

Abstract
The aim of the present study was to investigate whether the hypoxia responsive element (HRE) could be used to enhance suicide gene (HSV‐tk) expression and tumoricidal activity in radiation‐controlled gene therapy of human lung adenocarcinoma xenografts. A chimeric promoter, HRE–Egr, was generated by directly linking a 0.3‐kb fragment of HRE to a 0.6‐kb human Egr‐1 promoter. Retroviral vectors containing luciferase or the HSV‐tk gene driven by Egr‐1 or HRE–Egr were constructed. A human adenocarcinoma cell line (A549) was stably transfected with the above vectors using the lipofectamine method. The sensitivity of transfected cells to prodrug ganciclovir (GCV) and cell survival rates were analyzed after exposure to a dose of 2 Gy radiation and hypoxia (1%). In vivo, tumor xenografts in BALB/c mice were transfected with the constructed retroviruses and irradiated to a total dose of 6 Gy, followed by GCV treatment (20 mg/kg for 14 days). When the HSV‐tk gene controlled by the HRE–Egr promoter was introduced into A549 cells by a retroviral vector, the exposure to 1% O2 and 2 Gy radiation induced significant enhancement of GCV cytotoxicity to the cells. Moreover, in nude mice bearing solid tumor xenografts, only the tumors infected with the hybrid promoter‐containing virus gradually disappeared after GCV administration and radiation. These results indicate that HRE can enhance transgene expression and tumoricidal activity in HSV‐tk gene therapy controlled by ionizing radiation in hypoxic human lung adenocarcinoma. (Cancer Sci 2005; 96: 918–924)
Ionizing radiation is currently used in the treatment of many human tumors and its potential for eliciting the expression of exogenous therapeutic genes (radiogenetic therapy) is of considerable interest.( 1 ) Egr‐1 is a member of the radiation‐responsive gene family. The regions of the Egr‐l gene promoter that control its radiation response have been identified and consist of 10‐bp motifs known as CArG elements.( 2 ) These elements are activated by reactive oxygen species produced on exposure to ionizing radiation.( 2 ) In the experimental models developed so far, therapeutic genes are usually prodrug‐activating (‘suicide’) or immunomodulating genes, and their expression is controlled by the promoter regions of genes that are upregulated by ionizing radiation.( 3 , 4 , 5 , 6 ) For example, when the Egr‐1 promoter was used to control expression of the HSV‐tk gene, enhanced tumor cell killing was seen when cells were grown in the presence of the prodrug ganciclovir (GCV) following radiation.( 7 , 8 ) One of the problems associated with the use of ionizing radiation to regulate targeted gene expression is that the response is transient and increased gene expression is thus only maintained for a brief period after the radiation stimulus is withdrawn, resulting in low levels of therapeutic gene expression.( 3 , 4 , 9 ) Moreover, in most studies, the therapeutic responses usually came from a single fraction dose of 10–20 Gy, which was far higher than the clinical conventional dose of 2 Gy.( 9 )
Using tissue oxygen electrodes, it was found that oxygen tension in tumor tissue is 10 times lower than in normal tissue.( 10 ) A transcriptional response to hypoxia mediated by hypoxia inducible factor‐1 (HIF‐1) has been identified.( 10 ) HIF‐1 binds to DNA motifs known as hypoxia response elements (HRE), which have been found in the isoforms of a variety of genes that are overexpressed in cancer. These include vascular endothelium growth factor (VEGF), glucose transporters, enzymes of the glycolytic pathway and VEGF receptor l (flt‐1). HIF‐1 can be activated by local hypoxia in the tumor microenvironment.( 11 ) Furthermore, in some types of tumor HIF‐1 is constitutively active.( 12 ) The murine phosphoglycerate kinase‐1 (PGK‐1) HRE has been shown to transfer hypoxia inducibility to other promoters in vitro ( 13 ) and in vivo.( 11 ) Recently, similar HRE have been incorporated into several promoters that worked well in vitro, such as the SV40 minimal promoter in an adenoviral vector,( 14 ) VEGF receptor 2 (KDR)( 15 ) and human α‐fetoprotein (AFP)( 16 ) promoter in retroviral vectors.
Although the combination of HRE and CArG itself and its response to radiation and hypoxia have been reported elsewhere,( 17 , 18 ) the combination of the HRE/CArG promoter and HSV‐tk/GCV and its application in vivo have not been reported. In the present study, we constructed a hybrid promoter (HRE–Egr‐1) consisting of the hypoxia‐inducible enhancer of the human VEGF gene and Egr‐1 promoter. This hybrid promoter contained HRE directly linked to the Egr‐1 promoter. We evaluated the hypoxic/radiation‐specific enhancement of GCV‐mediated cytotoxicity that was induced by the HSV‐tk gene under the control of the HRE–Egr promoter in vitro and, more importantly, in vivo.
Materials and Methods
Cell culture and hypoxic intervention
The human lung adenocarcinoma cell line A549 (CCL‐185) was obtained from the American Tissue Culture Collection (ATCC; Manassas, VA, USA), and maintained in RPMI‐1640 (Sigma, St. Louis, MO, USA) supplemented with 10% heat‐inactivated fetal calf serum (Gibco‐BRL, Gaithersburg, MD, USA), 100 units/mL penicillin−100 µg/mL streptomycin (Sigma) and 2 mM l‐glutamine (Life Technologies, Gaithersburg, MD, USA), cultured in a well‐humidified incubator with 5% CO2 at 37°C. For gene transfection experiments, 5 × 105 exponentially growing cells were plated on a six‐well culture dish overnight. To expose cells to hypoxia, they were placed into an airtight prewarmed Lucite chamber that was evacuated and regassed with 1% O2/94% N2/5% CO2, and then the tightly sealed chambers were incubated at 37°C for 8 h.( 19 )
Construction of retroviral vectors
The self‐inactivating (SIN) retroviral vector pBABE puro SIN was used. The SIN vector was generated by deletion of a 299‐bp PvuII–SacI fragment within the 3′ long terminal repeats (LTR) of pBABE puro. The deletion in the U3 region of the 3′ LTR removed the CCAAT box, but not the retroviral TATA box.( 20 ) The 2.4‐kb cDNA of HSV‐tk was ligated into the SalI site of pBABE puro SIN (pBABE puro SIN‐tk). The 1.7‐kb cDNA of the firefly luciferase gene from pGL3 basic (Promega, Madison, WI, USA) was end‐filled and blunt‐end ligated into the SalI site of pBABE puro SIN (pBABE puro SIN‐luc) to generate the constructs with luciferase as the reporter gene. A 600‐bp fragment of the human Egr‐1 promoter was cloned as a BamHI–EcoRI fragment into pBABE puro SIN‐tk (SET) and pBABE puro SIN‐luc (SEL). The hypoxia‐inducible enhancer HRE was cloned into the BamHI site 5′ downstream of the promoter to generate HRE–SET and HRE‐SEL. The enhancer was completely sequenced (ABI377 automated sequencing apparatus, Applied Biosystems, Forster City, CA, USA) to confirm the orientation of the binding sites. The proviral structures of the vectors are shown in Fig. 1.
Figure 1.
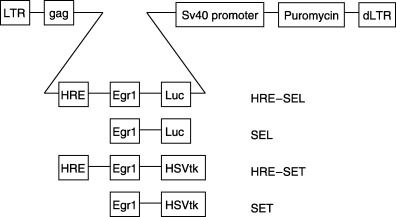
Retroviral constructs used in this study. A deletion in the U3 region of the 3′ long terminal repeats (LTR) of the retroviral vector pBABA puro was introduced (dLTR) to generate the self‐inactivating (SIN) vector pBABE puro SIN. The inserts consisting of the HSV‐tk or Luc genes with the Egr‐1 promoter and hypoxia responsive element (HRE) enhancer are indicated.
Packaging of retroviruses and infection of A549 cells
The retroviral packaging cell line am12 was transfected with 20 µg of each vector plasmid by calcium phosphate precipitation and selected in 1.25 µg/mL puromycin for 20 days. The colonies were pooled to generate a retrovirus‐producing cell line. At confluence, the culture medium was replaced with 50% of the standard volume. Twenty‐four hours later, the virus‐containing medium was filtered through a 0.45‐µm filter and stored at −70°C. For infection, A549 cells were exposed to the virus for 4 h in the presence of 8 µg/mL polybrene in a serum‐free medium. After 48 h cells were selected with 3.5 µg/mL puromycin and resistant colonies pooled to generate stable infected cell lines.
Analysis of luciferase expression
Firefly luciferase enzyme activity was measured following the standard single luciferase assay (Promega). Cells were cultured in six‐well plates and exposed to hypoxia (1%, 8 h) and/or radiation (2 or 20 Gy). They were then washed gently with phosphate‐buffered saline (PBS) twice and lysed by adding 500 µL passive lysis buffer (Promega). Samples were collected after 15 min incubation on a rocking table and 20‐µL aliquots were measured for luciferase enzyme activity for 10 s in a single‐sample luminometer. To calculate the cell numbers in different samples, the protein concentration of the lysates was measured using the bicinchoninic acid protein quantification kit (Dakewe, Shenzhen, China).
Analysis of HSV‐tk expression
Total RNA was extracted from parental and retrovirus‐infected cells by RNAzol B (TEL‐Test, Friendswood, TX, USA) according to the protocol provided by the manufacturer. Twenty micrograms of the RNA samples were electrophoresed in a 1% agarose/2.2 M formaldehyde gel and transferred to a nylon membrane. Northern blot analysis for the HSV‐tk RNA was carried out as described previously.( 21 ) The HSV‐tk and human glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) genes were used as probes to evaluate HSV‐tk expression and to monitor the loading of the RNA samples, respectively. The band densities were measured using the public‐domain National Institute of Health (NIH) Image Program and HSV‐tk expression levels were standardized based on GAPDH expression levels.
In vitro gene therapy
The G418‐selected pools of A549 cells transduced with virus (5 × 104 cells/well) were plated in 24‐well multiplates (day 0). In the preliminary experiment, to work‐out the optimal hypoxic condition, various concentrations of oxygen had been used in the treatment of cells (data not shown). The oxygen concentration used was the optimal hypoxic condition for promoter activity. In some experiments the cells were exposed to ionizing radiation of 2 Gy with an EXS‐300 γ irradiator (200 kV, 15 mA, filter 0.5 mm aluminum and 0.5 mm cuprum, 0.47 Gy/min) and/or incubated under hypoxia (1%) for 8 h previously on day 2. To test the sensitivity of A549 cells to GCV, GCV (Sigma) was added at 0–1000 µM 3 h after radiation. The medium in each well was replaced with fresh medium on days 4 and 6. On day 7, viable cells were counted by means of a trypan blue exclusion test. The mean inhibitory concentration (IC50), which represents 50% growth inhibition relative to the control, was calculated using curve‐fitting parameters.
In vitro gene therapy was carried out in various combinations as follows: group I, GCV (10 µM); group II, radiation (2 Gy) + GCV (10 µM); group III, hypoxia (1%) + GCV (10 µM); and group IV, radiation (2 Gy) + hypoxia (1%) + GCV (10 µM) for SET‐transfected A549 cells. For HRE–SET‐transfected cells, the combinations of interventions were the same as SET‐transfected A549 cells.
In vivo gene therapy
Female BALB/c nude mice, 5 weeks of age, were injected subcutaneously in both flanks with 200 µL of A549 cell suspension (1 × 107 cells) in RPMI‐1640. Three to five weeks later, when the tumor diameters reached 8–10 mm, 200 µL of retrovirus vector (2 × 107 plaque forming units [pfu]/µL) was injected intratumorally (day 1). In some experiments, xenografted tumors were irradiated (2 Gy) by focusing each tumor with an EXS‐300 γ irradiator (200 kV, 15 mA, filter 0.5 mm aluminum, 1.41 Gy/min) on days 2, 4 and 6. GCV administration (20 mg/kg injected intraperitoneally) was initiated 3 h after radiation and continued daily for 2 weeks.
We also divided the mice into groups: A549 cells injection only (control, n = 6); A549 injection and radiation (R, n = 6); SET transfection (2 × 107 pfu/µL) and radiation (S + R, n = 5); SET transfection (2 × 107 pfu/µL) and GCV (S + G, n = 6); SET transfection (2 × 107 pfu/µL), radiation and GCV (S + R + G, n = 5); HRE–SET transfection (2 × 107 pfu/µL) and radiation (H + R, n = 6); HRE–SET transfection (2 × 107 pfu/µL) and GCV (H + G, n = 6); and HRE–SET transfection (2 × 107 pfu/µL), radiation and GCV (H + R + G, n = 6). No mouse showed signs of wasting or other indications of toxicity. Care and treatment of the animals were in accordance with China Western Medical University Institutional Ethics Committee guidelines.
Apoptosis assays
Terminal deoxynucleotidyl transferase‐meditaed dUTP nick‐end labeling (TUNEL) assay to detect fragmented DNA in situ was carried out using the In situ Cell Death Detection kit (Boehringer Mannheim, Indianapolis, IN, USA). The treated cells were carefully removed from the cultures before experimentation by washing the cultures vigorously three times with PBS/0.02% ethylenediaminetetracetic acid (EDTA). Assays were carried out with two to three replicate plates of cells, and ≥ 10 randomly selected fields per plate were counted for TUNEL‐positive cells. Cells were harvested after treatment, washed in cold PBS, fixed in 1% paraformaldehyde for 1 h on ice, and permeabilized with 70% ethanol for 1 h on ice. Cells were then resuspended in 1 mL PBS to which 0.5 mL phosphate–citric acid buffer was added and incubated at room temperature for 5 min. The number of fluorescein‐isothiocyanate‐labeled cells per 100 cancer cells was used as a measure of the percentage of apoptosis.
Apoptosis in lung cancer xenografts was identified using the above‐mentioned kit according to the manufacturer's protocol. Briefly, sections were quenched in 2% hydrogen peroxide. The optimal dilution and incubation with the TdT enzyme was 1 : 54 for 1.5 h at 37°C. We used an antidigoxigenin antibody from Boehringer Manheim (Indianapolis, IN; 1 : 1000; 1 h at room temperature) in place of the antibody in the kit. The reaction was visualized using diaminobenzidine tetrahydrochloride (Vector Laboratories, Burlingame, CA, USA) as the chromogen, followed by a methyl green counterstain. The number of positive cells was determined by light microscopy at ×400 magnification.
Detection of hypoxia in tumor xenografts by immunohistochemistry
Excised tumors were fixed in paraffin. Slices (5 µm) from each axial section were deparaffinized with toluene and rehydrated by treatment with a series of alcohol and water mixtures and finally with water. To quench endogenous peroxidase, the tissue sections were exposed for 30 min to 0.3% hydrogen peroxide in absolute methanol. Microwave heating (4 min 500 W, 20 min defrost and 15 min room temperature) was used in the presence of Tris–EDTA buffer (0.01 M, pH 9.0) to achieve antigen retrieval prior to application of the primary monoclonal antibody. PBS (0.1 M, pH 7.3) plus Tween‐20 (polyoxyethylene sorbitan monolaurate) were used to wash slides between the two steps. The sections were incubated in anti‐CA IX (endogenous hypoxia marker) monoclonal antibody (1 : 100 dilution in PBS) for 30 min. Secondary incubation with peroxidase‐labeled antimouse Envision (Dako, Carpinteria, CA, USA) was also applied for 30 min. Bound peroxidase was developed using 0.033% hydrogen peroxide in 10% diaminobenzidine (DAB; Dako) for 7 min. After washing in distilled water, the sections were counterstained with hematoxylin for 1 min, dehydrated and mounted. Immunostained sections of the selected tumors were viewed by means of a Zeiss Axioskop 40 FL microscope (Carl Zeiss, Thornwood, NY, USA).
Statistical analysis
Results are expressed as mean ± standard deviation (SD). Standard descriptive statistics, Student's t‐test and Welch's t‐test, were used according to the distribution of experimental values. P‐values less than 0.05 were accepted as significant differences between groups.
Results
Hypoxia enhanced and lengthened radiation‐inducible transcriptional activity of the HRE–Egr promoter
To obtain higher levels and higher‐extension levels of radiation‐inducible transcription activity of the Egr‐1 promoter under hypoxia, we prepared a hybrid promoter (HRE–Egr) in which the 0.3‐kb fragment between −1073 and −775 5′–to the VEGF transcription initiation site was linked directly to the human Egr‐1 0.6‐kb downstream of the promoter (Fig. 1). The SIN retroviral vector was used to minimize interference from the viral LTR promoter with the exogenous promoter in the viral constructs. Retroviruses were constructed with the hypoxic enhancer 5′ of the Egr‐1 downstream of the promoter driving luciferase or HSV‐tk. A549 cells were infected and stably transfected cell lines were generated (all stable lines were pooled populations). By transient transfection assay, luciferase transcription induced by Egr‐1 or HRE–Egr was analyzed in A549 cells under radiation and hypoxic conditions.
As shown in Fig. 2, after exposure to hypoxia (1%, 8 h) and radiation (2 Gy), cells infected with the HRE‐containing virus expressed seven times as much luciferase as cells infected with enhancer‐less virus, and 4.5 times as much as cells exposed to radiation at normoxia. Cells stably transfected with virus containing the Egr‐1 promoter alone showed lower expression of luciferase under hypoxia compared with normoxia. The absolute expression levels in cells infected with HRE–Egr or Egr‐1 alone under normoxia were the same, indicating that the enhancer was silent without hypoxic stimulation. The orientation of the HRE enhancer did not affect the enhancer function (data not shown). Dynamic alterations in luciferase expression were observed following radiation, and this indicated that, despite both groups of Egr‐1 and HRE–Egr reaching peaks at 8 h post radiation, the HRE–Egr group declined at a slower rate, which returned to preradiation level at 72 h post radiation compared to the Egr‐1 group at 24 h post radiation.
Figure 2.
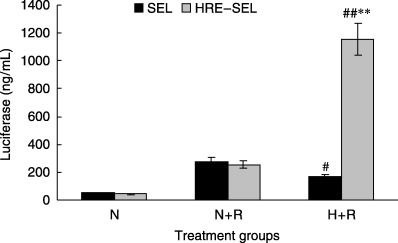
Enhancement of radio‐inducible transcriptional activity from the Egr‐1 promoter by hypoxic response elements (HRE) after transient transfection exposure to hypoxia. A549 cells were infected with the luciferase reporter constructs SEL or HRE‐SEL and incubated in normoxia (N) or hypoxia (H) (1%) for 8 h with exposure to 2 Gy of radiation (R). Twelve hours later, these cells were harvested and assayed for luciferase activity. The luciferase activity were normalized to that in non‐irradiated cells under aerobic conditions. The error bars in all of the figures show the standard deviation (SD) of at least five independent samples. Versus SEL: **P < 0.01; versus N + R : #P < 0.05, ##P < 0.01.
We then examined the levels of HSV‐tk expression in A549 cells infected with retroviruses exposed to radiation and hypoxia. As shown in Fig. 3, northern blot analysis revealed that the parental A549 cell line did not express the HSV‐tk gene, while SET and HRE–SET retrovirus‐infected A549 cells expressed the HSV‐tk gene. Furthermore, when the levels of HSV‐tk expression were standardized based on the corresponding GAPDH expression levels, it was shown that HRE–SET retrovirus‐infected A549 cells expressed approximately seven‐fold higher HSV‐tk RNA compared with SET retrovirus‐infected A549 cells with exposure of radiation and hypoxia. Conversely, SET retrovirus‐infected A549 cells exposed to 1% O2 expressed the HSV‐tk gene much more weakly than those exposed to normoxia, and the level of HSV‐tk expression under hypoxia was approximately 0.5‐fold lower than that under normoxia.
Figure 3.
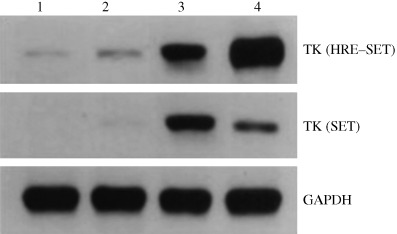
HSV‐tk expression in parental and infected A549 cells. The HSV‐tk of the SET and HRE–SET groups and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH)‐specific transcripts are shown. Lane 1, untreated infected cells at normoxia; lane 2, infected cells under hypoxia (1%); lanes 3–4, infected cells exposed to 2 Gy radiation with or without hypoxia (1%), respectively.
Specific and enhanced cytotoxicity of GCV by hypoxia in A549 cells infected with virus containing HRE
We constructed hybrid genes consisting of HSV‐tk genes under the control of either the Egr‐1 or HRE–Egr promoters and inserted them into retroviral vectors, thus producing SET and HRE–SET, respectively. The A549 cells were infected with these recombinant retroviruses. Because the transduction efficiency might contribute to the results, G418‐resistant pooled populations were used for in vitro studies of GCV‐mediated cytotoxicity to evaluate precisely the activity and specificity of the promoters.
Based on the IC50 value shown in Fig. 4, it was found that infection of HRE–SET after radiation and hypoxia significantly increased the sensitivity of cells to GCV (102−103‐fold), which was 55 times as much as SET‐infected cells. The IC50 for GCV in HRE–SET‐infected cells exposed to radiation at normoxia was the same as that of SET‐infected cells.
Figure 4.
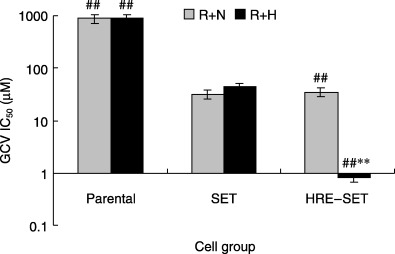
Concentration of ganciclovir (GCV) yielding 50% growth inhibition (IC50) in vitro in parental A549 cells, and in SET‐ or HRE–SET‐infected cells exposed to radiation with normoxia (R + N) or hypoxia (1%) (R + H). Cell viability was assayed by using the trypan blue exclusion test. The IC50 was calculated using curve‐fitting parameters. Versus SET: ##P < 0.01; versus R + N: **P < 0.01.
On the basis of these results, in vitro gene therapy was carried out in various combinations with 2 Gy of radiation, 1% O2 and 10 µM GCV. As shown in Fig. 5, HRE–SET‐infected cells became extremely sensitive to GCV after exposure to radiation and hypoxia compared with SET‐infected cells. Approximately 40% growth suppression was observed in the HRE–SET‐infected cells exposed to radiation and GCV without hypoxia, which was similar to SET‐infected cells. Using the TUNEL assay, increased cell apoptosis rates were found in HRE–SET‐infected cells exposed to radiation and GCV under hypoxia in comparison with normoxia (Table 1). These results demonstrate that the HSV‐tk/GCV system under the control of the HRE–Egr promoter is able to induce an enhanced and hypoxia‐specific cytotoxicity in tumor cells.
Figure 5.
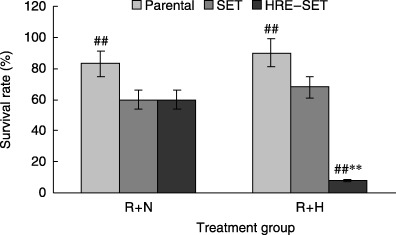
In vitro suicide gene therapy of A549 cells infected with either the SET or HRE–SET retrovirus. Cells infected with the recombinant retroviruses were incubated with 10 µM ganciclovir for 6 days, and exposed to 2 Gy radiation (R) and/or hypoxia (1%) (H) for 8 h previously every 2 days, followed by cell survival analysis. Data are representative of at least five separate experiments. Each bar represents the mean ± SD (n = 5–8).
Table 1.
Degree of apoptosis after treatment was quantitated by counting the percentage of terminal deoxynucleotidyl transferase‐meditated dUTP nick‐end labeling‐positive cells (mean ± SD)
| Parental | SET group | HRE–SET group | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | R | R | G | R + G | R | G | R + G | ||
| In vitro | Normoxia | 3.8 ± 0.6 | 16.2 ± 3.5 | 14.7 ± 3.7 | 19.6 ± 6.2 | 35.7 ± 6.3 | 15.8 ± 3.9 | 19.2 ± 5.1 | 37.6 ± 5.5 |
| Hypoxia | 4.2 ± 0.4 | 11.5 ± 2.1* | 10.0 ± 2.7* | 21.6 ± 3.5 | 29.2 ± 4.8* | 9.3 ± 3.2* | 20.8 ± 4.9 | 77.9 ± 9.5**, †† | |
| In vivo | 5.6 ± 1.2 | 12.7 ± 2.9 | 13.9 ± 3.6 | 15.8 ± 3.9 | 41.7 ± 5.9 | 14.8 ± 3.5 | 17.9 ± 4.1 | 82.8 ± 7.6†† | |
Versus normoxia: *P < 0.05, **P < 0.01; versus SET: † P < 0.05, †† P < 0.01. G, ganciclovir;. R, radiation.
Enhanced tumoricidal activity of the HSV‐tk/GCV system in tumor‐bearing athymic mice
In the present study, CA IX staining (endogenous hypoxia marker) indicated that cells in tumor xenografts were exposed to hypoxia, especially in the central part of xenografts (Fig. 6). Therefore, we analyzed SET or HRE–SET retrovirus‐mediated cytotoxicity to GCV in athymic mice bearing solid tumor xenografts. No significant differences were seen between groups of SET or HRE–SET virus‐infected animals that received radiation without GCV, and animals bearing A549 xenografts exposed to radiation only. Retardation of tumor growth was seen in the HRE–SET‐ and SET‐infected groups receiving GCV without radiation. SET‐infected tumors receiving radiation and GCV grew slowly compared with untreated animals. Only HRE–SET‐infected tumors receiving radiation and GCV were significantly decreased in size, and remained diminished almost 3 weeks after the gene therapy (Fig. 7). These results indicate that the HRE–Egr/HSV‐tk approach induces enhancement of the cytotoxicity of GCV in solid tumors with hypoxic microenvironments.
Figure 6.
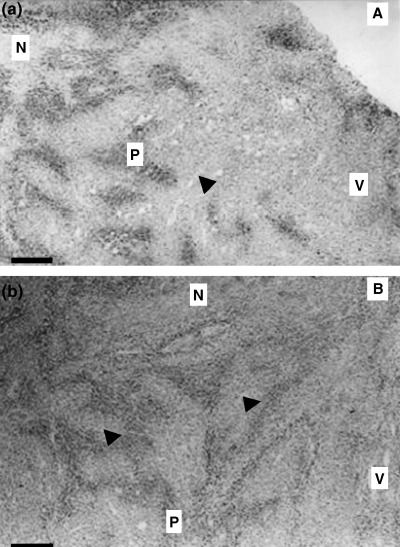
CA IX staining photographs. (a) Peripheral view. (b) Central view. Both slices are shown at ×25 magnification. Scale bar = 40 µm. N, necrosis; P, CA IX positive staining; V, viable, well‐oxygenated tumor tissue. The arrow indicates a blood vessel.
Figure 7.
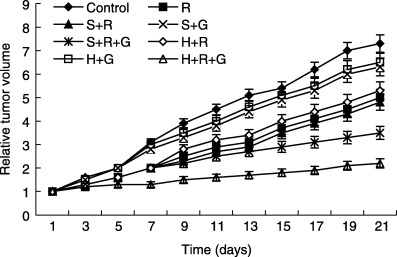
In vivo suicide gene therapy in solid tumor xenografts consisting of A549 cells. The nude mice bearing the subcutaneous tumors were infected with either SET or HRE–SET retroviruses, followed by daily intraperitoneal injection of 20 mg/kg ganciclovir (GCV) for 2 weeks, with exposure to 2 Gy radiation on days 2, 4 and 6. Tumor size was measured every 2 days. Control, A549 cells injection only; H + G, HRE–SET transfection and GCV; H + R, HRE–SET transfection and radiation; H + R + G, HRE–SET transfection, radiation and GCV; R, A549 injection and radiation; S + G, SET transfection and GCV; S + R, SET transfection and radiation; S + R + G, SET transfection, radiation and GCV.
Discussion
We have demonstrated here that the 300‐bp hypoxia enhancer HRE enhance radiation‐inducible transcriptional activity by the Egr‐1 promoter in hypoxic tumor cells. When the hypoxic enhancer region was placed in a sense or reverse‐sense orientation upstream of the expression cassette consisting of the Egr‐1 promoter and the luciferase gene in a retroviral construct, the luciferase expression in A549 cells infected with the retrovirus was enhanced seven‐fold compared with the corresponding cells infected with the same retrovirus without the hypoxic enhancer element when these cells were exposed to radiation and hypoxia. In the present study, we also transfected the HSV‐tk gene under the control of the human Egr‐1 promoter or a hybrid promoter (HRE–Egr) into human lung adenocarcinoma A549 cells using the SET and HRE–SET retroviruses, respectively. Subsequent northern blot analysis revealed that in A549 cells infected with retrovirus, HSV‐tk expression directed by the hypoxic enhancer and the Egr‐1 promoter was markedly higher than that directed only by the Egr‐1 promoter. The HRE–SET infection induced much more significant GCV cytotoxicity compared to SET infection in A549 cells exposed to 1% O2 and 2 Gy radiation, and when mice bearing solid tumors in which A549 cells were infected with HRE–SET were treated by GCV administration and radiation, marked regression of the tumors was observed without any signs of over toxicity.
Several previous studies have used radiation in tumor suicide gene therapy strategies. Kim et al.( 22 ) found that radiation enhanced the sensitivity of a glioma cell line constitutively expressing the HSV‐tk gene to the prodrug 5‐(2‐bromovinyl)‐2′‐deoxyuridine, presumably by synergy with the DNA‐damaging effects of radiation. Radiation‐responsive promoter‐mediated prodrug activation has been demonstrated in a number of in vitro systems including glioma,( 7 ) hepatocellular carcinoma( 23 ) and pancreatic( 8 ) cell lines transfected with plasmids in which the wild‐type Egr‐1 promoter directly controlled HSV‐tk production. Similarly, this promoter has been used to control the growth of tumor xenografts via expression of the tumor necrosis factor (TNF)‐α gene.( 6 , 7 ) However, in most studies the results were achieved using a total dose of 20–50 Gy (sometimes given as multiple 5‐Gy fractions), which was higher than the clinical conventional fraction dose 2 Gy.( 24 ) In the present study, substantial additional tumor cell growth inhibition was achieved by the hybrid promoter after a single exposure to doses as low as 2 Gy. Indeed this dose produced cell growth inhibition equivalent to that of 20 Gy with the Egr‐1 promoter, resulting in an exceptionally high radiation dose modifying factor of approximately 10. Therefore, the strategy presented in the present study can greatly decrease the required radiation dose for cancer treatment. While a suicide gene is likely to elicit the greatest killing effect in the cells that are induced to express it, we expect that more extensive and widespread prodrug‐mediated cell killing would be achieved under in vivo conditions where local (connexin‐mediated)( 25 ) or systemic (immune system‐mediated)( 26 ) bystander effects and cytotoxicity synergies will occur.
Aggressive tumors often have an insufficient blood supply, partly because tumor cells grow faster than the endothelial cells that make up the blood vessels, and partly because the newly formed vascular supply is disorganized. It induces the areas with reduced oxygen tension and deprived of nutrient in the tumor. It has been found that in solid tumors (but not in normal tissue), very low levels of oxygenation or hypoxia protect cells from killing by X‐radiation. We now know that hypoxia in solid tumors is not only a major cause of insensitivity to radiation therapy, but also leads to resistance to most anticancer drugs, and to accelerated malignant progression and increased metastasis.( 27 ) To date, efforts to overcome the hypoxia in tumors have had only limited advancement. Can the low oxygen levels in tumors be turned from a disadvantage to an advantage in cancer treatment?
Recently, several investigators have reported the potential exploitation of tumor‐specific conditions for the targeted expression of therapeutic genes in cancer therapy.( 11 , 14 , 28 , 29 ) Hypoxia is a powerful modulator of gene expression. An important mediator of these responses is the interaction of HIF‐1 with its cognate DNA recognizition site, HRE.( 16 ) Daths et al.( 11 ) fused three copies of HRE from the mouse PGK‐1 gene to the 9–27 promoter to control the expression of the bacterial cytosine deaminase or the marker cell differentiation (CD)2 gene. When HT1080 cells were transfected with the cytosine deaminase or CD 2 gene under the control of the PGK‐1 HRE, these cells were found to be more sensitive to the prodrug 5‐flurocytosine than were the parental cells after exposure to hypoxia, and CD 2 expression was upregulated in a solid tumor xenograft in nude mice. In another study, using a murine fibrosarcoma model, Gazit et al.( 28 ) reported on starvation‐inducible suicide gene therapy under the control of the glucose‐regulated protein 78 promoter. The strategies described in these reports are designed to use tumor‐specific conditions, such as hypoxia or starvation, that exist in almost all solid tumors regardless of their origin or location, to control the expression of heterologous genes. In the approach described in the present study, when the VEGF hypoxia‐inducible enhancer was linked directly to the Egr‐1 promoter, specific enhancement of transfected gene expression was induced in hypoxic A549 cells in vitro and in solid tumor xenografts consisting of A549 cells. Recently, Moeller et al. reported that radiation increased HIF‐1 expression in solid tumors,( 30 ) which might partly contribute to the enhancement of target gene expression in our study.
In conclusion, we believe this type of hybrid promoter (HRE–Egr) has the potential to enhance and lengthen the expression of a therapeutic gene (HSV‐tk). Our strategy will be particularly beneficial in situations where specific but relatively weak radiation‐responsive promoters are used to initiate target gene expression in tumors. The present study also provides an example of overcoming hypoxia‐caused radiation resistance in solid tumors by hypoxia‐enhanced gene expression, and the enhanced expression of HSV‐tk in tumors will therefore lead to more powerful tumoricidal activity and lower required radiation dose.
Acknowledgments
This work was supported by a national natural science fund of China grant (No. 30300097). We thank K. M. Sakamoto for kindly gifting the Egr‐1 promoter, Dr Y. Kurashima for the hypoxia‐inducible enhancer HRE, and Dr Yong Li for retroviral vector pBABE puro SIN.
References
- 1. Kufe D, Weichselbaum R. Radiation therapy: activation for gene transcription and the development of genetic radiotherapy‐therapeutic strategies in oncology. Cancer Biol Ther 2003; 2: 326–9. [DOI] [PubMed] [Google Scholar]
- 2. Datta R, Taneja N, Sukhatme VP et al. Reactive oxygen intermediates target CC (A/T) 6GG sequences to mediate activation of the early growth response 1 transcription factor gene by ionizing radiation. Proc Natl Acad Sci USA 1993; 90: 2419–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xia J, Xia K, Feng Y et al. The combination of suicide gene therapy and radiation enhances the killing of nasopharyngeal carcinoma xenographs. J Radiat Res (Tokyo) 2004; 45: 281–9. [DOI] [PubMed] [Google Scholar]
- 4. Hsu H, Rainov NG, Quinones A et al. Combined radiation and cytochrome CYP4B1/4‐ipomeanol gene therapy using the EGR1 promoter. Anticancer Res 2003; 23: 2723–8. [PubMed] [Google Scholar]
- 5. Hallahan DE, Weichselbaum RR. Role of gene therapy in radiation oncology. Cancer Treat Res 1998; 93: 153–67. [DOI] [PubMed] [Google Scholar]
- 6. Jin GH, Jin SZ, Liu Y et al. Therapeutic effect of gene‐therapy in combination with local X‐irradiation in a mouse malignant melanoma model. Biochem Biophys Res Commun 2005; 330: 975–81. [DOI] [PubMed] [Google Scholar]
- 7. Joki T, Nakamura M, Ohno T. Activation of the radiosensitive EGR‐1 promoter induces expression of the herpes simplex virus thymidine kinase gene and sensitivity of human glioma cells to ganciclovir. Hum Gene Ther 1995; 6: 1507–13. [DOI] [PubMed] [Google Scholar]
- 8. Takahashi T, Namiki Y, Ohno T. Induction of the suicide HSV‐TK gene by activation of the Egr 1 promoter with radioisotopes. Hum Gene Ther 1997; 8: 827–33. [DOI] [PubMed] [Google Scholar]
- 9. Advani SJ, Chmura SJ, WeichseIbaum RR. Radiogenetic therapy: On the interaction of viral therapy and ionizing radiation for improving local control of tumors. Seminars Oncol 1997; 24: 633–8. [PubMed] [Google Scholar]
- 10. Hellwig‐Burgel T, Stiehl DP, Wagner AE et al. Review: hypoxia‐inducible factor‐1 (HIF‐1): a novel transcription factor in immune reactions. J Interferon Cytokine Res 2005; 25: 297–310. [DOI] [PubMed] [Google Scholar]
- 11. Chau NM, Rogers P, Aheme W et al. Identification of novel small molecule inhibitors of hypoxia‐inducible factor‐1 that differentially block hypoxia‐inducible factor‐1 activity and hypoxia‐inducible factor‐1α induction in response to hypoxic stress and growth factors. Cancer Res 2005; 65: 4918–28. [DOI] [PubMed] [Google Scholar]
- 12. Maxwell PH, Hankinson O, Pugh CW et al. The tumour supressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolyses. Nature 1999; 399: 271–5. [DOI] [PubMed] [Google Scholar]
- 13. Firth JD, Ebert BL, Pugh CW et al. Oxygen‐regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3′ enhancer. Proc Natl Acad Sci USA 1994; 91: 6496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Binley K, Iqball S, Kingsman A et al. An adenoviral vector regulated by hypoxia for the treatment of ischaemic disease and cancer. Gene Ther 1999; 6: 1721–7. [DOI] [PubMed] [Google Scholar]
- 15. Modlich U, Paugh CW, Bicknell R. Increasing endothelial cell specific expression by the use of heterologous hypoxic and cytokine‐inducible enhancers. Gene Ther 2000; 7: 896–902. [DOI] [PubMed] [Google Scholar]
- 16. Ido A, Uto H, Moriuchi A et al. Gene therapy targeting for hepatocellular carcinoma: selective and enhanced suicide gene expression regulated by a hypoxia‐inducible enhancer linked to a human α‐fetoprotein promoter. Cancer Res 2001; 61: 3016–21. [PubMed] [Google Scholar]
- 17. Chadderton N, Cowen RL, Sheppard FC et al. Dual responsive promoters to target therapeutic gene expression to radiation‐resistant hypoxic tumor cells. Int J Radiat Oncol Biol Phys 2005; 62: 213–22. [DOI] [PubMed] [Google Scholar]
- 18. Greco O, Marples B, Dachs GU et al. Novel chimeric gene promoters responsive to hypoxia and ionizing radiation. Gene Ther 2002; 9 (20): 1403–11. [DOI] [PubMed] [Google Scholar]
- 19. Aragones J, Jones DR, Martin S et al. Evidence for the involvement of diacylglycerol kinase in the activation of hypoxia‐induced transcription factor 1 by low oxygen tension. J Biol Chem 2001; 276: 10 548–55. [DOI] [PubMed] [Google Scholar]
- 20. Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre vectors with multiple drug selection markers and a complementary helper‐free packaging cell line. Nucl Acids Res 1990; 18: 3587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao G, Kuriyama S, Gao J et al. Gene therapy for hepatocellular carcinoma based on tumor‐selective suicide gene expression using the alpha‐fetoprotein (AFP) enhancer and a housekeeping gene promoter. Euro J Cancer 2001; 37: 140–7. [DOI] [PubMed] [Google Scholar]
- 22. Kim JH, Kim SH, Brown SL et al. Selective enhancement by an antiviral agent of the radiation‐induced cell killing of human glioma cells transduced with HSV‐tk gene. Cancer Res 1994; 54: 6053–6. [PubMed] [Google Scholar]
- 23. Kawashita Y, Ohtsuru A, Kaneda Y et al. Regression of hepatocellular carcinoma in vitro and in vivo by radiosensitizing suicide gene therapy under the inducible and spatial control of radiation. Human Gene Ther 1999; 10: 1509–19. [DOI] [PubMed] [Google Scholar]
- 24. Scott SD, Marples B, Hendry JH et al. A radiation‐controlled molecular switch for use in gene therapy of cancer. Gene Ther 2000; 7: 1121–5. [DOI] [PubMed] [Google Scholar]
- 25. Kanczuga‐Koda L, Sulkowski S, Tomaszewski J et al. Connexins 26 and 43 correlate with Bak, but not with Bcl‐2 protein in breast cancer. Oncol Rep 2005; 14: 325–9. [PubMed] [Google Scholar]
- 26. Stefani Al, Barzon L, Castagliuolo I et al. Systemic efficacy of combined suicide/cytokine gene therapy in a murine model of hepatocellular carcinoma. J Hepatol 2005; 42: 728–35. [DOI] [PubMed] [Google Scholar]
- 27. Yeo EJ, Chun YS, Park JW. New anticancer strategies targeting HIF‐1. Biochem Pharmacol 2004; 68: 1061–9. [DOI] [PubMed] [Google Scholar]
- 28. Gazit G, Hung G, Chen X et al. Use of the glucose starvation‐inducible glucose‐regulated protein 78 Promoter in suicide gene therapy of murine fibrosarcoma. Cancer Res 1999; 59: 3100–6. [PubMed] [Google Scholar]
- 29. Shibata T, Giaccia AJ, Brown JM. Development of a hypoxia‐responsive vector for tumor‐specific gene therapy. Gene Ther 2000; 7: 491–8. [DOI] [PubMed] [Google Scholar]
- 30. Moeller BJ, Cao Y, Li C et al. Upregulation of hypoxia‐inducible factor‐1α following single‐dose radiotherapy: serial in vivo observations. Int J Radiat Oncol Biol Phys 2002; 54 (Suppl. 1): 220–1. [Google Scholar]