

Abstract
An inductive argument of metastasis with a metaphor of seed and soil was made by Stephen Paget in 1889. It is commonly held that metastasis is dependent on both the organ from which the primary tumors originate, and the organs to which the tumor cells travel. The assumption is based on the statistical observation of a number of autopsy samples. Here I attempt to establish a theory on the mechanisms of metastasis with experimental evidence. I propose that dysregulation of pro‐inflammatory Toll‐like receptor 4 signaling, stimulated by its endogenous ligands, establishes pre‐metastatic soil. Once specific parameters are established, deductive judgments could be possible to predict to which organ a given tumor metastasizes. (Cancer Sci 2009)
One of the traditional ideas regarding metastasis is that genetic heterogeneity in primary tumor cells by continuous acquisition of mutations during tumor progression creates a subset of tumor cells with high metastasizing abilities by clonal selection.( 1 ) Clonally selected populations of cells usually constitute a minority group in the whole tumor mass. Differential screening of cDNA libraries derived from murine melanoma cell lines of varying metastatic potentials accidentally revealed that the nm23 gene is associated with lower metastatic potential.( 2 ) In studies of human breast cancer, a significant correlation between decreased nm23 expression and tumor progression has been shown.( 3 ) Experimentally, in vivo‐selected populations of cells that are capable of metastasizing to specific organs have been established and subjected to detailed genetic studies.( 4 ) The system found a variety of genes involved in cellular signaling, transcription and its regulators, such as miR10b and miR335.( 4 , 5 , 6 ) The concept of tumor‐initiating cancer stem cells provided an idea that cells that can actually accomplish metastatic colony formation or re‐growth in secondary sites are those before differentiation.( 7 ) In addition, reversion to stem cells is now possible at least experimentally.( 8 ) Moreover, even non‐transformed cells can disseminate to secondary sites.( 9 ) In contrast to those ideas at the cellular levels, that minor or selected groups dictate metastasis, recent progress in gene profiling analysis has revealed that a majority of primary tumor cells express signatures that suggest their metastatic progression,( 10 ) indicating that molecular events required for metastasis exist even before clonal selection.
It remains to be elucidated to what extent genes or mechanisms that determine tissue tropism overlap with those that simply promote metastasis. Discrepancies are easily found, for example, CXCR4 in bone metastasis( 11 ) also plays an essential role in lung metastasis.( 12 ) Our experiment failed to observe upregulation of the CXCR4 system in the lungs.( 13 ) Taken together, we face difficulties in elucidating any cells or genes that can accurately determine future metastasis.
In addition to the information on tumor cells, evidence is accumulating as to how host cell factors contribute not only to primary tumors but also to metastasis. The most representative example is tumor angiogenesis in which tumor cells secrete growth factors including VEGF, TNFα and TGFβ to activate tumor( 14 ) and distant tissues to which they are destined to travel. Although lymph nodes can be assumed to be tissue, lymph node metastasis is achieved by invasion to lymphatic but not blood vessels.
In this mini‐review I first summarize our experimental results on the S100A8‐SAA3‐TLR4 system that have already been published.( 13 , 15 ) After describing the biological significance of TLR in metastasis, I refer to settings in published reports that are analogous to our findings. Finally, I compare our concept with the ideas on metastasis in Table 1 and try to give a theoretical insight into metastasis.
Table 1.
Comparison between three references and this article on the major issues of metastasis research
| Major issues | (1) | (55) | (62) | This article |
|---|---|---|---|---|
| Sequential steps | Cascade hypothesis | → | → | → |
| Metastatic phenotypes | Heterogeneity 1. Oncogene‐mediated mutations 2. Inductive by ME | Ras signaling | — | — |
| Migratory ability of tumor cells | Chemotactic factors (C5a, N‐fmlp) | 1. Circulatory 2. Chemokines | 1. Chemokines (CXCL12) 2. Its receptor (CXCR4) on tumor cells | 1. TLR4 agonists (S100A8, SAA3) 2. TLR4 on tumor cells 3. Oncogenes may not be required |
| Organotropism | 1. Mechanical 2. Seed and soil, assuming organ‐specific receptors | 1. Adhesion 2. Mechanical chemokine‐ receptor match | Pre‐metastatic soil formation by VEGFR1(+) BMDC and fibroblasts | Pre‐metastatic soil expansion by Mac1(+) BMDC and lung resident cells |
| Remote controls by primary tumors | Primary tumors inhibit metastatic growth | — | Tumor‐derived factors | Tumor‐derived VEGF, TNFα,TGFβ upregulate TLR4 agonists |
| Dormancy/re‐growth | 1. Avascularity 2. Immune (cytotoxic T) 1 and 2 are involved in dormancy | 1. ME‐dependent growth dictates organotropism (example:TGFβ‐ PTHrP in bone) 2. Chemokines activate Ras/MAPK | VEGFR1 on tumor cells | 1. Bidirectional signaling between BMDC and tumor cells 2. Sceptical about VEGFR1‐mediated growth |
| Angiogenesis | Heparin/cortisone inhibits metastasis | Anti‐angiogenic therapy may block metastatic tumor growth | VEGFR2(+) progenitor cells in the soil | Lung endothelial cells express TLR4 agonists |
| Roles of myeloid cells | Macrophages both inhibit and enhance metastasis | — | Soil formation | Amplification of TLR4 agonists expands soil |
In the 1980s the issues were already recognized phenomenologically. Around 2000, specific molecules were found that potentially govern both organ preference and re‐growth in metastasis and many genes involved in tumor angiogenesis were isolated and tested for their value as therapeutic targets. The outcome of anti‐VEGF treatments was not as efficient as expected and now provides resistance problems. Bone marrow‐derived myeloid cells (BMDC) are believed to be involved not only in the resistance but also in the development of the metastatic microenvironment (ME) before and after tumor cells arrive at metastatic sites. This gives the concept of pre‐metastatic soil. The discovery of the endogenous ligands for Toll‐like receptor 4 (TLR4), previously recognized as a sensor for extrinsic pathogens or danger signal after tumor invasion, raises a new issue of ‘an inflammation‐like state’ in metastasis.→, similar concept; –, not stated.
Experimental Results
Metastasis model of hematogenous dissemination. Metastasis is a complicated biological process consisting of at least five sequentially interrelated steps: (i) detachment of tumor cells from a primary tumor; (ii) intravasation; (iii) migration through the blood stream; (iv) extravasation; and (v) recruitment in metastatic sites (Fig. 1A). Development of primary tumors with subsequent distant metastasis can take place in rodents either spontaneously or by extrinsic factors such as retroviruses and chemicals. However, it is difficult to set up experiments in rodents with spontaneous metastasis.( 16 , 17 ) For example, human breast and prostate cancers most commonly display metastasis to bone. However, spontaneous mammary cancer in rodents has a very low incidence of spontaneous metastasis to bone.( 17 ) Spontaneous prostate cancer in rodents is also rare. Therefore, animal models cannot perfectly resemble clinical circumstances. One of the well‐considered models is the NOD/SCID–HAB model where HAB fragments were subcutaneously implanted into mice before injection of human prostatic cancer cell lines through the tail vein.( 18 ) Tumor cells can be found with a high frequency in HAB 8 weeks after injection. The immunocompromised background makes murine circulatory systems tolerant of the cross‐species engraftment that is accompanied by tumor cell migration.
Figure 1.
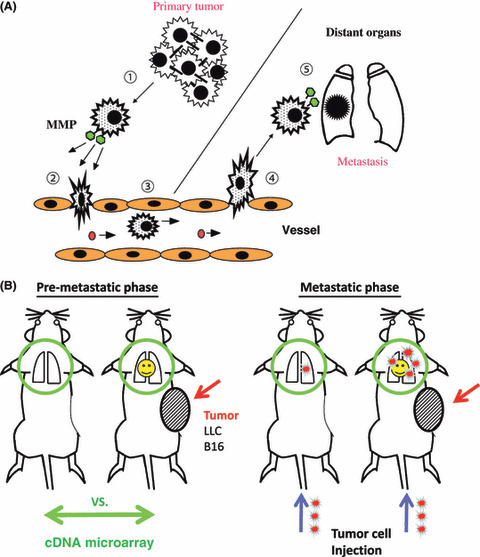
(A) Five steps in the metastatic progression of cancer: (1) detachment of tumor cells from a primary tumor; (2) intravasation; (3) migration through the blood stream; (4) extravasation; and (5) recruitment in metastatic sites. Biological significance of matrix metalloprotease (MMP) has been shown in both tumor cells, as represented here, and host cells.( 20 ) (B) Experimental pre‐metastasis model. Metastasis is divided into two phases, pre‐metastatic and metastatic. In the pre‐metastatic phase (left), we subcutaneously inject tumor cells until they give rise to a tumor, which we assume as a primary tumor. In the metastatic phase (right), we purposely inject labeled tumor cells through the tail vein and analyze the lungs or other organs for recruited tumor cells.
In our experimental system of lung metastasis, we injected labeled murine tumor cells, usually LLC cells or B16 melanoma cells, into the tail vein and count the number of tumor cells in the lungs or other organs 6–48 h after injection (Fig. 1B).( 13 ) The subcutaneous implantation of tumor cells, which gave rise to a tumor mass in 2 weeks, before the injection increased the number of recruited tumor cells in the lungs. The implanted tumor cells never reached the lungs, as judged by the copy number of the neomycin‐resistant gene retrovirally integrated into the genome of the injected tumor cells. This mimics a primary tumor with pre‐metastatic lungs without metastasis. The artificial hematogenous dissemination by itself is a demerit in the first place and therefore the system can hardly cover the lymph node metastasis. Second, the existence of a very small number of tumor cells in the assumed pre‐metastatic lungs cannot be completely excluded. The third concern is that injection into systemic circulation facilitates recruitment of tumor cells in the organ for them to initially encounter in the body. Injection into vessels that go to other organs, including portal vein, or splenic and cervical artery, requires a high level of technical training but occasionally needs to be carried out to control the experiment. Observation of tumor cells in the lungs during this short period of time does not guarantee their eventual colonization. The well‐known experiment by Dr Fidler in 1970 was carried out with B16 melanoma cells.( 19 ) Radiolabeled B16 cells were injected intravenously into C57BL/6J mice and the fate of tumor cells in circulation and each organ was examined at various intervals in a pharmacokinetic fashion. The experiment provides fundamental information of extreme importance that most tumor cells are found in the lungs and ∼1% of cells survived after 24 h to eventually form an average of 78 metastatic colonies. In other words, only a small fraction of tumor cells that enter the blood circulation can make metastatic foci.( 19 ) However, the primary merit of our system is that it provides a convenient and reproducible way of assessing the organotropic tumor cell migration in vivo in a short period of time by skipping both the first and second steps. The second merit is that we can purposely dissect pre‐metastatic and metastatic phases. Given the increased number of Mac1‐positive cells in the lungs of tumor‐bearing mice before lung metastasis actually takes place,( 20 ) macrophage‐defective conditions such as NOD mice should be avoided, particularly to study pre‐metastasis. Restriction of certain species of tumor cells due to immunological rejection is a demerit but we can leave the immune system intact.
Chemokine candidates. A cDNA microarray analysis of lungs was carried out between tumor‐bearing and non‐bearing mice. Although the upregulated top 50 genes in the pre‐metastatic lungs failed to include matrix metalloprotease 9 that has been shown to play a role in pre‐metastatic lungs,( 20 ) we found its upregulation by approximately 1.5–2.8‐fold. A pair of chemokine candidates, S100A8 and S100A9, was among the top 50 genes.( 13 ) Anti‐S100A8 neutralizing antibody treatment blocked metastasis and antibodies against VEGF, TNFα or TGFβ, individually or in combination, inhibited the upregulation of S100A8 in the lungs (Fig. 2). The contribution of these three growth factors in S100A8 upregulation is approximately 30% for each, as judged by the quantified PCR analysis under the antibody‐mediated blockade. Both S100A8 and S100A9 belong to the well‐conserved S100 family of EF‐hand calcium‐binding proteins and are highly produced in the synovial fluid of patients with rheumatoid arthritis.( 21 ) S100A8/A9 can also be detected in the serum of patients with inflammatory disorders, cancer and type I diabetes mellitus.( 22 , 23 ) They have been crystallized and shown to display either homo‐ or hetero‐dimeric form in a calcium‐dependent manner.( 24 ) The in vivo usage of the S100 family of proteins that share 25–65% homology appears to be dictated by their tissue‐specific expression and variable regions at the carboxy‐terminus that bind target proteins.( 22 ) The S100A8 knockout mice are 100% embryonic lethal at 9.5 days.( 25 ) S100A8 has been reported to activate NFκB and MAP kinases such as p38 and p44/p42Erk.( 13 , 26 ) Previously S100A8 was thought to bind unknown cell surface receptors on endothelial cells through heparan sulfate proteoglycans( 27 ) before a German group showed in 2007 that it binds to TLR4.( 28 ) Another candidate receptor for S100A8 is RAGE, a multiligand member of the immunoglobulin superfamily.( 29 ) AGE is enriched in the diabetic milieu and shown to accelerate atherogenesis.( 30 ) However, the presumable S100A8–RAGE interaction lacks direct biochemical evidence.
Figure 2.
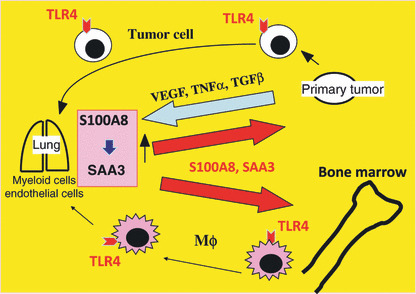
Ping‐pong mechanisms in metastasis. Primary tumor‐produced growth factors such as VEGF stimulate the S100A8–serum amyloid A3 (SAA3) paracrine system in the lungs, which in turn activates cell mobilization of bone marrow cells and tumor cells.
S100A8 can stimulate cell migration of myeloid cells and tumor cells. However, it still remains to be uncovered how S100A8 proteins are secreted into the extracellular space, because it lacks the signal sequence. A non‐secretory form of S100A8 has a positive regulatory role in the activation of NADPH oxidase (Nox2) by binding to its component Rac2 and p67.( 31 ) When cells are destroyed by, for example, cytotoxic agents in chemotherapy, S100A8 is passively released into the extracellular space. A similar pattern of release into extracellular space has been under controversy in heat shock cognate protein Hsc70 that is also assumed to be an endogenous ligand for TLR4.( 32 ) Therefore, we assume that S100A8 might play another role in the so‐called ‘danger signal’.( 33 ) However, an inflammatory caspase‐1 has recently been shown to participate in the active secretion of chemokines such as FGF‐2 lacking the signal sequence.( 34 ) Given that anti‐S100A8 antibody can abolish the chemotactic activity in the conditioned medium of lung organ cultures that are treated with TNFα, and that there is up‐regulation of S100A8 proteins in pre‐metastatic lungs before metastasizing tumor cells destroy the tissue, S100A8 is thought to be secreted by active mechanisms even prior to true danger or actual tissue destruction.
As mentioned above S100A8 expression was induced by primary tumors specifically in the lungs, although we could detect a small amount of S100A8 mRNA in liver. Conditioned medium from S100A8‐stimulated lungs in organ culture, but not other organs including liver, kidney or spleen, showed biological activity in the morphological transformation as well as cell migration of tumor cells in vitro.( 15 ) We also tried to facilitate hepatic recruitment of tumor cells by injecting labeled tumor cells through the portal vein in tumor‐bearing mice, but without success. We could still observe enhanced tumor cell homing in the lungs (S. Hiratsuka & Y. Maru, unpublished data, 2006). Thus all the phenomenological data support the lung specificity in our metastasis assay.
Looking back to our original cDNA microarray analysis we found another chemokine candidate, SAA3, in the top 50 genes.( 15 ) S100A8 induced SAA3 expression specifically in the lungs with the SAA3 promoter activation by approximately 2–4‐fold. Surface plasmon resonance analysis provided direct evidence for binding between SAA3 and the TLR4/MD2 complex with a surprising binding constant of 0.356 nM, which is one magnitude less than that of either S100A8 or lipid A.( 15 , 28 , 35 ) Thus it turns out that SAA3 is another endogenous ligand, and the strongest ligand ever found for TLR4. This establishes the S100A8‐SAA3 paracrine system in the lungs and both chemokine candidates are capable of acting as an agonist to activate TLR4 in our system of lung metastasis.
The downstream signaling events include p38 activation by S100A8 and NFκB activation by SAA3.( 15 ) TLR4 signaling mainly includes NFκB on which ATF3 has a negative regulatory role by bringing down HDAC1 to the promoter region.( 36 ) We observed that both LPS and S100A8 activate ATF3 at different levels (data not shown). SAA3 but not LPS induced morphological transformation of LLC in vitro.( 15 ) There may exist as yet unidentified differences between the two in the mode of TLR4 binding and/or cellular signaling. As activation of the p38 pathway can stimulate ATF3 expression by phosphorylating the transcription factor CREB,( 37 ) criss‐cross paracrine stimulation by both S100A8 and SAA3 in the lungs is complicated, and needs to be clarified in the future.
Biological Significance of TLR
TLR4 is a well‐studied molecule in innate immunity and believed to be a cell surface sensor receptor for extrinsic pathogens such as bacteria recognizing bacterial wall LPS.( 38 ) Defect or polymorphism in either TLR4 or RAGE results in altered responsiveness in the immune system, such as poor mobilization of white blood cells from bone marrow to the lungs under bacterial infection.( 39 , 40 ) Several endogenous ligands for TLR4 have been proposed, including Hsc70, fatty acid, S100A8, SAA3, and hyaluronan.( 15 , 28 , 32 , 41 , 42 ) Therefore, the self and non‐self discrimination in classical immunology can no longer be as strict as previously thought. TLR2 also recognizes bacterial peptidoglycans. Moreover, TLR2 has been shown to mediate lung metastasis by tumor‐producing versican.( 43 ) Versican and its binding partner hyaluronan are glycosaminoglycans. Hyaluronan is degraded by bacteria‐associated lung inflammation that produces superoxide or proteases. Hyaluronan fragments are not only cleared by its receptor CD44 but also stimulate TLR2 and TLR4 in the lungs for secretion of a variety of chemokines to rescue the tissue destruction (danger signal). Versican stimulates production of TNFα in a TLR2‐dependent manner. An individual genetic knockout of TLR4( 15 ) and TLR2( 43 ) or anti‐S100A8 neutralizing antibody( 13 ) efficiently suppressed lung recruitment of both Mac1‐positive cells and tumor cells in tumor‐bearing mice.
Previously anti‐tumor activity was thought to be achieved by activation of innate immunity against tumors such as that of NK cells. However, considering that both TLR4 and TLR2 are ectopically expressed in various tumor cells,( 44 ) it is not theoretically simple to apply pharmacological TLR agonists or antagonists for cancer therapy.( 45 )
Analogous Situations to SAA3‐TLR4 System
SDF‐1 (CXCL12) and its receptor CXCR4. SDF‐1 is expressed in lung, liver, bone marrow, and lymph nodes.( 11 , 46 ) SDF‐1‐null mice die in utero with poor bone marrow development. CXCR4 expression is low or absent in normal circumstances but CXCR4‐null mice also have defects in the hematopoietic system. This suggests the biological significance of SDF‐1 expression in osteoblasts that are localized specifically in bone marrow. CXCR4 is overexpressed in various tumors, presumably through activation of HIF1α, especially in prostate cancer, breast cancer and lung cancer. Tumor cells with CXCR4 can migrate toward its ligand SDF‐1. As CXCR4 is capable of activating NFκB to induce both CXCR4 and SDF‐1 mRNAs,( 11 , 47 , 48 ) a metastatic arrival of tumor cells in bone could result in amplification of this mobilization‐stimulating ligand.
RANKL and its receptor RANK. RANKL belongs to the TNF family and is expressed in bone marrow stroma cells. Physiologically, it participates in differentiation of osteoclasts that play an osteolytic role in bone morphogenesis.( 49 ) Its receptor RANK is ectopically expressed in prostate and breast cancer.( 50 ) By analogy to SDF‐1, tumor cells with receptor can move toward its ligand. When circulating breast cancer cells are stimulated by TGFβ, they secrete angiopoietin‐like 4, which then guides extravasation of the tumor cells in the pulmonary vascular beds, eventually leading to lung‐specific metastasis.( 51 ) A similar circumstance could take place when breast cancer cells approach bone that is highly abundant in TGFβ. However, angiopoietin‐like 4 failed to induce bone metastasis. TGFβ stimulates tumor cells to produce parathyroid hormone‐related protein, which in turn causes RANKL expression in osteoblasts.( 52 , 53 ) This induces osteoclast differentiation to release more TGFβ, making a vicious cycle in osteolysis.( 53 ) If tumor cells in the primary site are stimulated by TGFβ produced from interstitial cells in a tumor microenvironment, they secrete parathyroid hormone‐related protein that acts in an endocrine fashion to induce RANKL expression in bone which could serve as a pre‐metastatic soil.( 54 ) Once the tumor cells invade into bone TGFβ from the primary site is dispensable, which can worsen the clinical situation of bone destruction.
Theoretical Concepts and Prospects
Table 1 refers to three representative review articles as milestones of metastasis research. My challenge is to discriminate our ideas from theirs on the mechanisms of metastasis. In Advances in Cancer Research, published in 1985,( 1 ) metastasis was described as a sequence of interrelated steps that seems to be a non‐random process but rather selective. However, the authors had only a small amount of molecular evidence to refer to. In 2002 a detailed speculation of the possible molecular mechanisms of dissemination and growth of post‐metastatic tumor cells was made in Nature reviews.( 55 ) The authors explain ‘homing’ of tumor cells to specific organs by interactions between a chemokine and its receptor, both of which are expressed in specific cells or tissues. For example, breast cancer cells abundantly expressing CXCR4 metastasize to lungs that secrete the corresponding agonist CXCL12 (SDF‐1) but not to skin with unmatched CCL27 expression even at high levels. They suppose that efficient organ‐specific re‐growth after ‘homing’, rather than the preferential ‘homing’ by itself, is responsible for organ‐specific metastasis in clinical settings in which clinicians recognize metastasis only after it actually takes place. As malignant primary tumors might have spread onto a variety of secondary sites by the time primary tumors are clinically detected, the authors underline anti‐angiogenic strategies to inhibit the re‐growth step for treatment of metastasis. However, addition of anti‐VEGF antibody (bevacizumab) to oxaliplatin‐based chemotherapy, for example, significantly improved progression‐free survival in metastatic colorectal cancer patients, but the response rate was not improved.( 56 ) Resistance to anti‐VEGF therapy is mediated at least in part by bone marrow‐derived myeloid cells( 57 ) and carcinoma‐associated fibroblasts as sources of angiogenic factors or other bioactive molecules including, for example, CXCL12 that induces VEGF‐independent angiogenesis.( 58 , 59 , 60 ) Accumulating evidence of recent studies underscores the significant contribution of myeloid cells in metastasis. Hematologically, myeloid cells belong to white blood cells. They have been a clinically useful biomarker in patients with inflammatory disorders such as infection and include lymphoid cells such as T and B lymphocytes, and myeloid cells such as granulocytes and monocytes/macrophages and their progenitors, granulocyte–macrophage progenitor and common myeloid progenitor. Among them, monocytes/macrophages appear to play a pivotal role. Clodronate‐mediated depletion of monocytes/macrophages in an animal model of metastasis of lung cancer resulted in inhibition of bone metastasis.( 61 )
It was not until 2005 when the old concept of the seed and soil hypothesis in metastasis obtained molecular evidence( 12 ) and was reviewed the next year.( 62 ) Soil needs to be cultivated before seeds, as a metaphor of tumor cells, can be successfully spread. Therefore, the preparation of soil is the priming of secondary organs before metastasis occurs. VEGF has two high‐affinity receptors with catalytic tyrosine kinase activity, VEGFR1 (Flt‐1) and VEGFR2 (KDR),( 63 ) whose expression in endothelial cells is rather specific. The authors claimed that VEGFR1‐positive cells whose exit from bone marrow is regulated by VEGFR1 by itself participate in pre‐metastatic niche formation in the lungs. Although endothelial cells as well as their progenitors specifically express both receptors, VEGF exerts mitogenic activity through VEGFR2 on which VEGFR1 has an inhibitory effect, whether in a full‐length form or in a soluble form, encoding only the extracellular domain with a higher affinity to VEGF than VEGFR2. VEGFR1 is exceptionally expressed in myeloid cells as represented by macrophages.( 63 ) Formation of VEGFR1‐positive cell clusters, which is blocked by anti‐VEGFR1 antibody, precedes the arrival of VEGFR2‐expressing endothelial progenitors from bone marrow and tumor cells from primary tumors. The cultivated soil or pre‐metastatic niche secretes SDF‐1 abundantly, which in turn stimulates mobilization of CXCR4‐expressing tumor cells from the primary tumors.( 12 ) Then what is the mechanism of cluster formation or the driving force for VEGFR1‐positive cell mobilization? They speculate that certain tumor‐derived cytokines/chemokines appear to be responsible. Conditioned media of tumor cell cultures have metastasis‐inducing activity in tumor‐bearing mice in an organ‐specific manner, and VEGF signaling directly enhances tumor growth given the expression of VEGFR1 in some tumors. PlGF, a VEGFR1‐specific ligand, was shown to have weak mitogenic activity in one hematopoietic cell line.( 64 ) We should be cautious when addressing this issue. It is very difficult to experimentally overexpress VEGFR1 with usual expression vectors.( 63 , 65 ) Although we could achieve it with a papillomavirus‐based vector in NIH3T3 cells, we repeatedly failed to detect any mitogenic activity by VEGF or PlGF in those cells. Mutated forms of BCR‐FLT, in which the kinase domain of VEGFR1 was fused with BCR of the BCR‐ABL oncogene of CML, is transforming but the activity is due to phosphorylated tyrosine 177 of the BCR fragment that mediates Grb2/Ras signaling but not the VEGFR1 kinase domain.( 65 ) We assume that mitogenic activity of VEGFR1‐expressing cells could be too weak to allow re‐growth of tumor cells in metastatic sites.
The ability of VEGFR1 to mediate cell migration has been shown by many orthodox experimental systems in vitro ( 63 ) as well as ours.( 66 ) Migration of Mac1‐positive myeloid cells to lungs from bone marrow in LLC‐bearing mice is inhibited not completely but significantly (roughly 68%) by the specific knockout of the catalytic domain of VEGFR1 (tk−/−).( 20 ) VEGFR1‐dependent migration involves not only VEGF and PlGF but also a well‐known metabolic factor, low density lipoprotein.( 66 ) In addition, it is still unclear whether VEGFR1 mediates exit of bone marrow cells. CML is manifested by gross expansion and release of myeloid cells from bone marrow. Mating studies of VEGFR1 tk−/− mice( 63 ) and CML model mice resulted in no change in the leukemic cell count in the peripheral circulation (Y. Maru et al., unpublished data, 2008). Thus VEGFR1‐expressing cells can be mobilized by a variety of agonists or by VEGFR1‐independent factors (such as TLR4 agonists, see below) depending on the biological settings whose receptor has overlapped expression with VEGFR1.
If pre‐metastatic soil is discussed, any tumor cells should not be present in the metastatic sites. Then how do primary tumor‐derived factors determine organotropism? If SDF‐1 in the lung recruits tumor cells from the distant primary site, it is necessary for primary tumors to induce SDF‐1 expression specifically in the pre‐metastatic lungs. These critical issues are unanswered. In vivo selection of bone metastatic variants followed by their gene expression profiling analysis revealed a set of genes, most of which are secreted cytokines and cell surface receptors.( 4 ) Therefore it is assumed that growth factor/chemokine‐receptor systems working in a combined endocrine and paracrine fashion might be deterministic of the pre‐metastatic soil in their system.
In this mini‐review I give a theoretical concept in our lung‐specific metastasis. I propose that, while tumorigenesis is caused by genetic mutations with or without epigenetic modifications, inflammatory processes largely underlie metastasis. Cancer‐associated inflammation is caused by oncogenic signaling such as Ras and Myc, which induces production of inflammatory cytokines and chemokines.( 67 ) Hypoxic response through HIF‐1 is another cause. Different from this inflammation adjacent to or within the tumor, tumor cells appear to induce ‘an inflammation‐like state’ even in their absence. Figure 3 compares infection and metastasis. Microbial invasion through the airways makes a focus of pneumonia, which is defended by resident macrophages in the lungs that destroy bacterial walls generating endotoxin or LPS. LPS then stimulates mobilization of bone marrow cells, white blood cells that are positive in its receptor TLR4. LPS is shown to skip effects of cytokines such as granulocyte‐colony stimulating factor) on granulocyte–macrophage progenitor differentiating into monocytes/macrophages.( 68 ) When accumulating defenders defeat the enemy the infection is cleared, as shown in the algebraic image (Fig. 3). In metastasis, primary tumor cells make a remote control of distant organs, such as lungs, by activating the paracrine system of S100A8‐SAA3 in the lungs. Production of those endogenous TLR4 ligands is to be mis‐recognized as invasion of extrinsic pathogens by the bone marrow cells. However, when they reach the lungs they find no bacteria but instead contribute to the expansion of the paracrine field (Fig. 4). SAA3 can induce production of growth factors such as TNFα, secretion of which from the primary tumor serves as a trigger for the initiation of chemokine upregulation in the lungs. When the growth factors initially responsible for upregulation of S100A8 in the lungs are produced in the expanding field of the paracrine system in the lungs, it makes the growth factors from the primary tumor dispensable for preparation of pre‐metastatic soil. We suppose that the tissue tropism in metastasis might be determined by where an expanding vicious cycle of chemokine(s) and growth factor(s) exists. If any single tumor cell reaches the soil, bidirectional signaling between tumor cells and bone marrow‐derived cells is to be established. Tumor cells secrete VEGF and CSF‐1 to stimulate bone marrow‐derived cells, which in turn produce EGF to activate EGFR on the tumor cells for proliferation.( 69 ) This second vicious cycle may participate in re‐growth of metastatic tumor cells. There have been reports on the Ras‐Erk activation for mitogenic activity in the chemokine signaling, such as CXCR4.( 47 ) However, in the pre‐metastatic lungs in our metastasis system we could not observe up‐regulation of SDF‐1.( 13 )
Figure 3.

Comparison between infection and metastasis in the lungs. The bottom panels show algebraic images of defenders (bone marrow‐derived myeloid cells or macrophages) and invaders (microbes or tumor cells).
Figure 4.
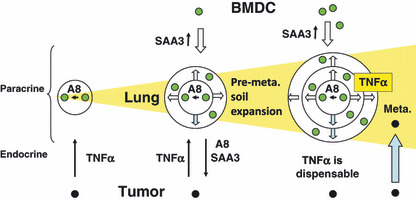
Expansion of the S100A8‐SAA3 paracrine field in the lungs. The S100A8–serum amyloid A3 (SAA3) paracrine system that is stimulated in an endocrine fashion by growth factors produced by primary tumors recruits bone marrow‐derived myeloid cells (BMDC). The paracrine system is supposed to expand until those Toll‐like receptor 4 (TLR4) agonists cause expression of TNFα in the lungs, making that derived from primary tumors dispensable for the preparation of pre‐metastatic (pre‐meta.) soil. Tumor cells from the primary site may arrive after maturation of pre‐metastatic soil. Alternatively, tumor cells and BMDC work in concert to prepare the soil. In this case, tumor cell arrival and soil preparation proceed simultaneously and it is beyond discussion which comes first, BMDC or tumor cells.( 72 )
Homeostatic TLR4 signals triggered by the endogenous ligands can cause spontaneous inflammation in the absence of molecules that restrict the signals, as exemplified by A20.( 70 ) This raises fundamental questions of what sort of homeostatic role the endogenous TLR4 agonists play in physiological settings, and whether or not elimination mechanisms, if any, of those restricting molecules can explain formation of a pre‐metastatic microenvironment.
Multi‐organ metastasis could consist of a subset of organ‐specific metastasis. It is not likely that metastatic potentials intrinsic to tumor cells uni‐directionally determine how many organs are affected by dissemination. Bi‐directional endocrine and paracrine signaling before and after metastasis, respectively, between tumor cells and host cells may establish metastasis specific to a single organ. Collection of this organotropic metastasis could constitute dissemination to multiple organs.
Antagonism of TLR4 either by genetic knockdown or neutralization of its endogenous agonists resulted in inhibition of metastasis.( 13 , 15 ) Osteoprotegerin is another example of suppression of bone metastasis through the RANKL system.( 54 ) A small molecule inhibitor of CXCR3 has also been reported to block lung metastasis in a murine model.( 71 ) Chemokines that are specific to organotropic metastasis need to be clarified for each organ. They might be the target of metastasis as a disease.
Conclusion
Growth factors produced from primary tumor cells stimulate the S100A8‐SAA3‐TLR4 paracrine cascade in the lungs. Mobilization and recruitment of bone marrow‐derived cells in the lungs may be achieved by their mis‐recognition of the endogenous TLR4 ligands as invasion by extrinsic pathogens. Tumors appear to hijack host defense mechanisms against infection. However, given as yet unidentified physiological roles of the endogenous TLR4 ligands, metastasis could be construed as a disease where homeostatic TLR4 signaling is impaired.
Abbreviations
- ATF
activating transcription factor
- BCR
breakpoint cluster region
- CCL27
CC chemokine ligand 27
- CML
chronic myeloid leukemia
- CREB
cAMP response element‐binding protein
- CSF1
colony stimulating factor 1
- CXCL
CXC chemokine ligand
- CXCR4
CXC chemokine receptor 4
- HAB
human adult bone
- HDAC1
histone deacetylase 1
- HIF
hypoxia‐inducible factor
- LLC
Lewis lung carcinoma
- LPS
lipopolysaccharide
- MΦ
macrophage
- NFκB
nuclear factor‐kappaB
- PlGF
placenta growth factor
- PTHrP
parathyroid hormone related protein
- RAGE
receptor for advanced glycation endproducts
- RANK
receptor activator of NFκB
- RANKL
receptor activator of NFκB ligand
- SAA
serum amyloid A
- SDF‐1
stromal cell‐derived factor‐1
- TGF
transforming growth factor
- TLR
Toll‐like receptor
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
Acknowledgments
I thank all the authors that contributed the references 13 and 15 for their help. This study was partly supported by Grants‐in‐Aid for Scientific Research (No. 20013041) to YM.
References
- 1. Schirrmacher V. Cancer metastasis: experimental approaches, theoretical concepts, and impacts for treatment strategies. Adv Cancer Res 1985; 43: 1–73. [DOI] [PubMed] [Google Scholar]
- 2. Steeg PS, De La Rosa A, Flatow U, MacDonald NJ, Benedict M, Leone A. Nm23 and breast cancer metastasis. Breast Cancer Res Treat 1993; 25: 175–87. [DOI] [PubMed] [Google Scholar]
- 3. Steeg PS, Bevilacqua G, Kopper L et al. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst 1988; 80: 200–4. [DOI] [PubMed] [Google Scholar]
- 4. Kang Y, Siegel PM, Shu W et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537–49. [DOI] [PubMed] [Google Scholar]
- 5. Ma L, Teruya‐Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA‐10b in breast cancer. Nature 2007; 449: 682–8. [DOI] [PubMed] [Google Scholar]
- 6. Tavazoie SF, Alarcón C, Oskarsson T et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008; 451: 147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reya T, Morrison SJ, Clarke MF, Weisman IL. Stem cells, acncer, and cancer stem cells. Nature 2001; 414: 105–11. [DOI] [PubMed] [Google Scholar]
- 8. Okita K, Ichisaka T, Yamanaka S. Generation of germline‐competent induced pluripotent stem cells. Nature 2007; 448: 313–7. [DOI] [PubMed] [Google Scholar]
- 9. Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science 2008; 321: 1841–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van‘t Veer LJ, Dai H, Van De Vijver MJ et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415: 530–6. [DOI] [PubMed] [Google Scholar]
- 11. Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF‐1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev 2006; 25: 573–87. [DOI] [PubMed] [Google Scholar]
- 12. Kaplan RN, Riba RD, Zacharoulis S et al. VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature 2005; 438: 820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour‐mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 2006; 8: 1369–75. [DOI] [PubMed] [Google Scholar]
- 14. Anderson ARA, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 2006; 127: 905–15. [DOI] [PubMed] [Google Scholar]
- 15. Hiratsuka S, Watanabe A, Sakurai Y et al. The S100A8‐serum amyloid A3‐TLR4 paracrine cascade establishes a pre‐metastatic phase. Nat Cell Biol 2008; 10: 1349–55. [DOI] [PubMed] [Google Scholar]
- 16. Rosol TJ, Tannehill‐Gregg SH, LeRoy BE, Mandl S, Contag CH. Animal models of bone metastasis. Cancer 2003; 97: 748–57. [DOI] [PubMed] [Google Scholar]
- 17. Blouin S, Baslé MF, Chappard D. Rat models of bone metastases. Clin Exp Metastasis 2005; 22: 605–14. [DOI] [PubMed] [Google Scholar]
- 18. Yonou H, Yokose T, Kamijo T et al. Establishment of a novel species‐ and tissue‐specific metastasis model of human prostate cancer in humanized non‐obese diabetic/severe combined immunodeficient mice engrafted with human adult lung and bone. Cancer Res 2001; 61: 2177–82. [PubMed] [Google Scholar]
- 19. Fidler IJ. Metastasis: guantitative analysis of distribution and fate of tumor embolilabeled with 125 I‐5‐iodo‐2′‐deoxyuridine. J Natl Cancer Inst 1970; 45: 773–82. [PubMed] [Google Scholar]
- 20. Hiratsuka S, Nakamura K, Iwai S et al. MMP9 induction by vascular endothelial growth factor receptor‐1 is involved in lung‐specific metastasis. Cancer Cell 2002; 2: 289–300. [DOI] [PubMed] [Google Scholar]
- 21. Santamaria‐Kisiel L, Rintala‐Dempsey AC, Shaw GS. Calcium‐dependent and ‐independent interactions of the S100 protein family. Biochem J 2006; 396: 201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ehrchen JM, Sunderkötter C, Foell D, Vogl T, Roth J. The endogenous Toll‐like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol 2009; 86. [DOI] [PubMed] [Google Scholar]
- 23. Bouma G, Lam‐Tse WK, Wierenga‐Wolf AF, Drexhage HA, Versnel MA. Increased serum levels of MRP‐8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes 2004; 53: 1979–86. [DOI] [PubMed] [Google Scholar]
- 24. Itou H, Yao M, Fujita I et al. The crystal structure of human MRP14 (S100A9), a Ca(2+)‐dependent regulator protein in inflammatory process. J Mol Biol 2002; 316: 265–76. [DOI] [PubMed] [Google Scholar]
- 25. Passey RJ, Williams E, Lichanska AM et al. A null mutation in the inflammation‐associated S100 protein S100A8 causes early resorption of the mouse embryo. J Immunol 1999; 163: 2209–16. [PubMed] [Google Scholar]
- 26. Hermani A, De Servi B, Medunjanin S, Tessier PA, Mayer D. S100A8 and S100A9 activate MAP kinase and NF‐κB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res 2006; 312: 184–97. [DOI] [PubMed] [Google Scholar]
- 27. Robinson MJ, Tessier P, Poulsom R, Hogg N. The S100 family heterodimer, MRP‐8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem 2002; 277: 3658–65. [DOI] [PubMed] [Google Scholar]
- 28. Vogl T, Tenbrock K, Ludwig S et al. Mrp8 and Mrp14 are endogenous activators of Toll‐like receptor 4, promoting lethal, endotoxin‐induced shock. Nat Med 2007; 13: 1042–9. [DOI] [PubMed] [Google Scholar]
- 29. Gebhardt C, Riehl A, Durchdewald M et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med 2008; 205: 275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stern DM, Yan SD, Yan SF, Schmidt AM. Receptor for advanced glycation endproducts (RAGE) and the complications of diabetes. Ageing Res Rev 2002; 1: 1–15. [DOI] [PubMed] [Google Scholar]
- 31. Kerkhoff C, Nacken W, Benedyk M, Dagher MC, Sopalla C, Doussiere J. The arachidonic acid‐binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac‐2. FASEB J 2005; 19: 467–9. [DOI] [PubMed] [Google Scholar]
- 32. Zou N, Ao L, Cleveland JC Jr et al. Critical role of extracellular heat shock cognate protein 70 in the myocardial inflammatory response and cardiac dysfunction after global ischemia‐reperfusion. Am J Physiol Heart Circ Physiol 2008; 294: H2805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fuchs EJ, Matzinger P. Is cancer dangerous to the immune system? Semin Immunol 1996; 8: 271–80. [DOI] [PubMed] [Google Scholar]
- 34. Keller M, Rüegg A, Werner S, Beer HD. Active caspase‐1 is a regulator of unconventional protein secretion. Cell 2008; 132: 818–31. [DOI] [PubMed] [Google Scholar]
- 35. Akashi S, Saitoh S, Wakabayashi Y et al. Lipopolysaccharide interaction with cell surface Toll‐like receptor 4‐MD‐2: higher affinity than that with MD‐2 or CD14. J Exp Med 2003; 198: 1035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gilchrist M, Thorsson V, Li B et al. Systems biology approaches identify ATF3 as a negative regulator of Toll‐like receptor 4. Nature 2006; 441: 173–8. [DOI] [PubMed] [Google Scholar]
- 37. Lu D, Chen J, Hai T. The regulation of ATF3 gene expression by mitogen‐activated protein kinases. Biochem J 2007; 401: 559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beutler BA. TLRs and innate immunity. Blood 2009; 113: 1399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bhide MR, Mucha R, Mikula I Jr et al. Novel mutations in TLR genes cause hyporesponsiveness to Mycobacterium avium subsp. paratuberculosis infection. BMC Genet 2009; 10: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Zoelen MA, Schouten M, De Vos AF et al. The receptor for advanced glycation end products impairs host defense in pneumococcal pneumonia. J Immunol 2009; 182: 4349–56. [DOI] [PubMed] [Google Scholar]
- 41. Suganami T, Tanimoto‐Koyama K, Ogawa Y et al. Role of the Toll‐like receptor 4/NF‐κB pathway in saturated fatty acid‐induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol 2007; 27: 84–91. [DOI] [PubMed] [Google Scholar]
- 42. Jiang D, Liang J, Fan J et al. Regulation of lung injury and repair by Toll‐like receptors and hyaluronan. Nat Med 2005; 11: 1173–9. [DOI] [PubMed] [Google Scholar]
- 43. Kim S, Takahashi H, Lin WW et al. Carcinoma‐produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009; 457: 102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their Toll – the function and regulation of Toll‐like receptors in cancer cells. Oncogene 2008; 27: 225–33. [DOI] [PubMed] [Google Scholar]
- 45. Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll‐like receptor agonists and antagonists. Nat Med 2007; 13: 552–9. [DOI] [PubMed] [Google Scholar]
- 46. Broxmeyer HE. Chemokines in hematopoiesis. Curr Opin Hematol 2008; 15: 49–58. [DOI] [PubMed] [Google Scholar]
- 47. Lu DY, Tang CH, Yeh WL et al. SDF‐1α up‐regulates interleukin‐6 through CXCR4, PI3K/Akt, ERK, and NF‐κB‐dependent pathway in microglia. Eur J Pharmacol 2009; 613: 146–54. [DOI] [PubMed] [Google Scholar]
- 48. Han Y, He T, Huang DR, Pardo CA, Ransohoff RM. TNF‐α mediates SDF‐1α‐induced NF‐κ B activation and cytotoxic effects in primary astrocytes. J Clin Invest 2001; 108: 425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ang E, Pavlos NJ, Rea SL et al. Proteasome inhibitors impair RANKL‐induced NF‐κB activity in osteoclast‐like cells via disruption of p62, TRAF6, CYLD, and IκBα signaling cascades. J Cell Physiol 2009; 220: 450–9. [DOI] [PubMed] [Google Scholar]
- 50. Mikami S, Katsube KI, Oya M et al. Increased RANKL expression is related to tumour migration and metastasis of renal cell carcinomas. J Pathol 2009; 218: 530–539. [DOI] [PubMed] [Google Scholar]
- 51. Padua D, Zhang XH, Wang Q et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin‐like 4. Cell 2008; 133: 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guise TA. Parathyroid hormone‐related protein and bone metastases. Cancer 1997; 80: 1572–80. [DOI] [PubMed] [Google Scholar]
- 53. Massagué J. TGFβ in Cancer. Cell 2008; 134: 215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jones DH, Nakashima T, Sanchez OH et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006; 440: 692–6. [DOI] [PubMed] [Google Scholar]
- 55. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2: 563–72. [DOI] [PubMed] [Google Scholar]
- 56. Saltz LB, Clarke S, Díaz‐Rubio E et al. Bevacizumab in combination with oxaliplatin‐based chemotherapy as first‐line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008; 26: 2013–9. [DOI] [PubMed] [Google Scholar]
- 57. Stockmann C, Doedens A, Weidemann A et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 2008; 456: 814–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Orimo A, Gupta PB, Sgroi DC et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF‐1/CXCL12 secretion. Cell 2005; 121: 335–48. [DOI] [PubMed] [Google Scholar]
- 59. Deshane J, Chen S, Caballero S et al. Stromal cell‐derived factor 1 promotes angiogenesis via a heme oxygenase 1‐dependent mechanism. J Exp Med 2007; 204: 605–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shojaei F, Wu X, Zhong C et al. Bv8 regulates myeloid‐cell‐dependent tumor angiogenesis. Nature 2007; 450: 825–31. [DOI] [PubMed] [Google Scholar]
- 61. Hiraoka K, Zenmyo M, Watari K et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci 2008; 99: 1595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre‐metastatic niche’: within bone and beyond. Cancer Metastasis Rev 2006; 25: 521–9. [DOI] [PubMed] [Google Scholar]
- 63. Shibuya M. Differential roles of vascular endothelial growth factor receptor‐1 and receptor‐2 in angiogenesis. J Biochem Mol Biol 2006; 39: 469–78. [DOI] [PubMed] [Google Scholar]
- 64. Fragoso R, Pereira T, Wu Y, Zhu Z, Cabeçadas J, Dias S. VEGFR‐1 (FLT‐1) activation modulates acute lymphoblastic leukemia localization and survival within the bone marrow, determining the onset of extramedullary disease. Blood 2006; 107: 1608–16. [DOI] [PubMed] [Google Scholar]
- 65. Maru Y, Yamaguchi S, Shibuya M. Flt‐1, a receptor for vascular endothelial growth factor, has transforming and morphogenic potentials. Oncogene 1998; 16: 2585–95. [DOI] [PubMed] [Google Scholar]
- 66. Usui R, Shibuya M, Ishibashi S, Maru Y. Ligand‐independent activation of VEGF receptor 1 by low density lipoprotein. EMBO Rep 2007; 8: 1155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature 2008; 454: 436–44. [DOI] [PubMed] [Google Scholar]
- 68. Nagai Y, Garrett KP, Ohta S et al. Toll‐like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006; 24: 801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pollard JW. Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol 2008; 84: 623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Turer EE, Tavares RM, Mortier E et al. Homeostatic MyD88‐dependent signals cause lethal inflammation in the absence of A20. J Exp Med 2008; 205: 451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Walser TC, Rifat S, Ma X, Kundu N et al. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res 2006; 66: 7701–7. [DOI] [PubMed] [Google Scholar]
- 72. Maru Y. Which came first, tumor cells or macrophages? Cell Adh Migr 2007; 1: 107–9. [DOI] [PMC free article] [PubMed] [Google Scholar]