

Abstract
The efficacy of epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitors such as gefitinib and erlotinib in non‐small cell lung cancer (NSCLC) is often limited by the emergence of drug resistance conferred either by a secondary T790M mutation of EGFR or by acquired amplification of the MET gene. We now show that the extent of activation of the tyrosine kinase Src is markedly increased in gefitinib‐resistant NSCLC (HCC827 GR) cells with MET amplification compared with that in the gefitinib‐sensitive parental (HCC827) cells. In contrast, the extent of Src activation did not differ between gefitinib‐resistant NSCLC (PC9/ZD) cells harboring the T790M mutation of EGFR and the corresponding gefitinib‐sensitive parental (PC9) cells. This activation of Src in HCC827 GR cells was largely abolished by the MET‐TKI PHA‐665752 but was only partially inhibited by gefitinib, suggesting that Src activation is more dependent on MET signaling than on EGFR signaling in gefitinib‐resistant NSCLC cells with MET amplification. Src inhibitors blocked Akt and Erk signaling pathways, resulting in both suppression of cell growth and induction of apoptosis, in HCC827 GR cells as effectively as did the combination of gefitinib and PHA‐665752. Furthermore, Src inhibitor dasatinib inhibited tumor growth in HCC827 GR xenografts to a significantly greater extent than did treatment with gefitinib alone. These results provide a rationale for clinical targeting of Src in gefitinib‐resistant NSCLC with MET amplification. (Cancer Sci 2009)
Upregulation of the EGFR occurs frequently and is negatively correlated with prognosis in many types of human malignancy.( 1 , 2 ) Recognition of the role of EGFR in carcinogenesis has prompted the development of EGFR‐targeted therapies.( 3 ) TKI of EGFR, such as gefitinib and erlotinib, both of which compete with ATP for binding to the tyrosine kinase pocket of the receptor, have been extensively studied in patients with NSCLC.( 4 ) Sensitivity to these drugs has been correlated with the presence of somatic mutations that affect the kinase domain of EGFR, such as deletions in exon 19 and the L858R mutation in exon 21 of the EGFR gene.( 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 ) However, the acquisition of an additional mutation (T790M) in exon 20 of EGFR results in the development of resistance to EGFR‐TKI.( 16 , 17 , 18 , 19 ) Irreversible EGFR‐TKI are thought to be a potential therapeutic option for overcoming such resistance.( 20 , 21 ) Amplification of the gene for the receptor tyrosine kinase MET has also recently been identified as a mechanism of gefitinib resistance, being detected in 22% of tumor samples from NSCLC patients with EGFR mutations who acquired gefitinib resistance.( 22 , 23 ) Exposure of gefitinib‐resistant NSCLC cells with MET amplification to the MET‐TKI PHA‐665752 or to gefitinib alone did not inhibit cell growth or survival signaling, given that both EGFR and MET signaling were found to be activated and to be mediated by ErbB3 (also known as Her3) in these cells.( 22 , 23 ) However, the combination of gefitinib and PHA‐665752 overcame gefitinib resistance attributable to MET amplification.( 22 , 23 ) No single agent that overcomes such resistance has been identified to date.
The proto‐oncogene SRC has been implicated in the development and poor clinical prognosis of several types of solid tumor as a result of the mediation by its product of signaling between integrins or receptor tyrosine kinases and their downstream effectors.( 24 , 25 , 26 ) We have examined the potential role of Src in EGFR or MET signaling and whether Src inhibitors might block these signaling pathways in gefitinib‐resistant NSCLC cells with MET amplification. We also evaluated the potential antitumor effect of Src inhibitors in order to provide insight into the mechanism by which such inhibitors might overcome gefitinib resistance in NSCLC cells with MET amplification.
Materials and Methods
Cell lines and reagents.
The human NSCLC cell lines H1299, H460, HCC827, HCC827 GR5, HCC827 GR6, and PC9 were obtained as described previously.( 22 , 27 ) H1838 and H820 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). EBC‐1 cells were obtained from the Health Science Research Resources Bank (Tokyo, Japan). PC9/ZD cells were established as a gefitinib‐resistant clone from PC9 cells as previously described( 28 ) and were shown to harbor the T790M mutation of EGFR by both PCR invader and PCR clamp assays carried out as previously described.( 29 , 30 ) HCC827, PC9, and PC9/ZD cells were cultured under a humidified atmosphere of 5% CO2 at 37°C in RPMI‐1640 medium (Sigma, St Louis, MO, USA) supplemented with 10% FBS. HCC827 GR5 and HCC827 GR6 cells were cultured in RPMI‐1640 medium supplemented with 10% FBS and 1 μm gefitinib. Dasatinib was kindly provided by Bristol‐Myers Squibb (New York, NY, USA), gefitinib was obtained from AstraZeneca (Macclesfield, UK), PP1 was from Biomol Research Laboratories (Plymouth Meeting, PA, USA), and PHA‐665752 was from Tocris Bioscience (Bristol, UK).
Immunoblot analysis.
Immunoblot analysis was carried out as described previously.( 27 ) Antibodies to the Y845‐phosphorylated form of EGFR, to EGFR, to phosphorylated Erk, to Erk, to phosphorylated Akt, to Akt, and to β‐actin as well as HRP‐conjugated goat antibodies to mouse or rabbit IgG were obtained as described previously.( 27 ) Antibodies to the Y1234/Y1235‐phosphorylated form of MET, to the Y1289‐phosphorylated form of ErbB3, to the Y416‐phosphorylated form of Src, to Src, and to PARP were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies to MET were from Zymed (South San Francisco, CA, USA) and those to ErbB3 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Immunoprecipitation assay.
Total cell lysates (500 μg protein) were incubated overnight at 4°C with 5 μg of a mouse monoclonal antibody (H‐12) to total Src (Santa Cruz Biotechnology) in a final volume of 200 μL. The immune complexes were precipitated by further incubation for 2 h at 4°C with a suspension of protein G‐ and protein A‐conjugated agarose (Calbiochem, Darmstadt, Germany). The immunoprecipitates were resolved by SDS‐PAGE on a 7.5% gel, and the separated proteins were subjected to immunoblot analysis as described previously,( 27 ) with the exception that the incubation with primary antibodies was carried out for 48 h.
Cell growth inhibition assay.
Cells were plated in 96‐well flat‐bottomed plates and cultured for 24 h before exposure to various concentrations of tested drugs for 72 h. TetraColor One (5 mm tetrazolium monosodium salt and 0.2 mm 1‐methoxy‐5‐methyl phenazinium methylsulfate; Seikagaku, Tokyo, Japan) was then added to each well, and the cells were incubated for 3 h at 37°C before measurement of absorbance at 490 nm with a Multiskan Spectrum instrument (Thermo Labsystems, Boston, MA, USA). Absorbance values were expressed as a percentage of that for untreated cells.
Assessment of tumor growth inhibition in vivo.
Tumor cells (2 × 106) were injected s.c. into the right hind leg of 7‐week‐old female athymic nude mice. The mice were divided into three treatment groups of five animals: those treated over 28 days by oral gavage daily of vehicle, gefitinib (50 mg/kg), or dasatinib (15 mg/kg). Treatment was initiated when tumors in each group achieved an average volume of 200 mm3, with tumor volume being determined twice weekly after the onset of treatment from caliper measurement of tumor length (L) and width (W) according to the formula LW 2/2.
Statistical analysis.
Data are presented as means ± SE as indicated and were analyzed by Student’s t‐test. A P‐value of <0.05 was considered statistically significant.
Results
Src is activated in gefitinib‐resistant NSCLC cells with MET amplification.
Amplification of MET is one mechanism for the acquisition of resistance to EGFR‐TKI in NSCLC.( 22 , 23 ) To explore approaches that might overcome such resistance, we examined the activation status of several signaling molecules in sublines of the gefitinib‐sensitive, EGFR mutation‐positive human NSCLC cell line HCC827 that have acquired MET amplification and gefitinib resistance. Immunoblot analysis revealed that the level of phosphorylation (activation) of both MET and ErbB3 was markedly increased in the HCC827 GR5 and GR6 sublines compared with the parental HCC827 cells (Fig. 1A), consistent with previous observations.( 22 , 23 ) Furthermore, we found that the level of Src activation was also markedly increased in HCC827 GR cells compared with HCC827 cells (Fig. 1A). Such Src activation was not observed in PC9/ZD cells (Fig. 1A), a subline of the gefitinib‐sensitive, EGFR mutation‐positive human NSCLC cell line PC9 that has acquired a secondary T790M mutation of EGFR and consequent gefitinib resistance. These results thus suggested that Src might contribute to gefitinib resistance in NSCLC cells with MET amplification. We also found that H1838, EBC‐1, and H820 NSCLC cells with MET amplification( 31 , 32 , 33 ) have higher activation of Src than that in NSCLC cells without MET amplification (H1299 and H460) (Fig. 1B). These results suggested that Src activation is associated with MET amplification in NSCLC cells.
Figure 1.
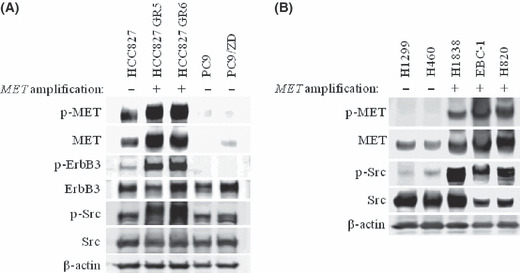
Activation of Src in non‐small cell lung cancer cells with or without MET amplification. (A) HCC827 cells, their gefitinib‐resistant clones with MET amplification (HCC827 GR5 and GR6), PC9 cells, and their gefitinib‐resistant clone with a secondary T790M mutation of epidermal growth factor receptor (PC9/ZD) were incubated for 24 h in medium containing 10% serum. Cell lysates were then prepared and subjected to immunoblot analysis with antibodies to phosphorylated (p‐) or total forms of MET, ErbB3, and Src as well as with those to β‐actin (loading control). (B) H1299 and H460 cells without MET amplification, and H1838, EBC‐1, and H820 cells with MET amplification were incubated for 24 h in medium containing 10% serum. Cell lysates were then prepared and subjected to immunoblot analysis with antibodies to phosphorylated (p‐) or total forms of MET and Src as well as with those to β‐actin (loading control).
Src activation blocked by a MET inhibitor in gefitinib‐resistant NSCLC cells with MET amplification.
Src associates with many receptor tyrosine kinases including EGFR and MET and transduces signals to a variety of downstream effectors of these receptors.( 24 , 25 , 26 , 34 , 35 , 36 ) To examine whether Src participates in MET or EGFR signaling in cells with EGFR mutations and with or without MET amplification, we examined the effects of the MET inhibitor PHA‐665752 or the EGFR‐TKI gefitinib on Src activation in HCC827 and HCC827 GR cells. In the parental HCC827 cells, Src activity (phosphorylation) was reduced by PHA‐665752 and was abolished by gefitinib (Fig. 2A). In contrast, Src activation was partially reduced by PHA‐665752 and was inhibited to a much lesser extent by gefitinib in HCC827 GR5 cells (Fig. 2A). Combined treatment with gefitinib and PHA‐665752 resulted in complete suppression of Src activation in both the parental and GR cells (Fig. 2A). These results suggested that Src activation is dependent on MET signaling to a greater extent than on EGFR signaling in gefitinib‐resistant cells with MET amplification, whereas the opposite is the case for cells without MET amplification.
Figure 2.
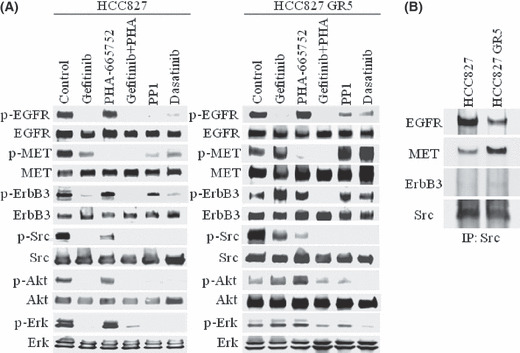
Effects of various inhibitors on epidermal growth factor receptor (EGFR) and MET signaling in gefitinib‐resistant non‐small cell lung cancer cells with MET amplification. (A) HCC827 cells and a gefitinib‐resistant clone with MET amplification (HCC827 GR5) were incubated for 12 h in the absence (control) or presence of gefitinib alone (1 μm), PHA‐665752 alone (1 μm), gefitinib and PHA‐665752 combined, PP1 (10 μm), or dasatinib (500 nm) in medium containing 10% serum. Cell lysates were then subjected to immunoblot analysis with antibodies to phosphorylated (p‐) or total forms of EGFR, MET, ErbB3, Src, Akt, and Erk. (B) HCC827 and HCC827 GR5 cells were incubated for 24 h in medium containing 10% serum, lysed, and subjected to immunoprecipitation (IP) with an antibody to Src. The resulting precipitates were subjected to immunoblot analysis with antibodies to EGFR, MET, ErbB3, and Src.
Effects of Src inhibitors on EGFR, ErbB3, and MET activation in gefitinib‐resistant NSCLC cells with MET amplification.
Src activates EGFR by phosphorylating the Y845 residue of the receptor,( 37 ) and it also interacts with MET.( 35 ) We therefore examined the effects of the Src inhibitors PP1 and dasatinib on EGFR, ErbB3, and MET activation. Both PP1 and dasatinib abolished EGFR activation and inhibited ErbB3 and MET activation in parental HCC827 cells (Fig. 2A). In contrast, these Src inhibitors did not suppress ErbB3 or MET activation and induced only partial inhibition of EGFR activation in HCC827 GR5 cells (Fig. 2A), suggesting that MET amplification affects the interactions of EGFR, ErbB3, and MET with Src.
Increased association between MET and Src in gefitinib‐resistant NSCLC cells with MET amplification.
We examined the effects of MET amplification on the physical association of Src with EGFR, MET, and ErbB3. Src was immunoprecipitated from both HCC827 and HCC827 GR5 cell lysates, and the resulting precipitates were subjected to immunoblot analysis with antibodies to EGFR, MET, ErbB3, or Src. The amount of MET associated with Src was greater for HCC827 GR5 cells than for HCC827 cells, whereas the amount of EGFR associated with Src was greater for HCC827 cells than for HCC827 GR5 cells (Fig. 2B). No association of ErbB3 with Src was apparent for either cell type. These results suggested that MET amplification results in an increase in the association between MET and Src, as well as a concomitant decrease in that between EGFR and Src, in HCC827 GR cells.
Src inhibitors block Akt and Erk signaling in gefitinib‐resistant NSCLC cells with MET amplification.
We next examined the effects of the Src inhibitors PP1 and dasatinib on Akt and Erk signaling pathways, both of which are activated by EGFR and MET. Both PP1 and dasatinib induced complete inhibition of Akt and Erk activation, as did gefitinib, in the parental HCC827 cells (Fig. 2A). Consistent with previous observations,( 22 , 23 ) the combination of gefitinib and PHA‐665752 inhibited Akt and Erk activation in HCC827 GR5 cells, whereas neither agent alone had such an effect (Fig. 2A). Both PP1 and dasatinib inhibited Akt and Erk activation to similar extents as the combination of gefitinib and PHA‐665752 in HCC827 GR5 cells (Fig. 2A). A single agent (Src inhibitor) was thus sufficient to block Akt and Erk signaling, which is important for cell survival and proliferation, respectively, in gefitinib‐resistant NSCLC cells with MET amplification.
Src inhibitor dasatinib suppresses growth of gefitinib‐resistant NSCLC cells with MET amplification.
The combination of gefitinib and PHA‐665752 was recently shown to inhibit the growth of, and to induce apoptosis in, HCC827 GR cells with MET amplification, whereas neither agent alone had such an effect.( 22 , 23 ) Given that we found that Src inhibitors block Akt and Erk signaling pathways as effectively as the combination of gefitinib and PHA‐665752 in such cells, we examined whether dasatinib might overcome gefitinib resistance in HCC827 GR cells. In the parental HCC827 cells, both gefitinib and dasatinib as well as the combination of gefitinib and PHA‐665752 effectively inhibited cell growth, but PHA‐665752 alone had less inhibitory effect (Fig. 3A). Dasatinib inhibited cell growth in a concentration‐dependent manner by the same marked extent as the combination of gefitinib and PHA‐665752, even in HCC827 GR5 cells, whereas neither gefitinib nor PHA‐665752 alone had a substantial effect (Fig. 3B). We also examined the effect of dasatinib on apoptosis as assessed on the basis of cleavage of the enzyme PARP in both HCC827 and HCC827 GR5 cells. Dasatinib (but not gefitinib) induced apoptosis in HCC827 GR5 cells to the same marked extent as did the combination of gefitinib and PHA‐665752, whereas dasatinib and gefitinib each induced apoptosis in the parental HCC827 cells (Fig. 3C). These results suggested that Src inhibitors efficiently induce growth inhibition and apoptosis in gefitinib‐resistant NSCLC cells with MET amplification.
Figure 3.
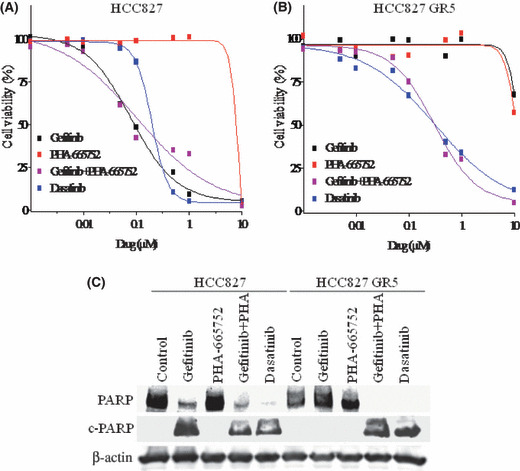
Effects of dasatinib on growth and apoptosis in gefitinib‐resistant non‐small cell lung cancer cells with MET amplification. (A) HCC827 cells or (B) HCC827 GR5 cells were treated for 72 h with increasing concentrations of gefitinib alone, PHA‐665752 alone, gefitinib and PHA‐665752 in combination, or dasatinib alone in medium containing 10% serum, after which cell viability was assessed. Data are means of triplicates from a representative experiment and are expressed as a percentage of the value for untreated cells. (C) HCC827 and HCC827 GR5 cells were incubated for 72 h with gefitinib (1 μm) alone, PHA‐665752 (1 μm) alone, gefitinib plus PHA‐665752, or dasatinib (1 μm) in medium containing 10% serum. Cell lysates were then prepared and subjected to immunoblot analysis with antibodies to poly(ADP‐ribose) polymerase (PARP) and to β‐actin. The positions of intact PARP (116 kDa) and the 85‐kDa cleavage fragment (c‐PARP) are shown.
Src inhibitor dasatinib inhibits tumor growth in gefitinib‐resistant NSCLC xenografts with MET amplification.
To determine whether the efficacy of dasatinib in gefitinib‐resistant NSCLC cells with MET amplification observed in vitro might also be apparent in vivo, we examined the antitumor effects of dasatinib in nude mice with solid tumors formed by HCC827 GR5 cells injected into the right hind leg. Gefitinib (50 mg/kg) could not reduce tumor size compared with vehicle treatment (Fig. 4). In contrast, dasatinib (15 mg/kg) inhibited tumor growth in HCC827 GR5 xenografts to a significantly greater extent than did treatment with gefitinib or vehicle alone (Fig. 4). These results indicated that Src inhibitor effectively exerts antitumor effects in gefitinib‐resistant NSCLC xenografts with MET amplification.
Figure 4.
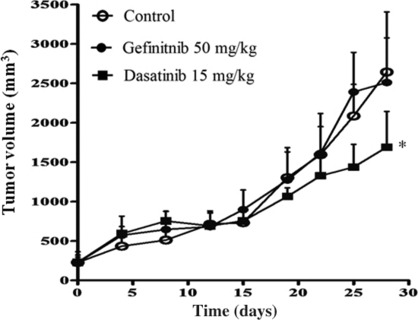
Effects of dasatinib on the growth of gefitinib‐resistant non‐small cell lung cancer cells with MET amplification in vivo. Nude mice with tumor xenografts established by s.c. implantation of HCC827 GR5 cells were treated daily for 28 days with vehicle (control), gefitinib (50 mg/kg), or dasatinib (15 mg/kg) by oral gavage. Tumor volume was determined at the indicated times after the onset of treatment. Points indicate the mean of values from five mice per group; bars indicate SE. *P < 0.05 for dasatinib versus control or gefitinib alone (Student’s t‐test).
Discussion
The emergence of MET amplification induces ErbB3‐dependent downstream signaling mediated by Akt and Erk that is important for cell survival and proliferation, ultimately leading to the development of gefitinib resistance, in NSCLC cells with EGFR mutations.( 22 , 23 ) Although the combination of the specific MET inhibitor PHA‐665752 and gefitinib is considered promising for overcoming gefitinib resistance due to MET amplification, a single‐agent therapy to overcome such resistance would be more desirable.( 22 , 23 ) We have shown that, in addition to MET activation, Src is markedly activated in NSCLC cells with MET amplification, including HCC827 GR cells. Forced expression of Src has previously been shown to result in gefitinib resistance in gallbladder adenocarcinoma cells( 38 ) and to promote tumorigenesis in EGFR‐overexpressing mammary epithelial cells.( 39 ) In addition, MET and Src cooperate to mediate proliferation of breast cancer cells in the presence of EGFR‐TKI.( 34 ) Consistent with these previous observations, our results now suggest that Src contributes to gefitinib resistance in NSCLC cells with MET amplification and is a potential target molecule for overcoming such resistance.
To explore how Src activation affects MET or EGFR signaling in gefitinib‐resistant NSCLC cells with MET amplification, we examined the effects of Src inhibitors on EGFR, ErbB3, and MET activation in both HCC827 and HCC827 GR5 cells. Gefitinib was previously shown to inhibit ErbB3 and MET activation as well as EGFR activation in the parental HCC827 cells,( 22 , 23 , 40 ) suggestive of a functional interaction between EGFR and both ErbB3 and MET in EGFR‐mutant NSCLC cells without MET amplification (Fig. 5A). In contrast, gefitinib did not inhibit ErbB3 or MET activation in HCC827 GR cells, with the combination of gefitinib and PHA‐665752 being necessary to achieve inhibition of ErbB3 activation in these cells with MET amplification.( 22 , 23 ) In addition, endogenous ErbB3 was co‐immunoprecipitated with MET from HCC827 GR cells but not from HCC827 cells.( 22 , 23 ) These previous results thus suggested that ErbB3 signaling becomes more dependent on MET than on EGFR after emergence of MET amplification, and that the MET‐ErbB3 signaling complex is largely independent of EGFR signaling (Fig. 5B).( 22 , 23 ) We have shown that Src inhibitors reduced the extent of EGFR activation in both HCC827 and HCC827 GR5 cells, consistent with previous observations showing that Src mediates EGFR activation by phosphorylating its Y845 residue.( 37 , 41 ) In HCC827 GR5 cells, however, Src inhibitors did not inhibit ErbB3 or MET activation, despite it doing so in the parental HCC827 cells. These results support the notion that MET signaling is independent of EGFR signaling as a result of the shift of the dependence of ErbB3 signaling from EGFR to MET in HCC827 GR cells (Fig. 5B).( 22 , 23 )
Figure 5.
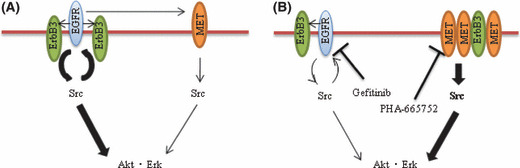
Models for signaling pathways in gefitinib‐sensitive non‐small cell lung cancer (NSCLC) cells (A) and gefitinib‐resistant NSCLC cells with acquired MET amplification (B). Src functions downstream of both epidermal growth factor receptor (EGFR) and MET as well as upstream of Akt and Erk signaling pathways and EGFR. However, the dependency of Src signaling is shifted from EGFR to MET and MET associates with ErbB3 after the acquisition of MET amplification. EGFR mediates, at least in part, activation of MET in gefitinib‐sensitive NSCLC cells, whereas EGFR and MET function independently of each other in gefitinib‐resistant NSCLC cells with acquired MET amplification. Pathways targeted by gefitinib or PHA‐665752 are indicated, and the relative activities of signaling pathways are denoted by the width of the arrows.
We examined whether MET amplification affects the physical association between Src and either EGFR, MET, or ErbB3 by immunoprecipitation. The association between MET and Src was increased in HCC827 GR5 cells compared with that in HCC827 cells, whereas the association between EGFR and Src was reduced in HCC827 GR5 cells. These findings are consistent with our results showing that PHA‐665752 blocks Src activation to a greater extent in HCC827 GR5 cells than in HCC827 cells, a pattern opposite to that for the effects of gefitinib (Fig. 5). The mechanism of increased association between MET and Src induced by acquired MET amplification has remained unclear. It is possible that MET amplification alters the protein expression which mediates binding of Src to MET. On the basis of the notion that Src is activated downstream of MET signaling in HCC827 GR cells, we examined the effects of Src inhibitors in these cells on Akt and Erk signaling pathways, both of which are known to be activated by Src.( 24 , 25 , 26 , 42 ) We have shown that Src inhibitors markedly inhibited Akt and Erk signaling pathways in gefitinib‐resistant NSCLC cells with MET amplification. Previous studies found that neither gefitinib nor PHA‐665752 alone blocked Akt or Erk pathways in HCC827 GR cells,( 22 , 23 ) with the combination of both of these agents being necessary for such inhibition, consistent with the notion that Akt and Erk pathways are dependent on both EGFR and MET signaling in these cells (Fig. 5B). We observed that gefitinib and PHA‐665752 each induced a slight increase in the phosphorylation levels of Akt in HCC827 GR5 cells (Fig. 2A), possibly because EGFR and MET pathways functionally compensate for each other when either is inhibited. Our results suggest that Src functions downstream of both EGFR and MET, but that it is mainly dependent on MET signaling in HCC827 GR cells. Together, our observations explain the ability of Src inhibitors to suppress Akt and Erk activation in gefitinib‐resistant NSCLC cells with MET amplification (Fig. 5B).
Finally, we found that Src inhibitor dasatinib also inhibited the growth of HCC827 GR5 cells as well as did combined treatment with gefitinib and PHA‐665752. HCC827 GR5 cells underwent apoptosis, as detected by PARP cleavage, after treatment with dasatinib. Furthermore, dasatinib inhibited tumor growth in HCC827 GR5 xenografts to a significantly greater extent than treatment with gefitinib alone. Our present data suggest that Src inhibitors might overcome gefitinib resistance in NSCLC patients with MET amplification. Our findings strengthen the rationale of the ongoing clinical trial of dasatinib for NSCLC patients who no longer respond to erlotinib or gefitinib (http://www.clinicaltrials.gov). The results of this clinical trial should provide insight into the relation between the efficacy of Src inhibitors and whether gefitinib resistance is attributable to the secondary T790M mutation of EGFR or to acquired MET amplification.
Abbreviations
- EGFR
epidermal growth factor receptor
- NSCLC
non‐small cell lung cancer
- PARP
poly(ADP‐ribose) polymerase
- TKI
tyrosine kinase inhibitor
References
- 1. Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull 1991; 47: 87–98. [DOI] [PubMed] [Google Scholar]
- 2. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor‐related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995; 19: 183–232. [DOI] [PubMed] [Google Scholar]
- 3. Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer 2004; 11: 689–708. [DOI] [PubMed] [Google Scholar]
- 4. Ettinger DS. Clinical implications of EGFR expression in the development and progression of solid tumors: focus on non‐small cell lung cancer. Oncologist 2006; 11: 358–73. [DOI] [PubMed] [Google Scholar]
- 5. Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 6. Paez JG, Janne PA, Lee JC et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 7. Pao W, Miller V, Zakowski M et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mitsudomi T, Kosaka T, Endoh H et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non‐small‐cell lung cancer with postoperative recurrence. J Clin Oncol 2005; 23: 2513–20. [DOI] [PubMed] [Google Scholar]
- 9. Takano T, Ohe Y, Sakamoto H et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non‐small‐cell lung cancer. J Clin Oncol 2005; 23: 6829–37. [DOI] [PubMed] [Google Scholar]
- 10. Han SW, Kim TY, Hwang PG et al. Predictive and prognostic impact of epidermal growth factor receptor mutation in non‐small‐cell lung cancer patients treated with gefitinib. J Clin Oncol 2005; 23: 2493–501. [DOI] [PubMed] [Google Scholar]
- 11. Tsao MS, Sakurada A, Cutz JC et al. Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med 2005; 353: 133–44. [DOI] [PubMed] [Google Scholar]
- 12. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004; 64: 8919–23. [DOI] [PubMed] [Google Scholar]
- 13. Shigematsu H, Lin L, Takahashi T et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97: 339–46. [DOI] [PubMed] [Google Scholar]
- 14. Tokumo M, Toyooka S, Kiura K et al. The relationship between epidermal growth factor receptor mutations and clinicopathologic features in non‐small cell lung cancers. Clin Cancer Res 2005; 11: 1167–73. [PubMed] [Google Scholar]
- 15. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–81. [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi S, Boggon TJ, Dayaram T et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 17. Pao W, Miller VA, Politi KA et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kosaka T, Yatabe Y, Endoh H et al. Analysis of epidermal growth factor receptor gene mutation in patients with non‐small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res 2006; 12: 5764–9. [DOI] [PubMed] [Google Scholar]
- 19. Yun CH, Mengwasser KE, Toms AV et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008; 105: 2070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kwak EL, Sordella R, Bell DW et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 2005; 102: 7665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Engelman JA, Mukohara T, Zejnullahu K et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR‐amplified lung cancer. J Clin Invest 2006; 116: 2695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Engelman JA, Zejnullahu K, Mitsudomi T et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 23. Arteaga CL. HER3 and mutant EGFR meet MET. Nat Med 2007; 13: 675–7. [DOI] [PubMed] [Google Scholar]
- 24. Alvarez RH, Kantarjian HM, Cortes JE. The role of Src in solid and hematologic malignancies: development of new‐generation Src inhibitors. Cancer 2006; 107: 1918–29. [DOI] [PubMed] [Google Scholar]
- 25. Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev 2003; 22: 337–58. [DOI] [PubMed] [Google Scholar]
- 26. Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res 2006; 12: 1398–401. [DOI] [PubMed] [Google Scholar]
- 27. Okabe T, Okamoto I, Tamura K et al. Differential constitutive activation of the epidermal growth factor receptor in non‐small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res 2007; 67: 2046–53. [DOI] [PubMed] [Google Scholar]
- 28. Koizumi F, Shimoyama T, Taguchi F, Saijo N, Nishio K. Establishment of a human non‐small cell lung cancer cell line resistant to gefitinib. Int J Cancer 2005; 116: 36–44. [DOI] [PubMed] [Google Scholar]
- 29. Tadokoro K, Kobayashi M, Yamaguchi T et al. Classification of hepatitis B virus genotypes by the PCR‐Invader method with genotype‐specific probes. J Virol Methods 2006; 138: 30–9. [DOI] [PubMed] [Google Scholar]
- 30. Nagai Y, Miyazawa H, Huqun et al. Genetic heterogeneity of the epidermal growth factor receptor in non‐small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid‐locked nucleic acid PCR clamp. Cancer Res 2005; 65: 7276–82. [DOI] [PubMed] [Google Scholar]
- 31. Jagadeeswaran R, Surawska H, Krishnaswamy S et al. Paxillin is a target for somatic mutations in lung cancer: implications for cell growth and invasion. Cancer Res 2008; 68: 132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lutterbach B, Zeng Q, Davis LJ et al. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res 2007; 67: 2081–8. [DOI] [PubMed] [Google Scholar]
- 33. Bean J, Brennan C, Shih JY et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007; 104: 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mueller KL, Hunter LA, Ethier SP, Boerner JL. Met and c‐Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res 2008; 68: 3314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bolanos‐Garcia VM. MET meet adaptors: functional and structural implications in downstream signalling mediated by the Met receptor. Mol Cell Biochem 2005; 276: 149–57. [DOI] [PubMed] [Google Scholar]
- 36. Mao W, Irby R, Coppola D et al. Activation of c‐Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 1997; 15: 3083–90. [DOI] [PubMed] [Google Scholar]
- 37. Sato K, Sato A, Aoto M, Fukami Y. c‐Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem Biophys Res Commun 1995; 215: 1078–87. [DOI] [PubMed] [Google Scholar]
- 38. Qin B, Ariyama H, Baba E et al. Activated Src and Ras induce gefitinib resistance by activation of signaling pathways downstream of epidermal growth factor receptor in human gallbladder adenocarcinoma cells. Cancer Chemother Pharmacol 2006; 58: 577–84. [DOI] [PubMed] [Google Scholar]
- 39. Dimri M, Naramura M, Duan L et al. Modeling breast cancer‐associated c‐Src and EGFR overexpression in human MECs: c‐Src and EGFR cooperatively promote aberrant three‐dimensional acinar structure and invasive behavior. Cancer Res 2007; 67: 4164–72. [DOI] [PubMed] [Google Scholar]
- 40. Guo A, Villen J, Kornhauser J et al. Signaling networks assembled by oncogenic EGFR and c‐Met. Proc Natl Acad Sci U S A 2008; 105: 692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang J, Kalyankrishna S, Wislez M et al. SRC‐family kinases are activated in non‐small cell lung cancer and promote the survival of epidermal growth factor receptor‐dependent cell lines. Am J Pathol 2007; 170: 366–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS‐354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res 2006; 66: 5542–8. [DOI] [PubMed] [Google Scholar]