuki R, Miyamoto S et al. A lipophilic statin, pitavastatin, suppresses inflammation‐associated mouse colon carcinogenesis. Int J Cancer 2007; 121: 2331–9. [DOI] [PubMed] [Google Scholar]
- 14. Cho SJ, Kim JS, Kim JM, Lee JY, Jung HC, Song IS. Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis‐associated colon cancer in mice. Int J Cancer 2008; 123: 951–7. [DOI] [PubMed] [Google Scholar]
- 15. Poynter JN, Gruber SB, Higgins PD et al. Statins and the risk of colorectal cancer. N Engl J Med 2005; 352: 2184–92. [DOI] [PubMed] [Google Scholar]
- 16. Hirose Y, Hata K, Kuno T et al. Enhancement of development of azoxymethane‐induced colonic premalignant lesions in C57BL/KsJ‐db/db mice. Carcinogenesis 2004; 25: 821–5. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki R, Kohno H, Yasui Y et al. Diet supplemented with citrus unshiu segment membrane suppresses chemically induced colonic preneoplastic lesions and fatty liver in male db/db mice. Int J Cancer 2007; 120: 252–8. [DOI] [PubMed] [Google Scholar]
- 18. Shimizu M, Shirakami Y, Sakai H et al. EGCG suppresses azoxymethane‐induced colonic premalignant lesions in male C57BL/KsJ‐db/db mice. Cancer Prev Res 2008; 1: 298–304. [DOI] [PubMed] [Google Scholar]
- 19. Shimizu M, Shirakami Y, Iwasa J et al. Supplementation with branched‐chain amino acids inhibits azoxymethane‐induced colonic preneoplastic lesions in male C57BL/KsJ‐db/db mice. Clin Cancer Res 2009; 15: 3068–75. [DOI] [PubMed] [Google Scholar]
- 20. Hayashi T, Yokote K, Saito Y, Iguchi A. Pitavastatin: efficacy and safety in intensive lipid lowering. Expert Opin Pharmacother 2007; 8: 2315–27. [DOI] [PubMed] [Google Scholar]
- 21. Yamada Y, Mori H. Pre‐cancerous lesions for colorectal cancers in rodents: a new concept. Carcinogenesis 2003; 24: 1015–9. [DOI] [PubMed] [Google Scholar]
- 22. Bird RP, Good CK. The significance of aberrant crypt foci in understanding the pathogenesis of colon cancer. Toxicol Lett 2000; 112‐113: 395–402. [DOI] [PubMed] [Google Scholar]
- 23. Mori H, Yamada Y, Kuno T, Hirose Y. Aberrant crypt foci and beta‐catenin accumulated crypts; significance and roles for colorectal carcinogenesis. Mutat Res 2004; 566: 191–208. [DOI] [PubMed] [Google Scholar]
- 24. Mori H, Hata K, Yamada Y, Kuno T, Hara A. Significance and role of early‐lesions in experimental colorectal carcinogenesis. Chem Biol Interact 2005; 155: 1–9. [DOI] [PubMed] [Google Scholar]
- 25. Tomita H, Yamada Y, Oyama T et al. Development of gastric tumors in Apc(Min/+) mice by the activation of the beta‐catenin/Tcf signaling pathway. Cancer Res 2007; 67: 4079–87. [DOI] [PubMed] [Google Scholar]
- 26. Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase‐2. Nat Rev Cancer 2001; 1: 11–21. [DOI] [PubMed] [Google Scholar]
- 27. Schonbeck U, Libby P. Inflammation, immunity, and HMG‐CoA reductase inhibitors: statins as antiinflammatory agents? Circulation 2004; 109: II18–26. [DOI] [PubMed] [Google Scholar]
- 28. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS‐1‐mediated inhibition of insulin receptor tyrosine kinase activity in TNF‐alpha‐ and obesity‐induced insulin resistance. Science 1996; 271: 665–8. [DOI] [PubMed] [Google Scholar]
- 29. Guclu F, Ozmen B, Hekimsoy Z, Kirmaz C. Effects of a statin group drug, pravastatin, on the insulin resistance in patients with metabolic syndrome. Biomed Pharmacother 2004; 58: 614–8. [DOI] [PubMed] [Google Scholar]
- 30. Koh KK, Quon MJ, Han SH et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis 2009; 204: 483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Freeman DJ, Norrie J, Sattar N et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103: 357–62. [DOI] [PubMed] [Google Scholar]
- 32. Ouchi N, Walsh K. Adiponectin as an anti‐inflammatory factor. Clin Chim Acta 2007; 380: 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vona‐Davis L, Howard‐McNatt M, Rose DP. Adiposity, type 2 diabetes and the metabolic syndrome in breast cancer. Obes Rev 2007; 8: 395–408. [DOI] [PubMed] [Google Scholar]
- 34. Wei EK, Giovannucci E, Fuchs CS, Willett WC, Mantzoros CS. Low plasma adiponectin levels and risk of colorectal cancer in men: a prospective study. J Natl Cancer Inst 2005; 97: 1688–94. [DOI] [PubMed] [Google Scholar]
- 35. Kaklamani VG, Wisinski KB, Sadim M et al. Variants of the adiponectin (ADIPOQ) and adiponectin receptor 1 (ADIPOR1) genes and colorectal cancer risk. JAMA 2008; 300: 1523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujisawa T, Endo H, Tomimoto A et al. Adiponectin suppresses colorectal carcinogenesis under the high‐fat diet condition. Gut 2008; 57: 1531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Molina A, Vendrell J, Gutierrez C et al. Insulin resistance, leptin and TNF‐alpha system in morbidly obese women after gastric bypass. Obes Surg 2003; 13: 615–21. [DOI] [PubMed] [Google Scholar]
- 38. Stattin P, Lukanova A, Biessy C et al. Obesity and colon cancer: does leptin provide a link? Int J Cancer 2004; 109: 149–52. [DOI] [PubMed] [Google Scholar]
- 39. Miyamoto S, Yasui Y, Tanaka T, Ohigashi H, Murakami A. Suppressive effects of nobiletin on hyperleptinemia and colitis‐related colon carcinogenesis in male ICR mice. Carcinogenesis 2008; 29: 1057–63. [DOI] [PubMed] [Google Scholar]
- 40. Hector J, Schwarzloh B, Goehring J et al. TNF‐alpha alters visfatin and adiponectin levels in human fat. Horm Metab Res 2007; 39: 250–5. [DOI] [PubMed] [Google Scholar]
- 41. Finck BN, Johnson RW. Anti‐inflammatory agents inhibit the induction of leptin by tumor necrosis factor‐alpha. Am J Physiol Regul Integr Comp Physiol 2002; 282: R1429–35. [DOI] [PubMed] [Google Scholar]
- 42. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev 2005; 26: 439–51. [DOI] [PubMed] [Google Scholar]
- 43. Fay JR, Steele V, Crowell JA. Energy homeostasis and cancer prevention: the AMP‐activated protein kinase. Cancer Prev Res 2009; 2: 301–9. [DOI] [PubMed] [Google Scholar]
- 44. Buzzai M, Jones RG, Amaravadi RK et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53‐deficient tumor cell growth. Cancer Res 2007; 67: 6745–52. [DOI] [PubMed] [Google Scholar]
- 45. Hwang JT, Ha J, Park IJ et al. Apoptotic effect of EGCG in HT‐29 colon cancer cells via AMPK signal pathway. Cancer Lett 2007; 247: 115–21. [DOI] [PubMed] [Google Scholar]
- 46. Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP‐activated protein kinase. Cancer Prev Res 2008; 1: 369–75. [DOI] [PubMed] [Google Scholar]
- 47. Sugiyama M, Takahashi H, Hosono K et al. Adiponectin inhibits colorectal cancer cell growth through the AMPK/mTOR pathway. Int J Oncol 2009; 34: 339–44. [PubMed] [Google Scholar]
- 48. Yamada Y, Yoshimi N, Hirose Y et al. Sequential analysis of morphological and biological properties of beta‐catenin‐accumulated crypts, provable premalignant lesions independent of aberrant crypt foci in rat colon carcinogenesis. Cancer Res 2001; 61: 1874–8. [PubMed] [Google Scholar]
- 49. Tan X, Hsueh W, Gonzalez‐Crussi F. Cellular localization of tumor necrosis factor (TNF)‐alpha transcripts in normal bowel and in necrotizing enterocolitis. TNF gene expression by Paneth cells, intestinal eosinophils, and macrophages. Am J Pathol 1993; 142: 1858–65. [PMC free article] [PubMed] [Google Scholar]
- 50. Hata K, Yamada Y, Kuno T et al. Tumor formation is correlated with expression of beta‐catenin‐accumulated crypts in azoxymethane‐induced colon carcinogenesis in mice. Cancer Sci 2004; 95: 316–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hirose Y, Kuno T, Yamada Y et al. Azoxymethane‐induced beta‐catenin‐accumulated crypts in colonic mucosa of rodents as an intermediate biomarker for colon carcinogenesis. Carcinogenesis 2003; 24: 107–11. [DOI] [PubMed] [Google Scholar]
- 52. Kuno T, Yamada Y, Hirose Y et al. Induction of apoptosis by sulindac in azoxymethane‐induced possible colonic premalignant lesions in rats. Jpn J Cancer Res 2002; 93: 242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]