

Abstract
The HSulf‐1 gene encodes an extracellular 6‐O‐endosulfatase and regulates the sulfation status of heparan sulfate proteoglycans (HSPG). We have demonstrated that promoter hypermethylation is correlated with the HSulf‐1 silencing in gastric cancer. To investigate the functional importance of HSulf‐1 silencing in gastric cancer, we restored HSulf‐1 expression in the gastric cancer cell line MKN28, which lacks endogenous HSulf‐1. Following restoration of expression, HSulf‐1 inhibited cell proliferation, motility, and invasion in vitro, as well as significantly suppressing the MKN28 xenograft model (P < 0.05). No noticeable changes in proliferation and motility were observed following restoration of HSulf‐1 in another gastric cancer cell line, namely AGS cells. Interestingly, in MKN28 cells, which have been reported to be dependent on extracellular Wnt signaling, we found that HSulf‐1 inhibited the transcriptional activity of the Wnt/β‐catenin pathway and downregulated its targeted genes. Conversely, in AGS cells, in the constitutive Wnt/β‐catenin pathway is active, HSulf‐1 had no effect on the activity of the Wnt/β‐catenin pathway. Furthermore, transfection of Wnt3a cDNA or β‐catenin shRNA resulted in rescue or enhancement, respectively, of the effects of HSulf‐1 in MKN28 cells. Furthermore, HSPG epitope analysis confirmed that HSulf‐1 affected the structure of heparan sulfate on the cell surface. Together, the results of the present study suggest that extracellular HSulf‐1 may function as a negative regulator of proliferation and invasion in gastric cancer by suppressing Wnt/β‐catenin signaling at the cell surface. (Cancer Sci 2011; 102: 1815–1821)
Gastric cancer is the second most common cause of cancer‐related deaths worldwide.( 1 , 2 ) Understanding the mechanisms involved in gastric tumorigenesis and metastasis is important for the development of new effective therapeutic agents.
The HSulf‐1 gene, characterized as Human ortholog of Qsulf‐1, can hydrolyze the sulfate ester bonds of heparan sulfate proteoglycans (HSPG), leading to removal of the sulfate at the 6‐O position of glucosamine.( 3 , 4 ) It is believed that changes in the sulfation status of HSPG can affect their interactions with signaling molecules and therefore modulate signal transduction.( 3 , 5 , 6 , 7 , 8 ) Recent evidence indicates that HSulf‐1 is downregulated in several types of cancer, including hepatocellular carcinoma (HCC), ovarian and breast cancer.( 5 , 7 ) When HSulf‐1‐transfected myeloma cells are implanted into SCID mice, the rate of tumor growth is significantly reduced.( 9 ) In addition, HSulf‐1 has been shown to enhance the suppression of tumorigenesis by histone deacetylase inhibitors in HCC.( 10 ) It has also been reported that HSulf‐1 suppresses carcinogenesis and angiogenesis by inhibiting the activation of the heparin‐binding growth factor pathway, including fibroblast growth factor (FGF)‐2, hepatocyte growth factor (HGF), epidermal growth factor (EGF),( 5 , 6 , 7 , 9 , 11 ) and vascular endothelial growth factor (VEGF) signaling.( 8 ) It has also been documented that the HSPG, which are coreceptors for cytokines,( 12 , 13 ) are required for Wnt‐dependent regulation in Drosophila, Xenopus laevis, and mammals.( 14 , 15 , 16 ) However, a role has been reported for HSulf‐1 as a positive regulator of the Wnt signaling pathway, promoting tumor growth, in pancreatic cancer.( 17 ) Therefore, the possible relationship between HSulf‐1 and Wnt signaling requires further exploration.
The Wnt signaling pathway plays a critical role in cell fate determination, tissue development, and cancer.( 18 , 19 , 20 ) Secreted Wnt proteins bind to Frizzled and lipoprotein receptor 5/6 (LPR 5/6), resulting in the stabilization of β‐catenin via inhibition of phosphorylation‐dependent degradation.( 19 , 20 ) The stabilized β‐catenin accumulates in the cytoplasm and further translocates to the nucleus, interacting with transcription factors in the TCF/LEF family to activate target genes.( 21 ) Accumulating evidence indicates a close correlation between the Wnt signaling pathway and the initiation and progression of gastric cancer.( 22 , 23 )
We have recently demonstrated downregulation of HSulf‐1 in human gastric cancer.( 24 ) To further evaluate the role of HSulf‐1 in the tumorigenesis and metastasis of gastric cancer, in the present study we restored HSulf‐1 expression in gastric cancer cell lines that lack endogenous HSulf‐1 expression and investigated its effects on the growth rate and invasiveness of the cells in vitro and in vivo. The results suggest that HSulf‐1 may inhibit tumor growth and invasion by suppressing canonical Wnt signaling in gastric cancer.
Materials and Methods
Constructs. The HSulf‐1 expression plasmid (Plasmid 13003) was purchased from Addgene (Cambridge, MA, USA). The TOPflash and FOPflash reporter plasmids containing wild‐type and mutant TCF/LEF binding sites, respectively, were kindly provided by Professor Yeguang Chen (Tsinghua University, Beijing, China). The CTNNB1 shRNA plasmid was purchased from Origene (Rockville, MD, USA). The targeted CTNNB1 gene sequence used in the present study was 5′‐GGTCCTCTGTGAACTTGCTCAGGACAAGG‐3′. The scrambled or target hairpin oligonucleotides were subcloned into pSliencer, provided by Professor Peng Li (Tsinghua University). The DKK1 and mWnt3a genes were subcloned into pCDNA3.1/myc‐his vectors (Invitrogen, Carlsbad, CA, USA). Transfections were performed using Lipofectamine 2000 (Invitrogen).
Cell culture. The human gastric cancer cell lines used in the present study (AGS and MKN28) were obtained from the China Center for Type Culture Collection (Wuhan, China). The MKN28 and AGS cells stably expressing HSulf‐1 or empty vector were selected with 0.5 and 1 mg/mL G418 (Invitrogen), respectively. Stable, pooled populations of individual clones were verified by immunoblotting analysis for HSulf‐1. Cells were maintained at 37°C in a humidified incubator with 5% CO2.
RNA extraction and semiquantitative RT‐PCR. RNA extraction and semiquantitative RT‐PCR were performed as described previously.( 24 ) The sequences of the primer pairs used in the present study are listed in Table 1.
Table 1.
Nucleotide sequences of the primers used in RT‐PCR
| Gene | Primers | |
|---|---|---|
| Forward | Reverse | |
| 18S rRNA | CAGCCACCCGAGATTGAGCA | TAGTAGCGACGGGCGGTGTG |
| Wnt3a | AAGCAGGCTCTGGGCAGCTA | GACGGTGGTGCAGTTCCA |
| Dvl1 | CATCCTTCCACCCTAATGTGTCC | TAAAGCCCGGGTCCTGGTAGGC |
| Dvl2 | CATCCTTCCACCCTAATGTGTCC | GTCCCCCAGGCTGGTACTCT |
| Dvl3 | CACGTGGTTGCTTCACATTGC | GACAAGTGGAAGTCGTCTAGG |
| Fzd2 | GCACTACACGCCGCGCATGTC | CCCACCCCGGGCGGAGGAAAG |
| Fzd3 | GTGAGTGTTCGAAGCTCATGG | ATCACGCACATGCAGAAAAG |
| Fzd5 | GACTGTCTGCTCTTCTCGGC | GACGCACACAGGCAGAGGAA |
| Fzd6 | ACTCTTGCCACTGTGCCTTTG | GTCGAGCTTTTGCTTTTGCCT |
| Fzd7 | GCCTCGACGCTCTTTACC | GAGGCCAACGTAGCACACC |
| HSulf‐1 | ACTGTACCAATCGGCCAGAG | CCTCCTTGAATGGGTGAAGA |
| Axin‐2 | GCAGACGACGAAGCATGTC | GCCTTTCCCATTGCGTTTGG |
| c‐Myc | TTCGGGTAGTGGAAAACCAG | CAGCAGCTCGAATTTCTTCC |
| CCND1 | AGCTCCTGTGCTGCGAAGTGGAAAC | AGTGTTCAATGAAATCGTGCGGGGT |
| DKK4 | AGCTCTGGTCCTGGACTTCA | CAACCCACGACATGTAGCAC |
| MMP‐2 | ATCATGATCAACTTTGGCCGCT | CAGCTGTTGTACTCCTTGCCAT |
| S100A4 | AGCTGATGAGCAACTTGGACAG | CATCAAGCACGTGTCTGAAGGA |
| S100P | CAGGAGGAAGGTGGGTCTGAA | TGTGACAGGCAGACGTGATTG |
Immunoblotting. The stable cell lines were cultured to confluence. Cells were then trypsinized, pelleted by centrifugation at 500g for 5 min. To obtain total cell lysates, cell pellets were resuspended in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X‐100, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate) and then vortexed for 5 s. After incubation on ice for 15 min, the suspension was centrifuged at 20,000g at 4°C for 5 min and the supernatant was collected for immunoblotting.
The cytosolic protein fraction was obtained as follows. Cell pellets were suspended in hypotonic buffer (20 mM Tris‐HCl, pH 7.5, 25 mM NaF, 1 mM EDTA) and put on ice for 30 min. The cell suspension was centrifuged in an ultracentrifuge (Optima MAX; Beckman Coulter, Fullerton, CA, USA) at 124,500g for 30 min at 4°C. The supernatant was collected as the cytosolic fraction.
Both total protein (20 μg) and cytosolic protein extracts (40 μg) were subjected to immunoblotting. The primary antibodies used were anti‐β‐actin (1:5000; Sigma‐Aldrich, St Louis, MO, USA), anti‐β‐catenin (1:2000; Sigma‐Aldrich), anti‐cyclin D1 (1:500; BD Biosciences, San Jose, CA, USA), anti‐c‐Myc (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐Dvl‐3 (1:200; Santa Cruz Biotechnology), and anti‐HSulf‐1 (1:250; Abnova, Taipei, Taiwan).
Cell proliferation assay. Cell growth rates were determined using the CellTiter 96 Aqueous Proliferation Assay Kit (also called MTS proliferation assay [Promega, Madison, WI, USA]). Stable cell lines were plated into 96‐well culture plates at a density of 5 × 102 cells/well. After the cells had been cultured for 0, 2, 3, and 4 days, the MTS solution was added to the culture medium and cells were incubated for a further 1.5 h. Absorbance was measured at 490 nm using a microplate reader (Model 680; Bio‐Rad, Hercules, CA, USA).
Colony formation and soft agar assays. Stable cells (1 × 103) were seeded in triplicate into 100‐mm dishes. The culture medium, containing G418, was replaced every 3–4 days. After cells had been cultured for 2–3 weeks, they were fixed with 4% paraformaldehyde for 10 min, stained with 0.1% crystal violet for 20 min, and then photographed.
The soft agar assay was performed in six‐well plates in triplicate using cells plated at a density of 1 × 104 cells/well. Cells were resuspended in medium with 0.35% agarose (Sigma‐Aldrich) and seeded onto plates precoated with 0.5% base agarose. Cells were then cultured for 2–3 weeks. The colonies were counted manually after cells had been stained with 0.005% crystal violet.
Analysis of HSPG epitopes. Stable cells (1 × 105) were harvested, washed twice with PBS, and then incubated at 4°C for 45 min with mouse monoclonal anti‐heparan sulfate (HS) antibody 10E4 (Seikagaku America, Falmouth, MA, USA), or IgM (Sigma‐Aldrich) as a control, at a dilution of 1:50. After cells had been washed twice with PBS, they were incubated in PBS containing FITC‐conjugated rabbit anti‐mouse Ig (DakoCytomation, Carpinteria, CA, USA) at a dilution of 1:50 for 30 min. Cells were the washed twice with PBS, fixed in 2% paraformaldehyde, and then analyzed by flow cytometry (FACScan; BD Biosciences).
Luciferase assay. Cells were seeded into 24‐well plates at a density of 5 × 103 cells/well prior to transfection. Cells were then transfected with the TOPflash or FOPflash plasmid in addition to the PRL‐TK plasmid (as an internal control; Promega). After a further 18 h culture, luciferase activity in the cells was measured using the Dual‐Glo Luciferase Assay (Promega).
Wound healing assay. Stable cell lines were seeded in six‐well plates and cultured until they reached confluence. Wounds were scratched on the monolayer of cells using 200‐μL pipette tips. The images of the wounds were photographed using an Olympus IX71 inverted microscope (Olympus Japan, Tokyo, Japan) after the cells had been cultured for 0, 24, and 48 h and wound size was measured.
Transwell migration and invasion assay. For the migration assay, 5 × 104 cells were suspended in serum‐free DMEM and plated on chambers (8‐μm pore size; Corning Costar, Cambridge, MA, USA) that were not coated with matrigel. For the invasion assay, the upper chamber was precoated with 5 mg/mL matrigel (Sigma‐Aldrich) before 5 × 105 cells in serum‐free DMEM were added to the chamber. For both assays, medium containing 10% FBS was added to the lower chamber as a chemoattractant. After 24 h culture, cells in the upper chamber were removed using a cotton swab and stained with 0.5% crystal violet. The motility and invasiveness of the cells were determined by dissolving the stained cells in 10% acetic acid and measuring absorbance at 560 nm.
Xenograft model. Experiments in mice were conducted in the Animal Facility of Tsinghua University and were approved by the Institutional Animal Care and Use Committee of Tsinghua University. Gastric cancer xenografts were established in 6‐week‐old female BALB/c nude mice. Briefly, MKN28 cells stably expressing empty vector or HSulf‐1 were trypsinized and resuspended in PBS (pH 7.4) and then mixed 1:1 (v/v) with matrigel (Vigorous, Beijing, China) at 4°C prior to injection into one mouse in a total volume of 100 μL. The mixture, containing 5 × 106 cells, was injected s.c. into the right flank of eight female mice (four in each group). Tumor size was determined using the formula (0.5 × width2 × length) at various times over the 5‐week period.
Five weeks after injection, tumors were collected and treated for RNA extraction, semiquantitative RT‐PCR, and western blot analysis, as described above.
Statistical analysis. Data were analyzed using GraphPad Prism 5.0 for Windows (GraphPad Software, Inc., San Diego, CA, USA). Data are presented as the mean ± SD of three independent experiments performed in triplicate. The significance of differences between groups was determined by Student’s t‐test. Two‐tailed P < 0.05 was considered significant.
Results
HSulf‐1 inhibits the proliferation of gastric cancer cells. First we examined the effect of HSulf‐1 on the growth of gastric cancer cells. The growth rate of MKN28 cells stably transfected with HSulf‐1 was significantly slower than that of cells stably transfected with empty vector (P < 0.001; Fig. 1a). However, stable transfection of HSulf‐1 did not affect the growth rate of AGS cells (P > 0.05; Fig. 1b). We also confirmed these observations in the colony formation (Fig. 1c,d) and soft agar (Fig. 1e,f) assays. Colony numbers were significantly less in MKN28 cells stably transfected with HSulf‐1 than in control cells (P < 0.01; Fig. 1c–f). To determine the effect of Hsulf‐1 expression on the sulfation status of HSPG in gastric cancer cells, we used the anti‐HS antibody 10E4, which has been reported to recognize HS 6‐O‐sulfated glucosamine residues as well as HS the N‐sulfated glucosamine moiety.( 3 , 25 , 26 , 27 ) Using flow cytometry, we found that the expression of the 10E4 epitope on the cell surface was significantly higher in MKN28 cells than in AGS cells (Fig. 1g,h), indicating possible differences in the HS structures between these two cell lines. Stable transfection of HSulf‐1 in MKN28 cells significantly decreased the 10E4 epitope on the cell surface compared with control cells (Fig. 1g). However, the same treatment of AGS cells had a relatively small effect on the cell surface 10E4 epitope (Fig. 1h).
Figure 1.

Effect of HSulf‐1 on cell proliferation in gastric cancer cell lines. A and B: Growth curves for (a) MKN28 and (b) AGS cells, as determined by the MTS assay, following transfection of cells with either empty vector ( ) or HSulf‐1 (
) or HSulf‐1 ( ). (c) Representative images and (d) quantification of the results of the colony formation assay in MKN28 and AGS cells stably transfected with either empty vector (□) or HSulf‐1 (▪). (e) Representative images and (f) quantification of the results of the soft agar assay in MKN28 cells stably transfected with HSulf‐1. (g,h) Flow cytometry analysis of the effects of HSulf‐1 expression on the cell surface expression of the 10E4 epitope in MKN28 (g) and AGS (h) cells. Cells stably expressing empty vector (red line) or HSulf‐1 (blue line) were stained with anti‐HS antibody or with an irrelevant antibody as a negative control (gray line). Data are the mean ± SEM. *P < 0.05, **P < 0.01 compared with empty vector.
). (c) Representative images and (d) quantification of the results of the colony formation assay in MKN28 and AGS cells stably transfected with either empty vector (□) or HSulf‐1 (▪). (e) Representative images and (f) quantification of the results of the soft agar assay in MKN28 cells stably transfected with HSulf‐1. (g,h) Flow cytometry analysis of the effects of HSulf‐1 expression on the cell surface expression of the 10E4 epitope in MKN28 (g) and AGS (h) cells. Cells stably expressing empty vector (red line) or HSulf‐1 (blue line) were stained with anti‐HS antibody or with an irrelevant antibody as a negative control (gray line). Data are the mean ± SEM. *P < 0.05, **P < 0.01 compared with empty vector.
HSulf‐1 the inhibits migration and invasion of gastric cancer cells. In the wound‐healing assay, we observed that 48 h after the wound had been made, the migration of MKN28 cells stably transfected with HSulf‐1 was markedly inhibited (∼40% reduction) compared with control cells (Fig. 2a). In contrast, no noticeable change in the migration of AGS cells was observed (Fig. 2a). We next performed the transwell assay to examine the mobility and invasion of those stable cells (Fig. 2b,c). In MKN28 cells stably transfected with HSulf‐1, an approximate 50% reduction in both mobility and invasion was seen compared with control cells (P < 0.001). However, there was no significant difference in the mobility of AGS cells stably transfected with HSulf‐1 and control cells. In accordance with these observations, we found that mRNA levels of metastasis‐related genes, such as DKK1, MMP‐2, S100A4, and S100P,( 22 , 28 , 29 , 30 ) were downregulated in HSulf‐1 transfected MKN28 cells, but that there were no noticeable change in the expression of these genes in AGS cells (Fig. 2d).
Figure 2.

Effect of HSulf‐1 on the migration and invasion of cells from the tow gastric cancer cell lines. (a) Wound healing assay. Wound size was monitored (top) and measured (bottom) at the time points indicated in MKN28 and AGS cells stably transfected with either empty vector ( ) or HSulf‐1 (
) or HSulf‐1 ( ). Scale bar, 200 μm. (b) Transwell migration assay of MKN28 and AGS cells stably transfected with empty vector or HSulf‐1. OD560, optical density at 560 nm. (c) The invasion assay for MKN28 cells stably transfected with empty vector or HSulf‐1. Data are the mean ± SEM percentage absorbance at 560 nm compared with control. *P < 0.05, **P < 0.01 compared with empty vector. (d) Semiquantitative RT‐PCR analysis of the mRNA expression of metastasis‐related genes in MKN28 and AGS cells stably transfected with empty vector or HSulf‐1.
). Scale bar, 200 μm. (b) Transwell migration assay of MKN28 and AGS cells stably transfected with empty vector or HSulf‐1. OD560, optical density at 560 nm. (c) The invasion assay for MKN28 cells stably transfected with empty vector or HSulf‐1. Data are the mean ± SEM percentage absorbance at 560 nm compared with control. *P < 0.05, **P < 0.01 compared with empty vector. (d) Semiquantitative RT‐PCR analysis of the mRNA expression of metastasis‐related genes in MKN28 and AGS cells stably transfected with empty vector or HSulf‐1.
HSulf‐1 regulates Wnt signaling in gastric cancer cells. HSulf‐1 can regulate signal transduction pathways by affecting interactions between HSPG and extracellular signals. In addition, it has been reported that HSulf‐1 is involved in the regulation of Wnt signaling in pancreatic cancer.( 17 ) Dysregulation of the Wnt signaling pathway has been implicated in gastric carcinogenesis and metastasis.( 22 , 23 , 31 , 32 , 33 )
Therefore, in the present study, we first assessed the expression profile of Wnt pathway genes in AGS and MKN28 cells. As shown in Figure 3(a), the expression of several Wnt ligands was elevated in both cell lines (i.e. Wnt3a in AGS cells; Wnt1 and Wnt3a in MKN28 cells). The expression of these Wnt ligands was not detectable in normal stomach tissue, used as a control. The expression of Frizzled (Fzd) family members (Fzd2, Fzd3, Fzd5 Fzd6, and Fzd7), receptors for Wnt ligands, was also increased in both AGS and MKN28 cells compared with expression in normal stomach tissue. In addition, the expression of a key effector of the Wnt pathway, namely Dishevelled (Dvl), and the downstream target genes CCND1 and c‐Myc was upregulated in the AGS and MKN28 cells (Fig. 3a). In accordance with these observations, the expression levels of the proteins of these Wnt pathway effectors (Dvl and cytosolic β‐catenin) and downstream target genes (cyclin D1 and c‐myc) were increased in AGS and MKN28 cells (Fig. 3b). Furthermore, transcription factor TCF/LEF reporter activity, another hallmark of Wnt signaling activation, was high in AGS and MKN28 cells (Fig. 4). These results confirm the aberrant activation of Wnt signaling in AGS and MKN28 cells.
Figure 3.
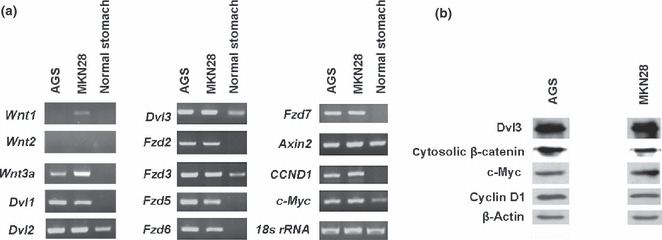
Analyses of gene expression profiles in the two gastric cancer cell lines. (a) Semiquantitative RT‐PCR analysis of the mRNA expression of Wnt signaling genes in MKN28 and AGS cells. Normal stomach tissue was used as a control, with 18s rRNA used as loading control. (b) Western blot analysis of β‐catenin in the cytosolic fraction and the Dvl3 and Wnt targets c‐Myc and Cyclin D1 in whole cell lysates. β‐Actin was used as a loading control.
Figure 4.
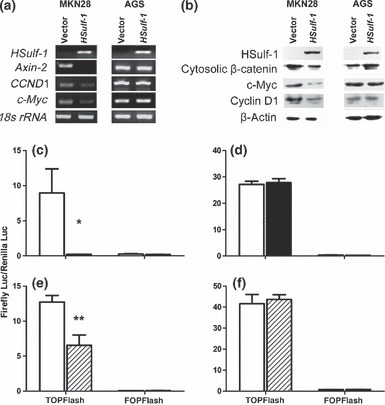
Effect of Hsulf‐1 on Wnt signaling pathway in the two gastric cancer cell lines. (a) Semiquantitative RT‐PCR analysis of the mRNA expression of HSulf‐1 and Wnt/β‐catenin downstream genes and (b) western blot analysis of protein levels of HSulf‐1, cytosolic β‐catenin, and Wnt targets in MKN28 and AGS cells stably transfected with empty vector or HSulf‐1. (c,d) TCF/LEF reporter activity and (e,f) TCF/LEF luciferase reporter activity in MKN28 (c,e) and AGS (d,f) cells stably transfected with empty vector (□), HSulf‐1 (▪), or DKK1 ( ). Data are the mean ± SEM. *P < 0.05, **P < 0.01 compared with empty vector.
). Data are the mean ± SEM. *P < 0.05, **P < 0.01 compared with empty vector.
To investigate the function of HSulf‐1 in Wnt activation in gastric cancer, we examined the mRNA expression of Wnt target genes in AGS and MKN28 cells stably transfected with HSulf‐1. In MKN28 cells stably transfected with HSulf‐1, the expression of all target genes examined was downregulated. In contrast, there were no obvious changes in the expression of these genes in AGS cells (Fig. 4a). We also determined the protein levels of cytosolic β‐catenin, a key component of the Wnt pathway, and cyclin D1 and c‐myc as downstream targets of Wnt.( 19 ) Consistent with our RT‐PCR results, cytosolic accumulation of β‐catenin and levels of cyclin D1 and c‐myc were markedly downregulated in MKN28 cells stably transfected with HSulf‐1, but not in AGS cells (Fig. 4b). Using the TOPflash/FOPflash reporter system to evaluate downstream transcription activity of the Wnt signaling pathway, we found that TCF/LEF reporter activity was significantly inhibited in MKN28 cells stably transfected with HSulf‐1 (P < 0.05; Fig. 4c). In contrast, HSulf‐1 transfection had no effect on TCF/LEF reporter activity in AGS cells (Fig. 4d). As a control, we transfected cells with DKK1, an extracellular antagonist for Wnt signaling,( 34 ) into MKN28 and AGS cells to confirm the regulation of upstream Wnt signaling on its downstream transcription activity. We found that normalized TOPflash reporter activity was significantly decreased in MKN28 cells (P = 0.006), but unaltered in AGS cells (P > 0.05), after DKK1 transfection (Fig. 4e,f). These results indicate that HSulf‐1 may be an extracellular suppressor inhibiting upstream Wnt signaling in gastric cancer cells.
To further test this hypothesis, we transfected Wnt3a into AGS and MKN28 cells stably transfected with HSulf‐1 and examined cell growth rates. Following Wnt3a transfection, the inhibitory effect of HSulf‐1 on MKN28 cell proliferation could be totally rescued (Fig. 5a). Conversely, neither HSulf‐1 nor Wnt3a had any effect on the proliferation of AGS cells (Fig. 5b). We also transfected CTNNB1 shRNA to knockdown β‐catenin in both cell lines (two different shRNA targeting CTNNB1 tested in the present study showed consistent results; one set of data are shown in Fig. 5). The efficiency of β‐catenin knockdown was confirmed by immunoblotting (Fig. 5d,f). We found that CTNNB1 shRNA alone inhibited cell proliferation in both MKN28 and AGS cells. In MKN28 cells, knocking down β‐catenin potentiated the inhibitory effects of HSulf‐1 on cell proliferation (Fig. 5c). However, in AGS cells, transfection of the combination of HSulf‐1 and CTNNB1 shRNA resulted in the same inhibitory effect on cell proliferation as that seen with transfection of CTNNB1 shRNA alone (Fig. 5e). Together, the results suggest that HSulf‐1 may inhibit MKN28 cell growth by suppressing upstream Wnt signaling.
Figure 5.
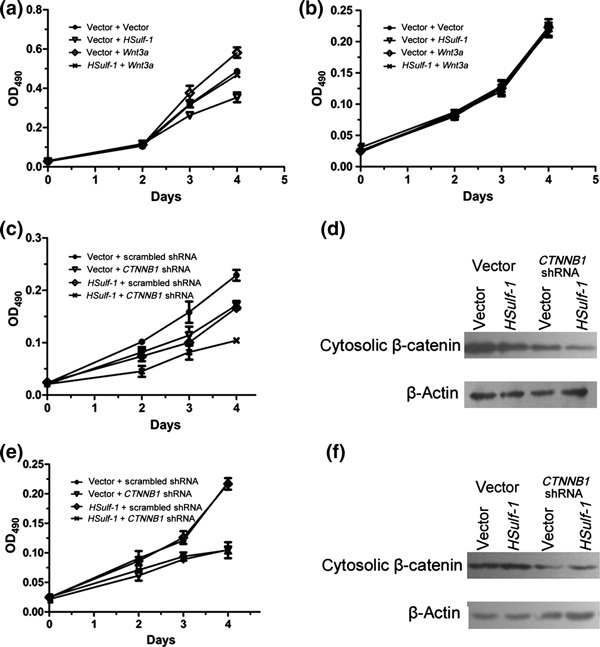
Effects of HSulf‐1 and the Wnt pathway on cell proliferation in two gastric cancer cell lines. (a,b,c,e) Results of the MTS assay after stable MKN28 (a,c) and AGS (b,e) cells were transfected with Wnt3a (a,b) or CTNNB1 shRNA (c,e). (d,f) Western blots of cytosolic β‐catenin in MKN28 (d) and AGS (f) cells transiently transfected with CTNNB1 shRNA. Data are the mean ± SEM.
HSulf‐1 inhibits growth of gastric cancer in vivo. Finally, MKN28 xenografts were established to evaluate the effect of HSulf‐1 on the growth of gastric cancer in vivo. Three weeks after inoculation in mice, we found that the tumor volume of tumors resulting from the injection of MKN28 cells stably transfected with HSulf‐1 was significantly less than that of the control (P < 0.05; Fig. 6a). In addition, at the completion of this experiment (i.e. 5 weeks after inoculation), the weight of the tumors arising after injection of MKN28 cells stably transfected with HSulf‐1 was significantly less than that of the control (P < 0.05; Fig. 6b,c). Furthermore, the expression of cytosolic β‐catenin and Wnt downstream target genes was downregulated in the tumors arising from injection of MKN28 cells stably transfected with HSulf‐1 (Fig. 6d), a finding that is consistent with the in vitro results. These data indicate that HSulf‐1 may inhibit the tumor growth of MKN28 cells in vivo by suppressing the Wnt signaling pathway.
Figure 6.

Effect of HSulf‐1 on the tumorigenicity of MKN28 cells in vivo. (a) Tumor volumes measured at different time points in MKN28 cells transfected with either empty vector ( ) or HSulf‐1 (
) or HSulf‐1 ( ). Data are the mean ± SEM. *P < 0.05, **P < 0.01 compared with empty vector. (b) Tumors harvested from mice 5 weeks after injection of MKN28 cells transfected with either empty vector or HSulf‐1. (c) Individual tumor weights for each mouse at the end of the experiment. (d) Semiquantitative RT‐PCR and western blot analyses of the Wnt signaling pathway in tumors arising in mice following the transfection of MKN28 cells transfected with either empty vector or HSulf‐1.
). Data are the mean ± SEM. *P < 0.05, **P < 0.01 compared with empty vector. (b) Tumors harvested from mice 5 weeks after injection of MKN28 cells transfected with either empty vector or HSulf‐1. (c) Individual tumor weights for each mouse at the end of the experiment. (d) Semiquantitative RT‐PCR and western blot analyses of the Wnt signaling pathway in tumors arising in mice following the transfection of MKN28 cells transfected with either empty vector or HSulf‐1.
Discussion
Although it has been shown that HSulf‐1 has a role in tumorigenesis and metastasis in several types of cancer,( 5 , 6 , 7 , 8 , 9 , 10 ) its role in gastric cancer had not been investigated. In the present study, we found that HSulf‐1 suppressed cell proliferation, tumor growth, and activity of the Wnt signaling pathway in MKN28 cells, but not in AGS cells. These results suggest a possible function for HSulf‐1 in the suppression of upstream Wnt signaling to inhibit gastric cancer cell growth. The mechanisms underlying aberrant Wnt activation in these two cell lines appear to be different. It has been reported previously that AGS cells have the G34E mutant allele in CTNNB1.( 35 ) We also confirmed this mutation in AGS cells by direct sequencing (data not shown). As a result, in AGS cells the Wnt pathway is constitutively active and independent of upstream Wnt signals. Conversely, MKN28 cells are dependent on upstream Wnt signaling. Consistent with these findings, we observed that low 10E4 epitope expression on the cell surface of AGS cells and a relatively small reduction in 10E4 epitope expression in response to HSulf‐1 in AGS cells compared with MKN28 cells. To confirm this differential cell surface regulation of Wnt signaling in MKN28 and AGS cells, we transfected DKK1, an extracellular antagonist of Wnt signaling,( 36 ) into AGS cells and demonstrated no noticeable reduction in the transcriptional activity of the downstream Wnt pathway. Conversely, DKK1 transfection did affect MKN28 cells. We also confirmed aberrant activation of Wnt pathway in the two cell lines. To further test our hypothesis, we performed gain‐of‐function and loss‐of‐function experiments (Fig. 5). We observed that Wnt3a transfection rescued the HSulf‐1 inhibition of MKN28 cell proliferation, but that it had no effect in AGS cells. In the loss‐of‐function experiment, although the proliferation of both AGS and MKN28 cells was inhibited by CTNNB1 shRNA knockdown alone, knocking down CTNNB1 only potentiated the inhibitory effects of HSulf‐1 on the proliferation of MKN28 cells, and not AGS cells. Together, these results indicate that HSulf‐1 may function as a negative extracellular regulator of upstream Wnt signaling important for cell growth in gastric cancer through its enzymatic effects on HSPG, consistent with a recent report that HSulf‐1 mediates the affinity of HSPG for extracellular ligands.( 36 )
In addition, we found that HSulf‐1 inhibited the migration and invasiveness of MKN28 cells, as well as the expression of several well‐known metastasis‐related genes (Fig. 2). Of these genes, DKK4, S100A4, and S100P have been identified as Wnt signaling downstream target genes.( 30 ) Although DKK4 is a Wnt antagonist, it is associated with the malignant properties of cancer cells.( 29 ) However, another metastasis‐related gene, namely MMP‐2, was not a target of Wnt signaling, indicating possible regulation of multiple signaling pathways by HSulf‐1 in the invasion of gastric cancer cells.
In summary, the results of the present study demonstrate that HSulf‐1 functions as a negative regulator of gastric carcinoma and metastasis, at least in part by regulating the sulfation status of HSPG and further suppression of upstream Wnt signaling.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported, in part, by funds from the National Key Basic Research Project (no. 2007CB914401), the National Key Basic Research and Development (973) Program of China (no. 2011CB910803), the China Natural Science Foundation (no. 30770475), and a research fund from Beijing ACCB Biotech.
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12: 354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ai X, Do AT, Lozynska O, Kusche‐Gullberg M, Lindahl U, Emerson CP Jr. QSulf1 remodels the 6‐O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol 2003; 162: 341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lai JP, Thompson JR, Sandhu DS, Roberts LR. Heparin‐degrading sulfatases in hepatocellular carcinoma: roles in pathogenesis and therapy targets. Future Oncol 2008; 4: 803–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lai J, Chien J, Staub J et al. Loss of HSulf‐1 up‐regulates heparin‐binding growth factor signaling in cancer. J Biol Chem 2003; 278: 23107–17. [DOI] [PubMed] [Google Scholar]
- 6. Lai JP, Chien J, Strome SE et al. HSulf‐1 modulates HGF‐mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene 2004; 23: 1439–47. [DOI] [PubMed] [Google Scholar]
- 7. Lai JP, Chien JR, Moser DR et al. hSulf1 sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin‐binding growth factor signaling. Gastroenterology 2004; 126: 231–48. [DOI] [PubMed] [Google Scholar]
- 8. Narita K, Staub J, Chien J et al. HSulf‐1 inhibits angiogenesis and tumorigenesis in vivo . Cancer Res 2006; 66: 6025–32. [DOI] [PubMed] [Google Scholar]
- 9. Dai Y, Yang Y, MacLeod V et al. HSulf‐1 and HSulf‐2 are potent inhibitors of myeloma tumor growth in vivo . J Biol Chem 2005; 280: 40066–73. [DOI] [PubMed] [Google Scholar]
- 10. Lai JP, Yu C, Moser CD et al. SULF1 inhibits tumor growth and potentiates the effects of histone deacetylase inhibitors in hepatocellular carcinoma. Gastroenterology 2006; 130: 2130–44. [DOI] [PubMed] [Google Scholar]
- 11. Lai JP, Sandhu DS, Shire AM, Roberts LR. The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J Gastrointest Cancer 2008; 39: 149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol 2005; 6: 530–41. [DOI] [PubMed] [Google Scholar]
- 13. Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development 2004; 131: 6009–21. [DOI] [PubMed] [Google Scholar]
- 14. De Cat B, Muyldermans SY, Coomans C et al. Processing by proprotein convertases is required for glypican‐3 modulation of cell survival, Wnt signaling, and gastrulation movements. J Cell Biol 2003; 163: 625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ohkawara B, Yamamoto TS, Tada M, Ueno N. Role of glypican 4 in the regulation of convergent extension movements during gastrulation in Xenopus laevis . Development 2003; 130: 2129–38. [DOI] [PubMed] [Google Scholar]
- 16. Capurro MI, Xiang YY, Lobe C, Filmus J. Glypican‐3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res 2005; 65: 6245–54. [DOI] [PubMed] [Google Scholar]
- 17. Nawroth R, van Zante A, Cervantes S, McManus M, Hebrok M, Rosen SD. Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE 2007; 2: e392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008; 8: 387–98. [DOI] [PubMed] [Google Scholar]
- 19. Clevers H. Wnt/beta‐catenin signaling in development and disease. Cell 2006; 127: 469–80. [DOI] [PubMed] [Google Scholar]
- 20. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004; 20: 781–810. [DOI] [PubMed] [Google Scholar]
- 21. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–50. [DOI] [PubMed] [Google Scholar]
- 22. Ganesan K, Ivanova T, Wu Y et al. Inhibition of gastric cancer invasion and metastasis by PLA2G2A, a novel beta‐catenin/TCF target gene. Cancer Res 2008; 68: 4277–86. [DOI] [PubMed] [Google Scholar]
- 23. Du R, Xia L, Sun S et al. URG11 promotes gastric cancer growth and invasion by activation of beta‐catenin signalling pathway. J Cell Mol Med 2010; 14: 621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Z, Fan JQ, Li J et al. Promoter hypermethylation correlates with the Hsulf‐1 silencing in human breast and gastric cancer. Int J Cancer 2009; 124: 739–44. [DOI] [PubMed] [Google Scholar]
- 25. van den Born J, Salmivirta K, Henttinen T et al. Novel heparan sulfate structures revealed by monoclonal antibodies. J Biol Chem 2005; 280: 20. [DOI] [PubMed] [Google Scholar]
- 26. Sala‐Newby GB, George SJ, Bond M, Dhoot GK, Newby AC. Regulation of vascular smooth muscle cell proliferation, migration and death by heparan sulfate 6‐O‐endosulfatase1. FEBS Lett 2005; 579: 6493–8. [DOI] [PubMed] [Google Scholar]
- 27. Hossain MM, Hosono‐Fukao T, Tang R et al. Direct detection of HSulf‐1 and HSulf‐2 activities on extracellular heparan sulfate and their inhibition by PI‐88. Glycobiology 2010; 20: 175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sherbet GV. Metastasis promoter S100A4 is a potentially valuable molecular target for cancer therapy. Cancer Lett 2009; 280: 15–30. [DOI] [PubMed] [Google Scholar]
- 29. Pukrop T, Klemm F, Hagemann T et al. Wnt 5a signaling is critical for macrophage‐induced invasion of breast cancer cell lines. Proc Natl Acad Sci USA 2006; 103: 5454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pendas‐Franco N, Garcia JM, Pena C et al. DICKKOPF‐4 is induced by TCF/beta‐catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25‐dihydroxyvitamin D3. Oncogene 2008; 27: 4467–77. [DOI] [PubMed] [Google Scholar]
- 31. Yamamoto H, Kitadai Y, Yamamoto H et al. Laminin gamma2 mediates Wnt5a‐induced invasion of gastric cancer cells. Gastroenterology 2009; 137: 242–52. [DOI] [PubMed] [Google Scholar]
- 32. Nojima M, Suzuki H, Toyota M et al. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 2007; 26: 4699–713. [DOI] [PubMed] [Google Scholar]
- 33. Sato H, Suzuki H, Toyota M et al. Frequent epigenetic inactivation of DICKKOPF family genes in human gastrointestinal tumors. Carcinogenesis 2007; 28: 2459–66. [DOI] [PubMed] [Google Scholar]
- 34. Zorn AM. Wnt signalling: antagonistic Dickkopfs. Curr Biol 2001; 11: R592–5. [DOI] [PubMed] [Google Scholar]
- 35. Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf‐1 interaction with LRP6/Arrow. Nat Cell Biol 2001; 3: 683–6. [DOI] [PubMed] [Google Scholar]
- 36. Dreyfuss JL, Regatieri CV, Jarrouge TR, Cavalheiro RP, Sampaio LO, Nader HB. Heparan sulfate proteoglycans: structure, protein interactions and cell signaling. An Acad Bras Cienc 2009; 81: 409–29. [DOI] [PubMed] [Google Scholar]