

Abstract
The prevalence of BRCA1/2 germline mutations in Japanese patients suspected to have hereditary breast/ovarian cancer was examined by a multi‐institutional study, aiming at the clinical application of total sequencing analysis and validation of assay sensitivity in Japanese people using a cross‐sectional approach based on genetic factors estimated from personal and family histories. One hundred and thirty‐five subjects were referred to the genetic counseling clinics and enrolled in the study. Full sequencing analysis of the BRCA1/2 gene showed 28 types of deleterious mutations in 36 subjects (26.7%), including 13 types of BRCA1 mutations in 17 subjects (12.6%) and 15 types of BRCA2 mutations in 19 subjects (14.1%). Subjects were classified into five groups and 22 subgroups according to their personal and family history of breast and/or ovarian cancer, and the prevalence of deleterious mutations was compared with previously reported data in non‐Ashkenazi individuals. Statistical analysis using the Mantel‐Haenszel test for groups I through IV revealed that the prevalence of Japanese subjects was significantly higher than that of non‐Ashkenazi individuals (P = 0.005, odds ratio 1.87, 95% confidence interval 1.22–2.88). Family history of the probands suffering from breast cancer indicated risk factors for the presence of deleterious mutations of BRCA1/2 as follows: (1) families with breast cancer before age 40 within second degree relatives (P = 0.0265, odds ratio 2.833, 95% confidence interval 1.165–7.136) and (2) families with bilateral breast cancer and/or ovarian cancer within second degree relatives (P = 0.0151, odds ratio 2.88, 95% confidence interval 1.25–6.64). (Cancer Sci 2008; 99: 1967–1976)
In Japan, breast cancer is the most frequent malignancy in women and estimates of new cases and deaths in 2002 were 32 245 and 9178, respectively.( 1 ) The standardized incidence ratio of breast cancer in Japan was approximately one‐third that of the US (32.7 vs 101.7 per 100 000 women).( 1 ) The incidence of breast cancer in Japanese women shows a steady increase; however, it is still much lower than in Western countries. In breast cancer, family history is the strongest risk factor for cancer predisposition. Epidemiological studies showed that 12% of women with breast cancer have one affected family member and 1% have two or more affected relatives.( 2 ) Women with one, two, and three or more first‐degree affected relatives have an increased breast cancer risk when compared with women who do not have an affected relative (risk ratios 1.8, 2.9, and 3.9, respectively).( 2 ) Recent advances in molecular genetics elucidated BRCA1 and BRCA2 (BRCA1/2) as two major susceptibility genes for breast cancer predisposition.( 3 , 4 ) Gene testing of BRCA1/2 is available as a routine clinical test for diagnosing hereditary breast/ovarian cancer (HBOC) in the US and other Western countries,( 5 , 6 ) while only a few reports have been published concerning the prevalence of BRCA1/2 mutations among Japanese people.( 7 , 8 , 9 , 10 , 11 , 12 ) The methods of genetic analysis employed in these studies varied, such as polymerase chain reaction (PCR)/ single strand conformational polymorphisms (SSCP), protein truncation test, and PCR/direct sequencing, but they were performed as preliminary in‐house tests in the research setting. In the US, commercial BRCA1/2 gene testing was initiated by Myriad Genetic Laboratories in 1996. The test is based on the exon‐by‐exon PCR/direct sequencing approach comprising sequence analysis of over 17 500 base pairs of the protein‐coding and adjacent non‐coding regions of BRCA1 and BRCA2 genes of which test results performed in more than 10 000 individuals were reported.( 5 , 6 ) Myriad Genetic Laboratories released some of the results to a public database, the Breast Cancer Information Core (BIC) and this test is now utilized as the de facto standard in the US.( 13 ) Examinees with positive test results or pathogenic mutations may undergo close surveillance of breast and ovarian cancers or there might be other options such as risk‐reducing surgeries including prophylactic salpingo‐oophorectomy and/or mastectomy.( 14 , 15 ) Chemoprevention using tamoxifen or oral contraceptives were also reported to reduce the risk for contralateral breast cancer or ovarian cancers.( 16 , 17 ) Clinical application of BRCA1/2 gene testing brought a paradigm shift in cancer prevention strategies targeted to at‐risk mutation carriers. The aim of the present study was to clarify the prevalence of germline BRCA1/2 mutations among Japanese patients suspected to have HBOC using the standardized method available in the clinical setting and to establish the genetic basis required for the clinical application of BRCA1/2 gene testing for Japanese.
Materials and Methods
The study was designed to validate the sensitivity of BRCA1/2 gene testing among Japanese with relevance to personal and family histories of breast and/or ovarian cancer in comparison with the previously reported data of non‐Ashkenazi individuals in the US.( 6 ) The study was performed as a contract research by FALCO biosystems. The study protocol was approved by the institutional review board of each participating institution before study initiation. All patients gave written informed consent before registration.
Recruitment of subjects. Candidates were recruited from surgical or gynecologic clinics in five major hospitals in the Tokyo metropolitan area (The Cancer Institute Hospital of Japanese Foundation for Cancer Research, National Cancer Center Hospital, Keio University Hospital, St. Lukes’ International Hospital, and Tochigi Cancer Center Hospital) or recruited through newspaper advertisements in the Tokyo metropolitan area. All participants were referred to genetic counseling clinics in these hospitals and checked for eligibility for enrollment in the study. As genetic counseling was mandatory in this study, they underwent genetic counseling before and after gene testing. All expenses for genetic counseling were covered by FALCO biosystems. From August 2003 through May 2006, 135 patients were enrolled in the study, among which 101 subjects were treated for breast and/or ovarian cancers in these hospitals, and 34 subjects were recruited through newspaper advertising. Patient records of their personal history of cancer, including diagnosis, treatment, and clinicopathological parameters, were obtained through a survey completed by their attending surgeons or physicians.
Patient selection. Patients were eligible for the study if they fulfilled the following conditions: (i) native Japanese over 20 years old; (ii) histological diagnoses of invasive breast cancer and/or ovarian cancer; and (iii) at least one first‐ or second‐degree relative diagnosed with either or both cancers. Eligible patients were assigned to the matrix chart according to their personal and family history of cancer (Fig. 1). This matrix chart was formulated based on the reported prevalence of BRCA1/2 mutations in 3011 non‐Ashkenazi individuals in the US.( 6 ) Enrolled subjects were classified into five groups, i.e. Group I through Group V. All subjects assigned to Groups I through IV had some form of risk factor estimated from their personal and family history, while the risk of those in Groups I and III was personal rather than familial. The proportion of familial risk increased in Groups II and IV, and Group IV had a combination of the highest risk of both personal and familial history. Reportedly, the prevalence of BRCA1/2 mutations in Group I is relatively low, 7.7% (64/828 cases), while those in Groups II, III, and IV were 21.3% (364/1709), 19.2% (10/52), and 48.7% (205/421) respectively.( 6 ) Group V included cases of male breast cancer at any age irrespective of their family history and sporadic cases of breast cancers with concomitant ovarian cancers. The prevalence of BRCA1/2 mutations in each matrix or group in the present study could be compared with those reported in the US, except for Group V that had no comparable counterpart.
Figure 1.

Classification and grouping of the enrolled subjects. *Numbers in parentheses indicate prevalence of BRCA1/2 mutations reported in non‐Ashkenazi individuals in the US.( 6 )
Exclusion criteria for enrollment were as follows: (i) informed consent was provided not by a principal, but by a representative; (ii) a precise history of breast and/or ovarian cancers was not available; (iii) the patient was undergoing bilateral oophorectomy due to non‐malignant disorders; (iv) correct diagnosis was not disclosed to the patient; (v) informed consent was not provided due to psychiatric problems; (vi) the patient had non‐invasive breast cancer; (vii) borderline ovarian neoplasms; (viii) the patient was undergoing allogeneic bone marrow transplantation; (ix) another relative had undergone BRCA1/2 gene testing; and (x) when gene testing and disclosure of genetic information might cause serious sociopsychological problems.
BRCA1 and BRCA2 gene testing. Analyses of BRCA1/2 were performed by direct sequencing, as described previously.( 5 , 6 ) Briefly, 7 mL of anticoagulated blood was sent to FALCO biosystems for DNA extraction. Aliquots of patient DNA were sent to Myriad Genetic Laboratories (Salt Lake City, UT, USA) and subjected to PCR/direct sequencing analysis for BRCA1/2. This analysis also included the detection of the following five specific large genomic rearrangements of the BRCA1 gene (five‐site rearrangement panel): 3.8‐kb deletion of exon 13 and 510‐bp deletion of exon 22 described in individuals of Dutch ancestry,( 18 ) 6‐kb duplication of exon 13 described in individuals of European (particularly British) ancestry,( 19 ) 7.1‐kb deletion of exons 8 and 9 described in individuals of European ancestry,( 20 ) and 26‐kb deletion of exons 14–20.( 21 ) Nucleotide positions of mutations were expressed according to GenBank entries U14680 and U43746. All variants were interpreted according to the following criteria.( 6 )
Positive for deleterious mutation. Mutations were interpreted as positive deleterious mutations if they prematurely terminated (truncated) the protein product of BRCA1 at least 10 amino acids from the C‐terminus or the protein product of BRCA2 at least 110 amino acids from the C‐terminus, based on the documentation of deleterious mutations in BRCA1/2. In addition, specific missense mutations and non‐coding intervening sequence mutations were interpreted as deleterious on the basis of data derived from linkage analysis of high‐risk families, functional assays, biochemical evidence, or demonstration of abnormal mRNA transcript processing.
Genetic variant of uncertain significance. This group includes missense mutations and mutations that occur in analyzed intronic regions whose clinical significance has not yet been determined, chain‐terminating mutations that truncate BRCA1 and BRCA2 distal to amino acid positions 1853 and 3308, respectively, and mutations that eliminate the normal stop codons of these proteins.
Mutational types were defined according to the international nomenclature system reported by Antonarakis et al.( 22 ) All of the detected mutations were searched for in the BIC database.( 13 ) The description of mutational types previously reported in the BIC database is indicated along with those defined by the international nomenclature system, in order to facilitate the comparison of the data with those reported in the database or other publications.
Multiplex ligation‐dependent probe amplification (MLPA) analysis. To search for unknown genomic rearrangements, we performed MLPA analysis for all samples in which no deleterious mutation was detected, using Salsa MLPA Kits P002 and P087, which are commercially available from MRC‐Holland (Amsterdam, The Netherlands). MLPA is a quantitative multiplex PCR approach to determine the relative copy number of each BRCA1/2 exon.( 23 , 24 ) Assay procedures were performed according to the manufacturer's instructions.
Statistical analysis. Statistical significance was analyzed by Fisher's exact test and unpaired t‐test using Prism 4 (GraphPad Software, San Diego, CA, USA). Comparisons of the prevalence of BRCA1/2 germline mutations divided by subgroups between Japanese and non‐Ashkenazi individuals were analyzed by Fisher's exact test or the Mantel‐Haenszel test using R package (version 1.1.2), available from the Comprehensive R Archive Network (CRAN) (http://strimmerlab.org/software/genets/). Cumulative incidence was analyzed by Kaplan–Meier plot (log‐rank test) using SAS software (SAS Institute Japan, Tokyo, Japan).
Results
Characteristics of the enrolled subjects. A total of 135 subjects were examined for BRCA1/2 germline mutations, and deleterious mutations were found in 36 subjects (17 for BRCA1 and 19 for BRCA2). Backgrounds of all subjects and those divided by carrier status are shown in Table 1. In the analysis of all subjects, average age at enrollment was 51.6 ± 12.4 years and the average number of counseling sessions per client was 3.36 ± 0.82. There was no significant difference as to the age at enrollment between non‐carriers and carriers with deleterious mutations. As for genetic counseling, significantly more sessions were performed for those carrying deleterious mutations of BRCA1/2 as compared to non‐carriers (3.75 ± 1.20 vs 3.22 ± 0.56, P = 0.0007). Of the 135 subjects examined, 131 were women and 4 were men. All of the male subjects developed breast cancer, while 113 women developed breast cancers (9 women developed ovarian cancers and 9 women developed both breast and ovarian cancers). There was no statistical significance in these variables between BRCA1/2 mutation carriers and non‐carriers. In the study, patient accrual was determined by personal and family histories within second‐degree relatives. Family history was precisely assessed in genetic counseling clinics and familial information was obtained for 2242 family members, including probands. There were no statistical differences in the numbers of ascertained relatives within the first‐ or second‐degree between non‐carriers and carriers. As for relatives beyond the second‐degree, significantly more relatives were ascertained in pedigrees with deleterious BRCA1/2 mutations as compared to those of non‐carriers (P < 0.0001) (Table 1). The clinical characteristics of breast and ovarian cancers that developed in enrolled subjects are listed in Table 2. In this protocol, non‐invasive cases, including pTis or DCIS, did not fulfill the eligibility, but two cases were enrolled in the study due to the subsequent occurrence of invasive breast cancers in the contralateral breast.
Table 1.
Background of subjects enrolled in the study
| Variables | All | Non‐carrier | Deleterious mutations in | ||
|---|---|---|---|---|---|
| BRCA1/2 | BRCA1 | BRCA2 | |||
| Number of patients | 135 | 99 | 36 | 17 | 19 |
| Age at enrollment | 51.6 ± 12.4 | 52.5 ± 12.7* | 49.1 ± 11.3* | 48.9 ± 10.1 | 49.2 ± 12.5 |
| Number of sessions for genetic counseling | 3.36 ± 0.82 (1–8) ‡ | 3.22 ± 0.56 (1–5)*** | 3.75 ± 1.20 (1–8)*** | – | – |
| Sex | |||||
| Female | 131 | 96 | 35 | 17 | 18 |
| Male | 4 | 3 | 1 | 0 | 1 |
| Types of affected cancer | |||||
| Female | |||||
| Breast | 113 | 83 | 30 | 12 | 18 |
| Ovarian | 9 | 8 | 1 | 1 | 0 |
| Both | 9 | 5 | 4 | 4 | 0 |
| Male | |||||
| Breast | 4 | 3 | 1 | 0 | 1 |
| Genealogical information; number of relatives ascertained | |||||
| Relatives ≤ 1st degree | 673 (4.99) † | 487 (4.92)** | 186 (5.16)** | 92 (5.41) | 94 (4.95) |
| Male | 305 (2.26) | 218 (2.20) | 87 (2.42) | 42 (2.47) | 45 (2.37) |
| Female | 368 (2.73) | 269 (2.72) | 99 (2.75) | 50 (2.94) | 49 (2.58) |
| 1st < Relatives ≤ 2nd degree | 1142 (8.46) | 825 (8.33)** | 317 (8.80)** | 166 (9.76) | 151 (7.95) |
| Male | 526 (3.90) | 383 (3.87) | 143 (3.97) | 72 (4.24) | 71 (3.74) |
| Female | 616 (4.56) | 442 (4.46) | 174 (4.83) | 94 (5.53) | 80 (4.21) |
| Relatives > 2nd degree | 427 (3.16) | 254 (2.57)** | 173 (4.81)** | 106 (6.24) | 67 (3.53) |
| Male | 192 (1.42) | 112 (1.13) | 80 (2.22) | 55 (3.24) | 25 (1.32) |
| Female | 235 (1.74) | 142 (1.43) | 93 (2.58) | 51 (3.00) | 42 (2.21) |
Numbers in parentheses indicate average number of relatives in a family.
‡Minimal and maximal.
P = 0.0007.
P = 0.1594, unpaired t‐test.
χ2 value 26.90, d.f. = 2, P < 0.0001.
Table 2.
Clinical characteristics of the subjects with breast or ovarian cancer
| Variables | Breast cancer † | Ovarian cancer ‡ | |
|---|---|---|---|
| Number of subjects | 126 | 18 | |
| Sex | Female | 122 | 18 |
| Male | 4 | – | |
| Age at diagnosis | Female | 46.2 ± 12.1 | 50.2 ± 11.8 |
| Male | 64.5 ± 3.7 | – | |
| Tumor size (T) | Tis | 2 ¶ | – |
| T1 | 66 | 11 | |
| T2 | 49 | 1 | |
| T3 | 4 | 4 | |
| Missing data (No.) | 5 | 2 | |
| Nodal status (N) | Negative | 95 | 10 |
| Positive | 24 | 5 | |
| Missing data (No.) | 7 | 3 | |
| Stage | 0 | 2 | – |
| I | 71 | 9 (IA 4, IC 5) | |
| II | 38 | 1 (IIA 1) | |
| III | 4 | 4 (IIIB 1, IIIC 3) | |
| IV | 1 | 1 | |
| Missing data (No.) | 13 | 3 | |
| Histology | Non‐invasive | 2 | Serous 3, mucinous 3, |
| Invasive | 111 | endmetrioid 2, clear cell 2, | |
| Missing data (No.) | 13 | undifferentiated 2, mixed‐cell type 2, others 2, sex‐cord stromal tumor 1, germ cell tumor 1 | |
| Estrogen receptor status | Positive | 72 | |
| Negative | 37 | ||
| Missing data (No.) | 17 | ||
| Progesterone receptor status | Positive | 64 | |
| Negative | 44 | ||
| Missing data (No.) | 18 | ||
| Histological grade | Grade 1 | 24 | |
| Grade 2 | 37 | ||
| Grade 3 | 24 | ||
| Missing data (No.) | 41 | ||
| Laterality | Unilateral | 104 | |
| Bilateral | 22 | ||
| Mutational status | No mutation | 91 | 13 |
| BRCA1/2 | 35 | 5 | |
| BRCA1 | 16 | 5 | |
| BRCA2 | 19 | 0 |
Includes subjects with ovarian cancer (n = 9).
Includes subjects with breast cancer (n = 9).
¶Subjects with multiple primary breast cancer, in which histology of the first primary cancer was pTis. They were enrolled in the study as the histology of the second primary breast cancer was ascertained to be invasive cancer.
Results of BRCA1/2 gene testing. In the analysis of BRCA1, 13 types of deleterious mutations were detected in 17 subjects. One mutation (L63X, c.188T > A) was found in five subjects. Genetic variants of uncertain significance were detected in nine subjects, among which three subjects had the same mutational types (S1557P, c.4729T > C), substituting cytosine for thymine (Table 3).
Table 3.
Deleterious mutations and genetic variants of uncertain significance detected in BRCA1
| BRCA1: Deleterious mutations | No. detected | Subgroup assigned | dbSNP ID | (Breast Cancer Information Core [BIC] mutation database) | ||||
|---|---|---|---|---|---|---|---|---|
| Exon | Designation | BIC designation | Type | No. reported | Ethnicity | |||
| 5 | L63X(c.188T > A) | 5 | II‐2, II‐6, IV‐3, V‐5, III‐1 | NR | L63X | N | 6 | Asian: 5 |
| 5 | c.190_193delTGTA | 1 | I‐2 | NR | (309del4) ‡ | F | 0 | |
| 7 | Y130X(c.390C > A) | 1 | II‐7 | NR | Y130X | N | 1 | Asian: 1 |
| 11 | c.1112delC | 1 | II‐2 | NR | (1231delC) | F | 0 | |
| 11 | K503X(c.1507 A > G) | 1 | IV‐2 | NR | (K503X) | N | 0 | |
| 11 | E908X(c.2722G > T) | 1 | II‐5 | NR | E908X | N | 58 | Asian: 0 |
| 11 | Q934X(c.2800C > T) | 1 | IV‐5 | NR | Q934X | N | 4 | Asian: 2 |
| 11 | c.3442delG | 1 | II‐2 | NR | 3561delG | F | 2 | Asian: 2 |
| 11 | c.3505_3509delGACAT | 1 | IV‐3 | NR | (3624del5) | F | 0 | |
| 11 | c.4041_4042delAG | 1 | I‐2 | NR | 4160delAG | F | 7 | Asian: 0 |
| 13 | IVS13 + 1G > T † | 1 | II‐5 | NR | IVS13 +1G > T | S | 1 | NR |
| 13 | R1443X(c.4327C > T) | 1 | IV‐3 | NR | R1443X | N | 126 | Asian: 1 |
| 24 | c.5533_5534insT | 1 | II‐2 | NR | (5652insT) | F | 0 | |
| BRCA1: Genetic variant of uncertain significance | (BIC Mutation Database) | |||||||
|---|---|---|---|---|---|---|---|---|
| Exon | Designation | No. detected | Subgroup assigned | dbSNP ID | BIC Designation | Type | No. reported | Ethnicity |
| 5 | L52F(c.154C > T) | 1 | II‐1 | NR | L52F | M | 5 | Asian: 2 |
| 10 | P209L(c.626C > T) | 1 | II‐1 | NR | (P209L) | M | 0 | |
| 11 | S1217P(c.3649T > C) | 1 | II‐1 | NR | (S1217P) | M | 0 | |
| 16 | S1577P(c.4729T > C) | 3 | I‐2, II‐2, II‐5 | NR | S1577P | M | 1 | Asian: 1 |
| 20 | R1753T(c.5258G > C) | 1 | II‐3 | NR | (R1753T) | M | 0 | |
| 21 | F1761S(c.5282T > C) | 1 | II‐2 | NR | F1761S | M | 1 | Asian: 0 |
| 24 | Y1853C(c.5558 A > G) | 1 | II‐5 | NR | Y1853C | M | 1 | Asian: 0 |
Mutation in the donor site of intron 13, suspected to be deleterious, resulting in a splicing error.
‡ Mutations in parentheses indicate mutational types of unreported cases represented according to the style of BIC nomenclature. F, frameshift mutation; M, missense mutation; N, nonsense mutation; NR, not reported; P, genetic polymorphism; S, splice site mutation; Syn, synonymous mutation.
In the analysis of BRCA2, 15 types of deleterious mutations were detected in 19 subjects. Each of four mutational types (c.1813delA, S1882X[c.5645C > A], c.5576_5579delTTAA, and R2318X[c.6952C > T]) was detected in two subjects. Eight types of genetic variants of uncertain significance were detected in 13 subjects, among which four types were detected in more than two subjects (Table 4). Of the deleterious mutations, five of 13 mutational types in BRCA1 and four of 15 mutational types in BRCA2 were not reported previously in the BIC database. As for genetic variants of uncertain significance, all of these variants were missense mutations, among which three in BRCA1 and four in BRCA2 were not reported in the BIC database. No genomic rearrangement of the BRCA1 gene was detected in analysis using a 5′‐site rearrangement panel. In analysis using MLPA, genomic rearrangements were not detected in BRCA1/2 in all subjects.
Table 4.
Deleterious mutations and genetic variants of uncertain significance detected in BRCA2
| BRCA2: deleterious mutation | No. detected | Subgroup assigned | dbSNP ID | (Breast Cancer Information Core [BIC] mutation database) | ||||
|---|---|---|---|---|---|---|---|---|
| Exon | Designation | BIC designation | Type | No. reported | Ethnicity | |||
| 3 | c.86_87delTT | 1 | I‐1 | NR | 314delTT | F | 1 | NR |
| 10 | c.1813delA | 2 | II‐2, II‐2 | NR | 2041delA | F | 16 | Asian: 0 |
| 11 | c.2612delCinsTTT | 1 | I‐2 | NR | (2840delC insTTT) ‡ | F | 0 | |
| 11 | c.3847_3848delGT | 1 | V‐6 | NR | 4075delGT | F | 61 | Asian: 0 |
| 11 | c.4021delT | 1 | II‐2 | NR | (4249delT) | F | 0 | |
| 11 | S1882X(c.5645C > A) | 2 | I‐2, II‐1 | NR | S1882X | N | 28 | Asian: 2 |
| 11 | c.5207_5208delAA | 1 | II‐5 | NR | (5435delAA) | F | 0 | |
| 11 | c.5576_5579delTTAA | 2 | I‐1, I‐1 | NR | 5804del4 | F | 29 | Asian: 0 |
| 11 | c.6445_6446delAT | 1 | II‐2 | NR | 6673delAT | F | 3 | Asian: 0 |
| 13 | R2318X(c.6952C > T) | 2 | II‐1, II‐5 | NR | R2318X | N | 5 | Asian: 2 |
| 18 | c.8064_8065delCT | 1 | II‐2 | NR | (8292delCT) | F | 0 | |
| 20 | S2835X(c.8504C > A) | 1 | II‐2 | NR | S2835X | N | 1 | Asian: 0 |
| 23 | Q3026X(c.9076C > T) | 1 | II‐2 | NR | Q3026X | N | 3 | Asian: 1 |
| 23 | P3039P (c.9117G > A) † | 1 | II‐2 | rs28897756 | P3039P | Syn | 14 | Asian: 0 |
| 25 | R3128X(c.9382C > T) | 1 | II‐2 | NR | R3128X | N | 50 | Asian: 0 |
| BRCA2: Genetic variant of uncertain significance | (BIC Mutation Database) | |||||||
|---|---|---|---|---|---|---|---|---|
| Exon | Designation | No. detected | Subgroup assigned | dbSNP ID | BIC designation | Type | No. reported | Ethnicity |
| 10 | K322Q(c.964 A > C) | 2 | I‐1, II‐1 | rs11571640 | K322Q | M | 11 | Asian: 7 |
| 10 | M524I(c.1572G > C) | 1 | I‐1 | NR | (M524I) | M | 0 | |
| 10 | K610Q(c.1828 A > C) | 1 | II‐2 | NR | (K610Q) | M | 0 | |
| 11 | I770V(c.2308 A > G) | 1 | II‐5 | NR | (I770V) | M | 0 | |
| 11 | K1132R(c.3395 A > G) | 1 | I‐2 | rs1801406 | K1132R | M | 1 | Asian: 1 |
| 11 | G2044V(c.6131G > T) | 3 | II‐2, II‐2, V‐5 | NR | G2044V | M | 10 | Asian: 10 |
| 11 | V2109I(c.6325G > A) | 2 | I‐1, I‐2 | NR | V2109I | M | 8 | Asian: 5 |
| 17 | S2616F(c.7847C > T) | 2 | I‐1, II‐1 | NR | (S2616F) | M | 0 | |
Synonymous mutation in the exon–intron junction of exon 23, suspected to be deleterious, resulting in a splicing error.
‡ Mutations in parentheses indicate mutational types of unreported cases represented according to the style of BIC nomenclature. F, frameshift mutation; M, missense mutation; N, nonsense mutation; NR, not reported; P, genetic polymorphism; S, splice site mutation; Syn, synonymous mutation.
Comparison of prevalence of BRCA1/2 germline mutations between Japanese and non‐Ashkenazi individuals. Deleterious mutations of BRCA1/2 were detected in 26.7% (36/135) of the subjects enrolled in the study. The prevalence of deleterious mutations in non‐Ashkenazi individuals in each matrix or group was calculated based on the data reported previously.( 6 ) The prevalence of mutations in Groups I through IV in the Japanese cohort was 27.2% (34/125), while that in non‐Ashkenazi individuals was 20.3% (590/2900), respectively (Table 5). The prevalence of BRCA1/2 mutations in each matrix or subgroup was compared with that in non‐Ashkenazi individuals. In the analysis of each subgroup, statistical difference was observed only in the subgroup I‐1 between Japanese and non‐Ashkenazi individuals, which was characterized as the presence of breast cancers in both the proband and her relatives at over 50 years of age. In Subgroup I‐1, the prevalence of BRCA1/2 mutations was 21.4% (3/14) in Japanese versus 2.3% (4/172) in non‐Ashkenazi individuals, showing significantly higher prevalence in Japanese (odds ratio [OR] 11.11, 95% confidence interval [CI] 1.450–75.21, P = 0.01). All three mutations detected in Group I‐1 were in BRCA2, of which two subjects showed the same mutational type, i.e. c.5576_5579delTTAA in exon 11 of the BRCA2 gene, formerly designated as ‘5804del4,’‘5804_5807delTTAA’, or ‘5802delAATT’ in the BIC database or references( 10 , 11 ) (Table 4). One subject developed breast cancer at 57 years of age and her sister developed breast cancer at 58 years of age, and they were found to be identical twins. In this pedigree, no other relatives suffered from breast and/or ovarian cancer. Another subject developed lt‐breast cancer at 52 years of age and her aunt developed lt‐breast cancer at 63 years of age. The prevalence of BRCA1/2 mutations in each group or all subgroups between Japanese and non‐Ashkenazi individuals was analyzed by the Mantel‐Haenszel test, which is an extended method of analyzing multiple 2 × 2 contingency tables in retrospective studies and can exclude the effect of confounding factors between subgroups.( 25 ) It was elucidated that the prevalence of BRCA1/2 mutations in Japanese was significantly higher than that in non‐Ashkenazi individuals in the analysis of all subjects enrolled in Groups I through IV (P = 0.005, OR 1.873, 95% CI 1.217–2.884) or subtotal for Group I (P = 0.0471, OR 2.613, 95% CI 1.108–6.162). Subtotals for Groups II and IV were not significant, although the prevalence was higher in Japanese than non‐Ashkenazi people in Group II (28.0%vs 21.3%) and in Group IV (71.4%vs 48.6%), respectively. In Group V, two deleterious mutations were detected in 10 subjects, comprising four subjects with male breast cancer and six subjects with concomitant breast and ovarian cancers. All subjects assigned to Group V were sporadic, without familial predisposition.
Table 5.
Prevalences of BRCA1/2 germline mutations between non‐Ashkenazi individuals and Japanese
| Group | Subgroup | Prevalence of mutations in BRCA1/2 | P‐values* | Odds ratio (95% CI)* | |
|---|---|---|---|---|---|
| Non‐Ashkenazi individuals (Myriad)( 6 ) | Japanese (FALCO) | ||||
| I | I‐1 | 4/172 (2.3%) | 3/14 (21.4%) | 0.010 | 11.11 (1.450–75.21) |
| I‐2 | 55/579 (9.5%) | 4/26 (15.4%) | 0.307 | 1.730 (0.418–5.360) | |
| I‐3 | 5/77 (6.5%) | 0/2 (0%) | ND | ND | |
| II | II‐1 | 34/315 (10.8%) | 2/16 (12.5%) | 0.689 | 1.180 (0.125–5.491) |
| II‐2 | 206/806 (25.6%) | 13/33 (39.4%) | 0.103 | 1.892 (0.848–4.079) | |
| II‐3 | 25/67 (37.3%) | 0/1 (0%) | ND | ND | |
| II‐4 | 4/87 (4.6%) | 0/5 (0%) | ND | ND | |
| II‐5 | 41/236 (17.4%) | 4/11 (36.4%) | 0.119 | 2.704 (0.554–11.22) | |
| II‐6 | 35/111 (31.5%) | 1/6 (16.7%) | 0.665 | 0.437 (0.009–4.112) | |
| II‐7 | 19/87 (21.8%) | 1/3 (33.3%) | 0.534 | 1.776 (0.029–35.87) | |
| III | – | 10/52 (19.2%) | 1/1 (100%) | ND | ND |
| IV | IV‐1 ‡ | 16/39 (41.0%) | 0/0 | ND | ND |
| IV‐2 | 9/20 (45.0%) | 1/1 (100%) | ND | ND | |
| IV‐3 | 126/267 (47.2%) | 3/5 (60%) | 0.671 | 1.675 (0.189–20.35) | |
| IV‐4 ‡ | 38/71 (53.5%) | 0/0 | ND | ND | |
| IV‐5 | 16/24 (66.7%) | 1/1 (100%) | ND | ND | |
| V | V‐1 | NR | 0/0 | ||
| V‐2 | NR | 0/0 | |||
| V‐3 | NR | 0/0 | |||
| V‐4 | NR | 0/0 | |||
| V‐5 | NR | 1/4 (25.0%) | |||
| V‐6 | NR | 1/6 (16.7%) | |||
| Subtotal for Group I | 64/828 (7.7%) | 7/42 (16.7%) | 0.0471 † | 2.613 (1.108–6.162) † | |
| Subtotal for Group II | 364/1709 (21.3%) | 21/75 (28.0%) | 0.134 † | 1.554 (0.915–2.640) † | |
| Subtotal for Group IV | 151/311 (48.6%) | 5/7 (71.4%) | 0.455 † | 2.589 (0.484–13.87) † | |
| Subtotal for Group V | NR | 2/10 (20.0%) | ND | ND | |
| Total for Groups I–IV | 589/2900 (20.3%) | 34/125 (27.2%) | 0.005 † | 1.873 (1.217–2.884) † | |
Fisher's exact test.
† Estimated by Mantel‐Haenszel test.
‡ Excluded from Mantel‐Haenszel test due to the absence of enrolled subjects. CI, confidence interval; ND, not done.
Clinical characteristics showing significant association with BRCA1/2 mutational status. Statistical analysis was carried out to search for clinical characteristics showing an association with BRCA1/2 mutational status (Table 6). In the analysis of female subjects with breast cancer, ages at diagnosis were significantly younger in BRCA1/2‐positive subjects than BRCA1/2‐negative ones (42.4 ± 11.0 years vs 47.3 ± 12.0 years, P = 0.0272), while in the analysis of ovarian cancer, there was no significant differences between BRCA1/2‐positive and ‐negative groups (52.2 ± 8.6 years vs 49.4 ± 13.1 years, P = 0.6647). In the analysis of four subjects with male breast cancer, mutation of the BRCA2 gene was detected in one subject. Age at onset was high (more than 60 years) in male breast cancer regardless of BRCA1/2 mutational status (Table 6). Statistical analysis of other clinicopathological indices showed significant results in stage (P = 0.026), estrogen receptor (ER) status (P = 0.0002), progesterone receptor (PgR) status (P = 0.0020), and histological grade (P = 0.0386) between non‐carriers and BRCA1/2 mutation carriers suffering from breast cancer (Table 7). There seemed to be more stage II or III tumors in BRCA1/2 positive cases (P = 0.026). In BRCA1 mutation carriers, ER or PgR status was negative in 84.6% (11/13) of subjects and all tumors were either Grade 2 (2/7) or Grade 3 (5/7). In BRCA2 mutation carriers, positivities for ER and PgR were 76.4% (13/17) and 56.2% (9/16), respectively, and higher than BRCA1. As for histological grades, frequencies of Grade 1 tumors were 0% (0/7) and 14.2% (2/14) in BRCA1‐positve or BRCA2‐positive subjects, significantly lower than in non‐carriers. In the analysis of ovarian cancer, five subjects showed mutation of BRCA1, while no subjects showed mutation of BRCA2. No statistical significance was observed in clinicopathological indices such as tumor size, nodal status, and stage by BRCA1 mutational status.
Table 6.
Ages at onset of participants with breast and/or ovarian cancer
| All | Breast cancer (female) | Breast cancer (male) | Ovarian cancer | |
|---|---|---|---|---|
| Number of subjects | 135 | 122 | 4 | 18 |
| Age at diagnosis | ||||
| Overall | 46.5 ± 12.1 | 46.2 ± 12.1 | 64.5 ± 3.7 | 50.2 ± 11.8 |
| No deleterious mutations | 47.7 ± 12.1 (n = 99) | 47.3 ± 12.0 (n = 86) | 64.0 ± 4.4 (n = 3) | 49.4 ± 13.1 (n = 13) |
| Deleterious mutations in BRCA1 or BRCA2 | 43.1 ± 11.3 (n = 36) | 42.4 ± 11.0 (n = 34) | 66 (n = 1) | 52.2 ± 8.6 (n = 5) |
| P‐values | 0.052 | 0.0272 | 0.7295 | 0.6647 |
| Deleterious mutations in BRCA1 | 42.0 ± 11.9 (n = 17) | 41.8 ± 9.6 (n = 16) | – | 52.2 ± 8.6 (n = 5) |
| Deleterious mutations in BRCA2 | 44.2 ± 13.1 (n = 19) | 42.9 ± 12.3 (n = 18) | 66 (n = 1) | – |
| P‐values | 0.574 | 0.757 | – | – |
Table 7.
Clinicopathological characteristics of the breast or ovarian cancers by BRCA1/2 mutational status
| Variables | Breast cancer | Ovarian cancer | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Non‐carrier* | Deleterious mutations in | P‐values* | Non‐carrier | Deleterious mutations in | P‐values | ||||
| BRCA1/2 | BRCA1* | BRCA2* | BRCA1 | ||||||
| No. of subjects | 91 | 35 | 16 | 19 | 13 | 5 | |||
| Tumor size (T) | Tis | 2 | 0 | 0 | 0 | 0.3884 | – | – | 0.8338 |
| T1 | 50 | 16 | 7 | 9 | 8 | 3 | |||
| T2 | 35 | 14 | 7 | 7 | 1 | 0 | |||
| T3 | 1 | 3 | 1 | 2 | 3 | 1 | |||
| Missing (No.) | (3) | (2) | (1) | (1) | (1) | (1) | |||
| Nodal status (N) | Negative | 70 | 25 | 11 | 14 | 0.7859 | 7 | 3 | 1.000 |
| Positive | 17 | 7 | 4 | 3 | 4 | 1 | |||
| Missing (No.) | (4) | (3) | (1) | (2) | (2) | (1) | |||
| Stage | 0 | 2 | 0 | 0 | 0 | 0.026 | – | – | 0.9875 |
| I | 56 | 15 | 6 | 9 | 6 | 3 | |||
| II | 25 | 13 | 6 | 7 | 1 | 0 | |||
| III | 1 | 3 | 3 | 0 | 3 | 1 | |||
| IV | 1 | 0 | 0 | 0 | 1 | 0 | |||
| Missing (No.) | (6) | (4) | (1) | (3) | (2) | (1) | |||
| ER status | Positive | 57 | 15 | 2 | 13 | 0.0002 | |||
| Negative | 22 | 15 | 11 | 4 | |||||
| Missing (No.) | (12) | (5) | (3) | (2) | |||||
| PgR status | Positive | 53 | 11 | 2 | 9 | 0.0020 | |||
| Negative | 26 | 18 | 11 | 7 | |||||
| Missing (No.) | (12) | (6) | (3) | (3) | |||||
| Histological grade | Grade 1 | 22 | 2 | 0 | 2 | 0.0386 | |||
| Grade 2 | 28 | 9 | 2 | 7 | |||||
| Grade 3 | 14 | 10 | 5 | 5 | |||||
| Missing (No.) | (27) | (14) | (9) | (5) | |||||
| Laterality | Unilateral | 75 | 29 | 13 | 16 | 0.9711 | |||
| Bilateral | 16 | 6 | 3 | 3 | |||||
χ2‐test was performed between non‐carriers, subjects with BRCA1 mutations, and BRCA2 mutations. ER, estrogen receptor; PgR, progesterone receptor.
In 122 subjects with female breast cancer, cumulative incidences were analyzed by Kaplan–Meier plot and log‐rank test (Fig. 2). In subjects with deleterious BRCA1/2 mutations, median ages at onset were 42.5 years (BRCA1/2), 42 years (BRCA1), and 42.5 years (BRCA2), respectively, indicating that breast cancer developed at a significantly younger age than in non‐carriers (P = 0.0144 between BRCA1/2‐positive and ‐negative subjects, P = 0.0338 between BRCA1‐positive, BRCA2‐positive, and ‐negative subjects) (Fig. 2). Similar analysis was extended to relatives and it was shown that the age at onset of breast cancer could be a significant predictor of BRCA1/2 mutational status (Table 8). Breast cancer developing before age 40 within second‐degree relatives indicated a significantly higher prevalence of BRCA1/2 mutations (P = 0.0265). Of the 122 pedigrees among which index patients suffered from female breast cancer, 26 pedigrees (21.3%) fulfilled this condition and the frequency of deleterious BRCA1/2 mutations was significantly higher than in the controls (46.2%[12/26]vs 22.9%[22/96], OR 2.833, 95% CI 1.165–7.136). This feature was further emphasized when the disease history of cousins was included in the family history, in which 29 of 122 pedigrees (23.8%) fulfilled the criteria and the prevalence of BRCA1/2 mutations showed a significant increase compared to the controls (48.3%[14/29]vs 21.5%[20/93], OR 3.407, 95% CI 1.412–8.217). When the index patient suffered from breast cancer, the presence of ovarian cancer and/or bilateral breast cancer within second‐degree relatives was shown to be a strong indicator of BRCA1/2 mutations. Half of the enrolled subjects (49.2%[60/122]) fulfilled this condition and the prevalence of BRCA1/2 mutations in this group was considerably high (38.3%[23/60]vs 17.7%[11/62], OR 2.882, 95% CI 1.252–6.637).
Figure 2.
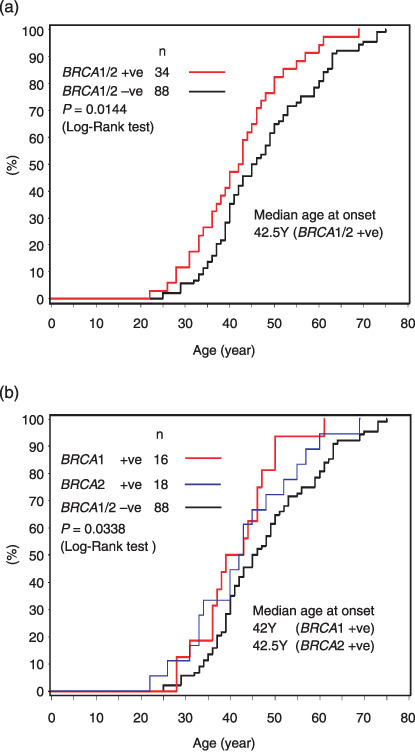
Kaplan–Meier plot of the cumulative incidence of breast cancer in female index patients examined for germline BRCA1/2 mutations. (a) Grouped in BRCA1/2 positive (n = 34) or negative cases (n = 88), (b) grouped in BRCA1 positive (n = 16), BRCA2 positive (n = 18) and BRCA1/2 negative cases (n = 88).
Table 8.
Univariate analysis of family‐based clinical variables associated with germline BRCA1/2 mutations in 122 pedigrees with female probands affected with breast cancers
| Variables | BRCA1/2 deleterious mutations | P‐values (χ2‐test) | Odds ratio (95% confidence interval) | ||
|---|---|---|---|---|---|
| Yes | No | ||||
| Breast cancer before age 40 | Yes | 12 | 14 | 0.0265 | 2.833 (1.165–7.136) |
| Within second‐degree relatives | No | 22 | 74 | ||
| Breast cancer before age 40 | Yes | 14 | 15 | 0.0084 | 3.407 (1.412–8.219) |
| Within second‐degree relatives and cousins | No | 20 | 73 | ||
| Ovarian cancer and/or bilateral breast cancer within second‐degree relatives | Yes | 23 | 37 | 0.0151 | 2.882 (1.252–6.637) |
| No | 11 | 51 | |||
Discussion
A total of 135 subjects were enrolled in the study, 131 women and four men, all of whom had a history of breast and/or ovarian cancers (Table 1). All patients were selected and enrolled in the study from genetic counseling clinics of the corresponding hospitals and the average number of sessions of genetic counseling was 3.36 ± 0.82. Patients usually underwent one or two sessions before and one session after gene testing when test results were negative. For patients with deleterious mutations, additional sessions were performed as follow‐up because of the risk of developing psychosocial issues; hence, the number of sessions for mutation carriers was significantly more than for non‐carriers (3.75 ± 1.20 vs 3.22 ± 0.56, P = 0.0007). In all subjects, the course was uneventful until the end of the study. Genealogical information was obtained in counseling sessions and the numbers of relatives greater than second‐degree were significantly more in mutation carriers than non‐carriers (P < 0.0001). Patient accrual was decided by family history within second‐degree relatives, and in greater than second‐degree relatives, family history might be investigated more precisely in subjects with deleterious BRCA1/2 mutations after disclosure of test results; therefore, genealogical information on relatives greater than second‐degree may be biased in carriers.
Among mutations detected in BRCA1, L63X (c.188T > A), Q934X (c.288C > T), and K503X (c.1507 A > G) were previously reported in Japanese.( 7 , 9 , 10 , 11 ) Sekine et al. reported L63X and Q934X as the two common founder mutations in Japanese.( 11 ) L63X was detected in five subjects and Q934X in only one subject in this study. In a search of the BIC database, five mutational types were previously unreported, of which K503X (c.1507 A > G) was detected in one Japanese subject,( 10 ) and the other four mutational types were considered to be previously unreported deleterious mutations. Genetic variants of unknown significance were detected in nine subjects; all were missense mutations and three types were thus far unreported in the BIC database (Table 3).
In the analysis of BRCA2, 5804del4 (c.5576_5579delTTAA), Q3026X (c.9076C > T), and R3128X (c.9382C > T) were previously reported in Japanese people (Table 4).( 9 , 10 , 11 ) As for genetic variants of uncertain significance, two mutational types (G2044V [c.6131G > T] and V2109I [c.6325G > A]) were reported in Japanese, and G2044V was found in at least one of 28 Japanese healthy volunteers.( 12 ) M524I (c.1572G > C), K610Q (c.1828 A > C), I770V (c.2308 A > G), and S2616F (c.7847C > T) were previously unreported mutational types.
The prevalence of BRCA1/2 mutations in Japanese subjects classified to each subgroup was compared with that of non‐Ashkenazi individuals, and statistical difference was observed in subgroup I‐1, in which two mutational types were detected in three subjects in Japanese. They were 314delTT in exon 3 of BRCA2 and two subjects with c.5576_5579delTTAA or 5804delTTAA in exon 11 of BRCA2. Two cases with 5804delTTAA have been reported so far in Japanese.( 9 , 11 ) Ikeda et al. reported seven cases of 5802del AATT,( 10 ) which is the same mutational type as 5804delTTAA as there is a repeated sequence in this region; therefore, this mutational type seems to be rather common in Japanese. In this study, c.5576_5579delTTAA were detected in two subjects. In the former, breast cancer developed in identical twins at 57 and 58 years of age, respectively. Reportedly, statistically significant effects of heritable factors were observed for breast cancers coincidentally developing in identical twins, some of which would be attributed to polygenic inheritance.( 26 ) In the latter case, lt‐breast cancer developed at 52 years of age in the proband and her aunt developed lt‐breast cancer at 63 years. No other relatives suffered from breast and/or ovarian cancer in these pedigrees and we strongly suspected that c.5576_5579delTTAA might be a relatively common mutation with low penetrance in Japanese. If these two subjects were excluded, no statistical significance was observed between Japanese subjects and non‐Ashkenazi individuals in the subgroup I‐1. Further studies are required to elucidate the prevalence of this particular mutational type in a Japanese healthy cohort.
A significantly higher frequency of mutation was found in patients with breast cancer older than 50 years of age who had a family history of breast cancer at older than 50 years within second‐degree relatives (Subgroup I‐1) compared to the corresponding non‐Ashkenazi individuals (Table 5). This may imply that Japanese carriers of BRCA1/2 mutation suffer from breast cancer with later onset than non‐Ashkenazi carriers. As for non‐Ashkenazi individuals, the clinical backgrounds of the enrolled subjects, such as age at onset, were not available except for the data so far reported;( 6 ) therefore, it is hard to produce a Kaplan–Meier plot of cumulative incidences in non‐Ashkenazi individuals. Likewise, few reports have shown the prevalence of BRCA1/2 mutations analyzed by full sequencing in non‐Ashkenazi individuals as a population‐based study or cross‐sectional study. Recently, John et al. estimated the prevalence of BRCA1 mutations in white, non‐Hispanic breast cancer patients without Ashkenazi ancestry younger than 65 years at diagnosis. They analyzed 508 breast cancer patients enrolled in the Breast Cancer Family Registry and pathogenic mutations of BRCA1 were found in 14 subjects, of which six subjects (42.8%) and 11 (78.6%) developed breast cancer before age 35 and age 50, respectively.( 27 ) Not exclusive to non‐Ashkenazi individuals, Metcalfe et al. analyzed 927 women with unilateral breast cancer and with positive BRCA1/2 mutations from eight countries (Austria, Canada, France, Israel, Italy, Norway, Poland, and the US) and their average age at diagnosis of the first breast cancer was 42.2 years.( 28 ) In our study, average age at onset of breast cancer in BRCA1/2 carriers was 42.4 years and these data look similar to those reported in Western countries.
The numbers of subjects classified into each group were 42 in Group I, 75 in Group II, one in Group III, seven in Group IV, and 10 in Group V (Table 5). Originally, we assumed that more subjects would be enrolled into Group I than Group II, as the first recruitment of all eligible patients was made by the attending doctors, and a considerable number of the breast cancer patients fulfilling the eligibility criteria had a modest family history. It seemed likely that patients with a higher risk wished to be enrolled in the study and those with a modest risk did not visit the clinic for genetic counseling. This may be why fewer subjects were enrolled in Group I than in Group II. We designed the study as an unbiased hospital‐based, cross‐sectional study in which all subjects with a family history of breast and/or ovarian cancer were enrolled, but the results seemed closer to a family‐based study rather than a hospital‐based study. This trend was similar to the subjects enrolled in the study through press advertising.
The results of the Mantel‐Haenszel test showed that the prevalence of BRCA1/2 mutations in Japanese was significantly higher than those reported in non‐Ashkenazi individuals. As the sample size was too small to reach a conclusion, one reason may be that all gene tests were carried out in genetic counseling clinics, where clients were more likely to be prone to hereditary cancers. It should be noted that the prevalence of BRCA1/2 mutations in Japanese was as high or even higher than that of non‐Ashkenazi individuals reported in the US.
Ikeda et al. examined 113 Japanese breast cancer patients showing a modest to minimal familial risk, i.e. those with at least one breast cancer or one ovarian cancer patient within their first‐degree relatives.( 10 ) They reported that families with early onset patients diagnosed at younger than 40 years of age showed a higher frequency (38%, 19/50 subjects) of BRCA1/2 mutations than those without early onset patients, and families with bilateral breast cancer patients showed a higher frequency (40%, 6/15 subjects) than those with only unilateral breast cancer patients, but all these differences were statistically insignificant.( 10 ) In the present study, we found statistical significance in families with breast cancer before age 40 within second‐degree relatives (46.2%, 12/26 subjects) and within second‐degree relatives and cousins (48.3%, 14/29 subjects). Families with ovarian cancer and/or bilateral breast cancer within second‐degree relatives exhibited statistically significant BRCA1/2 mutation frequency (38.3%, 23/60 subjects). Predisposition to breast cancer in cousins seems informative for assessing familial risk, particularly in cases where the responsible genes are likely to be transmitted from the paternal side. In genetic counseling, precise family history is a key point in assessing genetic risk and the inclusion of familial risk within second‐degree relatives or cousins would be helpful for proper risk assessment.
In conclusion, this is the first cross‐sectional study elucidating the prevalence of BRCA1/2 mutations among Japanese people with varying genetic susceptibility. Genetic counseling performed prior to gene testing in genetic counseling clinics is an effective approach to assess the risk for cancer predisposition and subsequent indication for gene testing. Full sequencing analysis of BRCA1/2 genes would be a useful modality for diagnosing HBOC and the results of the present study provide a basis for the clinical application of a cancer prevention strategy targeted to BRCA1/2 mutation carriers in Japanese.
Acknowledgments
The authors thank Ms. Satoko Ando, Mr Takafumi Ishiguro, Izumi Ohki, MD, Ph.D, and Hirohiko Totsuka, MS, for their assistance and advice on statistical analysis. We also express our deepest gratitude to the late Professor Shiro Nozawa in the Department of Gynecology, School of Medicine, Keio University; we appreciate his support and contribution to this study. We also appreciate the continuous support of Mr Shigeru Shoji and Mr Hiroshi Yamane from FALCO biosystems who participated in launching of the project.
References
- 1. Ferlay J, Bray F, Pisani P, Parkin DM. GLOBOCAN 2002: Cancer Incidence, Mortality and Prevalence Worldwide. IARC Cancerbase No. 5, Version 2.0. Lyon, France: IARC Press, 2004. [Google Scholar]
- 2. Collaborative Group on Hormonal Factors in Breast Cancer . Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 2001; 358: 1389–99. [DOI] [PubMed] [Google Scholar]
- 3. Miki Y, Swensen J, Shattuck‐Eidens D et al . A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science, 1994; 266: 66–71. [DOI] [PubMed] [Google Scholar]
- 4. Wooster R, Bignell G, Lancaster J et al . Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378: 789–92. [DOI] [PubMed] [Google Scholar]
- 5. Frank TS, Critchfield GC. Identifying and managing hereditary risk of breast and ovarian cancer. Clin Perinatol 2001; 28: 395–406. [DOI] [PubMed] [Google Scholar]
- 6. Frank TS, Deffenbaugh AM, Reid JE et al . Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol 2002; 20: 1480–90. [DOI] [PubMed] [Google Scholar]
- 7. Inoue R, Fukutomi T, Ushijima T, Matsumoto Y, Sugimura T, Nagao M. Germline mutation of BRCA1 in Japanese breast cancer families. Cancer Res 1995; 55: 3521–4. [PubMed] [Google Scholar]
- 8. Katagiri T, Emi M, Ito I et al . Mutations in the BRCA1 gene in Japanese breast cancer patients. Hum Mutat 1996; 7: 334–9. [DOI] [PubMed] [Google Scholar]
- 9. Noguchi S, Kasugai T, Miki Y, Fukutomi T, Emi M, Nomizu T. Clinicopathologic analysis of BRCA1‐ or BRCA2‐associated hereditary breast carcinoma in Japanese women. Cancer 1999; 85: 2200–5. [PubMed] [Google Scholar]
- 10. Ikeda N, Miyoshi Y, Yoneda K et al . Frequency of BRCA1 and BRCA2 germline mutations in Japanese breast cancer families. Int J Cancer 2001; 91: 83–8. [DOI] [PubMed] [Google Scholar]
- 11. Sekine M, Nagata H, Tsuji S et al . Mutational analysis of BRCA1 and BRCA2 and clinicopathologic analysis of ovarian cancer in 82 ovarian cancer families: two common founder mutations of BRCA1 in Japanese population. Clin Cancer Res 2001; 7: 3144–50. [PubMed] [Google Scholar]
- 12. Kawahara M, Sakayori M, Shiraishi K et al . Identification and evaluation of 55 genetic variations in the BRCA1 and the BRCA2 genes of patients from 50 Japanese breast cancer families. J Hum Genet 2004; 49: 391–5. [DOI] [PubMed] [Google Scholar]
- 13. Szabo C, Masiello A, Ryan JF, Brody LC. The breast cancer information core; database design, structure, and scope. Hum Mutat 2000; 16: 123–31. [DOI] [PubMed] [Google Scholar]
- 14. Kauff ND, Satagopan JM, Robson ME et al . Risk‐reducing salpingo‐oophorectomy (RRSO) in women with a BRCA1 or BRCA2 mutation. N Engl J Med 2002; 346: 1609–15. [DOI] [PubMed] [Google Scholar]
- 15. Uyei A, Peterson SK, Erlichman J et al . Association between clinical characteristics and risk‐reduction interventions in women who underwent BRCA1 and BRCA2 testing: a single‐institution study. Cancer 2006; 107: 2745–51. [DOI] [PubMed] [Google Scholar]
- 16. Metcalfe K, Lynch HT, Ghadirian P et al . Contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2004; 22: 2328–35. [DOI] [PubMed] [Google Scholar]
- 17. McLaughlin JR, Risch HA, Lubinski J et al . Hereditary Ovarian Cancer Clinical Study Group . Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case‐control study. Lancet Oncol 2007; 8: 26–34. [DOI] [PubMed] [Google Scholar]
- 18. Petrij‐Bosch A, Peelen T, Van Vliet M et al . BRCA1 genomic deletions are major founder mutations in Dutch breast cancer patients. Nat Genet 1997; 17: 341–5. [DOI] [PubMed] [Google Scholar]
- 19. The BRCA1 Exon 13 Duplication Screening Group . The Exon 13 duplication in the BRCA1 gene is a founder mutation present in geographically diverse population. Am J Hum Gen 2000; 67: 207–12. [PMC free article] [PubMed] [Google Scholar]
- 20. Rohlfs EM, Puget N, Graham ML et al . An Alu‐mediated 7.1 kb deletion of BRCA1 exons 8 and 9 in breast and ovarian cancer families that results in alternative splicing of exon 10. Genes Chromosomes Cancer 2000; 28: 300–7. [DOI] [PubMed] [Google Scholar]
- 21. Ward BD, Hendrickson BC, Judkins T et al . A multi‐exonic BRCA1 deletion identified in multiple families through single nucleotide polymorphism haplotype pair analysis and gene amplification with widely dispersed primer sets. J Mol Diagn 2005; 7: 139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Antonarakis SE and the Nomenclature Working Group . Recommendations for a nomenclature system for human gene mutations. Hum Mutat, 1998; 11: 1–3. [DOI] [PubMed] [Google Scholar]
- 23. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation‐dependent probe amplification. Nucleic Acids Res 2002; 30: e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bunyan DJ, Eccles DM, Sillibourne J et al . Dosage analysis of cancer predisposition genes by multiplex ligation‐dependent probe amplification. Br J Cancer 2004; 9: 1155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959; 22: 719–48. [PubMed] [Google Scholar]
- 26. Lichtenstein P, Holm NV, Verkasalo PK et al . Environmental and heritable factors in the causation of cancer – analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343: 78–85. [DOI] [PubMed] [Google Scholar]
- 27. John EM, Miron A, Gong G et al . Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial/ethnic groups. JAMA 2007; 298: 2869–76. [DOI] [PubMed] [Google Scholar]
- 28. Metcalfe KA, Lubinski J, Ghadirian P et al . Predictors of contralateral prophylactic mastectomy in women with a BRCA1 or BRCA2 mutation: the Hereditary Breast Cancer Clinical Study Group. J Clin Oncol 2008; 26: 1093–7. [DOI] [PubMed] [Google Scholar]