

Abstract
It was hypothesized that if dendritic cells (DC) could be efficiently manipulated in vivo, this might enable functional maturation and retention of their potent functions and might represent a more promising approach in DC immunotherapy. The present study focused on the modulation of DC in tumor microenvironment using Fms‐like thyrosine kinase 3 ligand (Flt3L) combined with interferon‐γ‐inducing factor (IL‐18). Tumor‐inoculated mice were treated with in vivo electroporation (IVE) of expression plasmids carrying complementary DNA of Flt3L. As a combination therapy, mice in the other group were treated with intra‐tumoral injection of adenoviral vector carrying IL‐18 gene (Ad.IL‐18). Significant antitumor effect was observed in mice treated with Ad.IL‐18 alone when compared with that of control (P < 0.01). Complete eradication was observed more frequently (100%versus 33%: P < 0.05) in the mice treated with Flt3L and Ad.IL‐18 when compared with the mice treated with Ad.IL‐18 alone. In un‐injected distant tumor, significant antitumor responses were observed only in the mice treated with combination therapy. Lymphoid cells in lymph nodes of mice treated with combination therapy showed significant cytolytic activity against inoculated tumor cells and YAC‐1 cells when compared with the lymphoid cells in other groups. In the tumor microenvironment, combination therapy resulted in the recruitment of mobilized DC into the tumor bed, although Flt3L–IVE alone had an effect in the peri‐tumoral area. Tumor‐infiltrating DC in mice treated with combination therapy showed higher CD86 expression and more potent allogeneic T‐cell stimulatory capacity. These results may suggest that local expression of IL‐18 combined with in vivo DC mobilization with Flt3L is clinically applicable as a new strategy of DC immunotherapy. (Cancer Sci 2008; 99: 2028–2036)
Dendritic cells (DC) are the most potent antigen‐presenting cells (APC) of the immune system and have been used in cancer immunotherapy to induce tumor‐specific antitumor immunity; however, the clinical outcome has not been entirely satisfactory. The lack of efficacy for tumor regression could be due to the unfavorable tumor microenvironment, which renders it incapable of propagating a robust antitumor immune response.( 1 ) Furthermore, most DC immunotherapy uses DC propagated in vitro from the monocytes or CD34+ cells prepared using leukocyte apheresis from cancer patients; however, these cultured DC may not have sufficient functions that are critical to generate antitumor‐specific immunity after administration to the patient. Alternatively, if DC could be efficiently manipulated in vivo, they might exhibit potent functions and may represent a more rational approach to DC immunotherapy.
Fms‐like thyrosine kinase 3 ligand (Flt3L), a cytokine that augments the number of functional DC and natural killer cells (NK‐cells) in vivo, is considered to be one of the candidates to effectively manipulate DC in vivo.( 2 , 3 )>In vivo over‐expression of Flt3L induces the expansion and activation of natural killer dendritic cells (NKDC), which are a novel subtype of DC with NK‐cell properties, in situ.( 4 ) Although Flt3L alone has been reported to induce the regression of chemically induced sarcomas in mice,( 5 ) these results have not been supported in other research, including ours.( 6 , 7 , 8 ) In clinical trials, Flt3L is capable of mobilizing DC into the peripheral blood of cancer patients( 9 ) but no clinical responses have been reported. Thus, it appears necessary to develop combination therapy to induce potent and durable antitumor immune responses along with in vivo manipulation of DC using Flt3L.
Interferon‐γ‐inducing factor (IL‐18), which induces interferon (IFN)‐γ production and enhances the cytolytic activity of NK‐cells, shares several biological activities with IL‐12 and has led to a series of studies on its effects on T‐cells and NK‐cells.( 10 ) Our in vitro study has shown that IL‐18 induces tumor‐specific immunity by enhancing NK activity, which mediates tumor cell death and activates and primes DC. This induction of an antitumor response by IL‐18 has the capacity to be superior to IL‐2 and IL‐12 in some situations.( 11 ) Furthermore, IL‐18 has recently been shown to activate NKDC to secrete IFN‐γ.( 12 )
From this information, we hypothesized that combination therapy with Flt3L and IL‐18 may work synergistically in the tumor microenvironment to induce potent antitumor immunity, primarily mediated through NK activity. We here report that intra‐tumoral gene transfer of IL‐18 combined with systemic Flt3L administration induces potent and systemic antitumor responses that mediate the activation of not only NK‐cells, but also tumor‐infiltrating DC mobilized by Flt3L in the tumor microenvironment.
Materials and Methods
Animals. Female C57BL/6 J (B6; H2b) and BALB/c (BALB/c; H2d) mice were purchased from CLEA JAPAN (Tokyo, Japan) and Charles River Japan (Tokyo, Japan) respectively. All mice were housed in a specific pathogen‐free facility at the Institute of Medical Science of the University of Tokyo, and were used at 8–10 weeks of age for all experiments.
Cell lines. MCA205, a methylcholanthrene‐induced murine fibrosarcoma cell line, and MC38, a murine colon adenocarcinoma, were generous gifts from Dr S. A. Rosenberg (National Cancer Institute, Bethesda, MD, USA). MCA205 cell line expressed more than 80% of major histocompatability complex (MHC) class I expression. AC‐1 was a generous gift from W. Chambers (University of Pittsburgh, Pittsburgh, PA, USA). These cell lines were cultured in complete medium (CM), which included Roswell Park Memorial Institute medium (RPMI)‐1640 supplemented with 10% of heat‐inactivated fetal bovine serum, 0.1 mM‐non‐essential amino acid, 1 mM sodium pyruvate (Invitrogen, Tokyo, Japan), 5 × 10−5 M 2‐mercaptoethanol (Nacalai Tesque, Kyoto, Japan), 100 µg/mL streptomycin and 100 unit/mL penicillin (Invitrogen).
Cytokines. Recombinant human IL‐2 was purchased from Chiron (CA, USA), recombinant human Flt3L was from PeproTech (London, UK), and recombinant mouse IL‐18 was from MBL (Nagoya, Japan). Cytokines were reconstituted in phosphate‐buffered saline (PBS).
Plasmid DNA. Plasmid pNGVL–hFlt3L containing the full‐length human Flt3L gene was obtained from the National Gene Vector Laboratory (University of Michigan at Ann Arbor, MI, USA). Complementary DNA (cDNA) of hFlt3L was subcloned into CMV (cytomegalovirus promoter)‐intron A (enhancer)‐poly A backbone. pEGFP (enhanced green fluorescent protein), with the same promoter and enhancer as the control, was purchased from Clontech (CA, USA). These plasmid vectors were prepared by E. coli transfection and purified by the general cesium chloride ultra‐centrifugation method.
Adenoviral vector. A recombinant adenoviral vector encoding both IL‐1 receptor antagonist (IL‐1ra) and IL‐18 fragment (Ad.IL‐18) was constructed by the cosmid–adenoviral DNA terminal protein complex method (Takara Shuzo, Kyoto, Japan). Secretion of bioactive IL‐18 was facilitated by fusing the leader sequences of IL‐1ra to the 5′ end of mature murine IL‐18 cDNA as described previously.( 13 ) An adenovirus‐vector expressing EGFP (Ad.EGFP) was constructed in the same manner by subcloning the respective fragments obtained from pEGFP (Clontech, CA, USA).
In vivo electroporation. In vivo electroporation (IVE) was performed to obtain systemic transgene expression in the blood, instead of the administration of high‐dose protein. Each plasmid conditioned with 1.0 µg/µL in dH2O and 30 µL of plasmid solution was administered into the bilateral tibialis anterior muscles or gastrocnemius muscles using a disposable insulin syringe with a 27‐gauge needle under general anesthesia. After 90 s, a pair of stainless steel electrode needles with a 5 mm gap was inserted into the muscles to encompass the DNA injection sites, and electric pulses were given using an electric pulse generator (CUY‐21, Tokiwa Science, Tokyo, Japan). Four pulses of 100 V each were delivered to each injection site at a rate of one pulse per second, each pulse lasting for 50 ms, followed by four pulses of the opposite polarity. After IVE with Flt3L plasmid (Flt3L–IVE), Flt3L was detected in the serum for 10 days after Flt3L–IVE at significant levels and we also confirmed the bioactivity of Flt3L by Flt3L–IVE as described previously.( 6 )
Quantitation of transgene expression for IL‐18 in the tumor. To confirm the transgene expression of IL‐18 in the tumor, the supernatant of dissociated tumor cells with PBS 72 h after intra‐tumoral gene transfer with Ad.IL‐18 was examined using enzyme‐linked immunosorbent assay (ELISA) for IL‐18. Control groups included PBS or adenovirus containing the gene for EGFP (Ad.EGFP), which functions as a reporter gene and does not have a therapeutic effect. ELISA for IL‐18 was performed with reagents purchased from PharMingen (San Diego, CA, USA), using the procedure recommended by the manufacturer.
Tumor treatment model. Mice were inoculated intradermally into the right flank with 1 × 105 MCA205 on day 0. On day 5, when tumors became palpable, mice were electroporated with plasmid DNA to the bilateral tibialis anterior muscles or gastrocnemius muscles and on day 12, this treatment was repeated. As a combination therapy, each mouse also treated with an intra‐tumoral injection (i.t.) of 1 × 108 pfu of adenoviral vector on day 9 and 16. The perpendicular diameters of individual tumors were measured every 3–4 days and the tumor area was calculated as the product of the tumor diameters.
Depletion of immune‐responding cells with antibodies. To deplete asialoGM1‐positive cells in mice, 20 µL of antiasialo GM1 antiserum (ASGM1; Wako Bioproducts, Richmond, VA, USA) was administered intraperitoneally 5, 3 and 1 days before tumor inoculation, and subsequently once every 4 days for an additional 20 days (eight times in total). This dose was confirmed to be effective in suppressing more than 95% of lytic activity against YAC‐1 cells mediated by spleen cells harvested from animals receiving antiasialo GM1 antiserum injections. Monoclonal antibody against CD8+ cells (clone 2.43; American Type Culture Collection) was prepared and treated intraperitoneally (200 mg per mouse) 5 and 1 days before tumor cell inoculation and repeated once per week thereafter (six times in total). Depletion of CD8+ T‐cells after the administration of antibody was confirmed by flow cytometric analysis of spleen cells.
Isolation of tumor‐infiltrating cells. Tumors were surgically removed, minced into small fragments and incubated in collagenase D (1 mg/mL: Roche Diagnostics, Germany) for 45 min at 37°C. Each tumor was then dissociated into a single‐cell suspension, passed over a 70 µm nylon mesh and washed twice in PBS for analysis. In some experiments, CD11c+ cells were immunobead‐sorted by labeling with a bead‐conjugated antimouse CD11c mAb (Miltenyi Biotec, Auburn, CA, USA) followed by passage through a positive selection paramagnetic column (Miltenyi Biotec). To count the number of tumor‐infiltrating cells, samples were analyzed by flow cytometry by counting 50 000 lymphocyte‐sized events based on splenocytes as an internal control. CD3e−NK1.1+ cells were determined as NK‐cells. As the tumor sizes were different, data were normalized to the tumor volume using the volume equation: V (in mm3) = 0.4(ab, ( 2 )), where a is the long diameter and b is the short diameter.
Phenotypic and functional analyzes of bone‐marrow‐derived DC with Flt3L in the presence or absence of IL‐18 in vitro. Bone‐marrow‐derived DC were propagated from Flt3L using the procedure described initially by Gilliet et al.( 14 ) In brief, bone marrow cells (BM) were flushed from the tibia and femur of C57BL/6 mice with CM, centrifuged, and resuspended in Tris‐ammonium chloride buffer to lyze red blood cells. After centrifugation, BM were resuspended at 5 × 106 cells/mL in CM supplemented with 100 ng/mL recombinant human Flt3L in 24‐well plates (Costar Corning, Cambridge, UK) at 37°C for 9 days. To evaluate the influence of IL‐18 on DC propagated from Flt3L, different concentrations of IL‐18 (0.1, 1 and 10 ng/mL) were added at day 7 for the last 48 h of culture. In some experiments, anti‐IFN‐γ antibody (XMG1.2; PharMingen) was also added to the culture medium including 10 ng/mL of rIL‐18 at day 7 for the last 48 h of the culture.
On day 9, floating cells (CD11c‐positive cell fraction was >90%) were harvested, and phenotypic and functional analyzes were performed.
Flow cytometry. DC were characterized by cytofluorography using a panel of monoclonal antibodies (mAbs), including those directed against mouse DC‐restricted Ag CD11c, MHC class I (H2Kb), MHC class II (IAb), CD40, CD80, or CD86 (all mAbs from PharMingen). NK‐cells were determined as CD3e−NK1.1+ using mAbs against mouse CD3 and NK1.1, and both mAbs were purchased from PharMingen. Two‐color analysis was performed using the Cell Quest software package with FACSCAN (Becton‐Dickinson, Franklin Lakes, NJ, USA).
Cytotoxicity assays. Lymphoid cells were obtained from the drained inguinal lymph nodes, centrifuged and resuspended in a Tris‐ammonium chloride buffer to lyze red blood cells. After centrifugation, these lymphoid cells were re‐stimulated in vitro with γ‐irradiated MCA205 cells in the presence of rhIL‐2 10 IU/mL for 4 days, and then the cells were used as cytotoxic effectors in a 4 h 51Cr release assay. MCA205 was used as the source of a specific cytolytic T‐lymphocyte (CTL) target, MC38 as a non‐specific CTL target and YAC‐1 cells as non‐specific NK targets. In brief, 106 of each target cell were labeled with 3.7 MBq of Na2 51CrO4 for 45–60 min. After washing three times, effector and target cells were plated at an appropriate effector : target ratio in 96‐well round‐bottomed plates. The supernatant (100 µL) was collected after 4‐h incubation, and radioactivity was measured with a γ‐counter. Maximum 51Cr release was determined by the osmotic lysis of target cells. Spontaneous release was always less than 15% of the maximum. The percentage of specific lysis was calculated using the following formula:% lysis = 100× (experimental release – spontaneous release)/(maximal release – spontaneous release). The results are expressed as the means ± 1 SD of percentage‐specific 51Cr release in triplicate cultures.
Mixed leukocyte reaction (MLR). Primary MLR assay was used to examine the ability of tumor‐infiltrating DC to stimulate freshly isolated, naive, allogeneic T‐cells. Splenic T‐lymphocytes of BALB/c (H‐2Kd) were purified by passing through nylon wool columns (30 min, 37°C), and then cultured with graded numbers of irradiated CD11c+‐enriched tumor‐infiltrating DC (H‐2Kb) in 96‐well microculture plates for 5 days. For the final 18 h of culture, 37 KBq [3H]thymidine was added to each well. The cultures were harvested onto glass‐fiber filter disks using a multiple cell harvester and thymidine incorporation was quantified using a liquid scintillation counter (Wallac, Turku, Finland).
Immunohistochemistry. At the indicated time points, mice were sacrificed and tumors were dissected, embedded in optical cutting temperature and frozen in cold hexane. Cryostat‐cut tumor sections (5 µm) were mounted on slides, air dried and desiccated at –80°C until use. Immediately before use, sections were fixed for 10 min in cold acetone, air dried for 20 min and hydrated in PBS. To block endogenous peroxidase activity, sections were incubated with 1% H2O2 for 10 min, followed by washing with PBS. Sections were then incubated for 60 min at room temperature with purified hamster antimouse CD11c antibody or purified rat antimouse CD8 antibody. After rinsing three times in PBS for 5 min, the sections were incubated for 30 min with a biotinylated goat antihamster or antirat antibody, followed by the avidin–biotin–peroxidase complex method. The color reaction was developed for 4–5 min using diaminobenzidine (DAB) Nuclei were lightly counterstained with hematoxylin. Negative controls included staining with the relevant antibodies. All antibodies were purchased from PharMingen, and were evaluated by preliminary titration against normal spleen and tumor for appropriate use.
Statistical analysis. Statistical analysis was performed using the Mann–Whitney U‐test and repeated measures ANOVA, as appropriate. Differences were considered significant when the P‐value was less than 0.05. Data are presented as the means ± SE.
Results
Transgene expression of IL‐18 was detected in the tumor with intra‐tumoral injection of Ad.IL‐18. As shown in Table 1, the tumor with intra‐tumoral injection (i.t) of Ad.IL‐18 at 1 × 108 and 1 × 109 pfu produced IL‐18 at the levels of 67.09 ± 5.00 and 148.78 ± 4.26 pg/mL/mm3, respectively. These IL‐18 expressions were significantly higher than the tumor with PBS (34.40 ± 0.99 pg/mL/mm3) and Ad.EGFP at 1 × 108 pfu (36.56 ± 3.35 pg/mL/mm3) as internal controls. IL‐18 production in serum was not detected in any experimental groups. The detection limit of the IL‐18 ELISA kit was 15.6 pg/mL.
Table 1.
Interferon‐γ‐inducing factor (IL‐18) expression in tumor transfected with adenovirus‐vector expressing (Ad.) enhanced green fluorescent protein (EGFP) or Ad. IL‐18 in vivo
| Groups | IL‐18 expression | |
|---|---|---|
| Tumor (pg/mm3) | Serum (pg/mL) | |
| Phosphate‐buffered saline i.t. | 34.40 ± 0.99 | <15.6 |
| 1 × 109 Ad.EGFP i.t. | 36.56 ± 3.35 | <15.6 |
| 1 × 108 Ad.IL‐18 i.t. | 67.09 ± 5.00 | <15.6 |
| 1 × 109 Ad.IL‐18 i.t. | 148.78 ± 4.26 | <15.6 |
i.t. = intra‐tumoral injection.
Synergistic antitumor effects by intra‐tumoral injection of Ad.IL‐18 in combination with Flt3L–IVE. Mice received intradermal inoculation of 1 × 105 MCA205 fibrosarcoma on day 0 in the bilateral flanks. On day 5 and 12, mice were treated with in vivo electroporation (IVE) with the expression plasmid carrying cDNA of human Flt3L or EGFP to bilateral hind legs. As the combination therapy, some of the mice were also treated with intra‐tumoral injection of Ad.IL‐18 (1 × 108 p.f.u.) or Ad.EGFP (1 × 108 pfu) on day 9 and 16. Significant antitumor effect was observed in mice treated with Ad.IL‐18 alone and in those treated with combination therapy of Flt3L–IVE and Ad.IL‐18 when compared with the control (P < 0.01). Combination treatment with Flt3L–IVE and Ad.IL‐18 resulted in a more potent antitumor response when compared with Flt3L–IVE treatment alone (P < 0.01), and complete eradication was observed more frequently (100%versus 33%: P < 0.05) in mice treated with the combination therapy when compared with those receiving Ad.IL‐18 treatment alone. In un‐injected tumors in other flank, only intra‐tumoral injection of Ad.IL‐18 and Flt3L–IVE combination therapy showed a significant antitumor effect when compared with control treatment (P < 0.01) (Fig. 1a).
Figure 1.

(a) Combination therapy of Fms‐like thyrosine kinase 3 ligand (Flt3L)–in vivo electroporation (IVE) and adenovirus‐vector expressing (Ad.) interferon‐γ‐inducing factor (IL‐18) i.t. mediated significant growth suppression of MCA205 in both tumors. MCA205 cells were inoculated into the right and left flanks of mice on day 0. On day 5, mice were electroporated with expression plasmid into bilateral hind legs and 4 days later, each mouse received 1 × 108 pfu of adenoviral vector as combination therapy. This treatment protocol was performed for two cycles. The mean tumor area on each flank was determined for all mice bearing tumors (left panel; treated tumor growth, right panel; contralateral, un‐injected tumor growth). Data represent the mean tumor size + SE. *P < 0.05, **P < 0.01 Results are representative of two separate experiments. (b) Combined therapy significantly enhanced cytotoxicity against MCA205 and YAC‐1. Lymphoid cells were obtained from draining inguinal lymph nodes harvested from 2 mice/group. After 4 days’ coculture with irradiated MCA205, cytotoxic activity was assessed against each cell line (MCA205, YAC‐1, and MC38). The results shown are the mean of three separate experiments. *P < 0.05, **P < 0.01.
Similar antitumor responses were observed in another tumor model using MC38 adenocarcinoma. (data not shown)
Intra‐tumoral injection of Ad.IL‐18 in combination with Flt3L–IVE enhanced the generation of both CTL and NK activity in regional lymph nodes. To evaluate the mechanism responsible for this antitumor effect, lymphoid cells were collected from regional lymph nodes of treated mice 4 days after second intra‐tumoral injection of adenovirus, and they were cocultured with irradiated MCA205 cells for 4 days in the presence of rhIL‐2 10 IU/mL (Fig. 1b). Lymphoid cells from mice treated with combination therapy of Ad.IL‐18 and Flt3L–IVE showed significant cytolytic activity against MCA205 compared with the other treatment groups. Furthermore, cytolytic activity of combination therapy against YAC‐1 (NK target) was also significantly higher than that of Ad.IL‐18 treatment alone or other treatment (P < 0.05). On the other hand, cytolytic activity against MC38 as a third party control was significant but modest when compared with those of all other groups.
AsialoGM1 positive cells and CD8‐positive T‐cells significantly involved in the antitumor effects of this combination therapy. To analyze antitumor immune responses, asialoGM1 or CD8‐positive cells were depleted in vivo using specific antibodies administered before and during treatment with combination therapy of intra‐tumoral injection of Ad.IL‐18 and Flt3L–IVE (Fig. 2a). In non‐depleted animals, the combination therapy of Ad.IL‐18 i.t. and Flt3L–IVE significantly suppressed growth of tumor not only the injected tumors but also the un‐injected tumor site. Depletion of asialoGM1 positive cells completely abrogated the antitumor effect of combination therapy of intra‐tumoral injection of Ad.IL‐18 and Flt3L–IVE for both tumors. Moreover, tumor growth in mice administered with anti‐CD8 antibody completely abrogated the antitumor effects of the combination therapy for un‐injected tumors but only partially for the injected tumors and remained statistically significant when compared with the control treatment. To confirm the involvement of NK‐cells, we quantified the number of tumor‐infiltrating NK‐cells by fluorescence‐activated cell sorting (FACS). In the mice treated with Flt3L combined with Ad.IL‐18, the number of tumor‐infiltrating NK‐cells was significantly increased (Fig. 2b). Interestingly, by the depletion of asialoGM1‐positive cells, the number of CD8 T‐cells in the un‐injected tumor was markedly decreased even with the combination therapy (Fig. 2c).
Figure 2.

Inhibitory effects of combination therapy on tumor growth were mainly dependent on natural killer (NK)‐cells. (a) Mice were pretreated with specific antibodies to abrogate AsialoGM1+ or CD8+ cells before tumor inoculation. Treatment protocols are described in ‘Material and Methods’. The mean tumor area on each flank was determined for all mice bearing tumors. Data represent the mean tumor areas + SE. Results are representative of two separate experiments. (b) Single‐cell suspensions from tumor were prepared by enzymatic digestion. Cells were stained for the presence of CD3e−NK1.1+ cells. The specific cell number was analyzed as described in ‘Materials and Methods’ and is presented as cells/mm3 of tumor. The results are the mean of six separate experiments. *P < 0.05, **P < 0.01. (c) Immunohistochemical analysis of CD8+ cells infiltrating into un‐injected tumor under combination therapy (left panel; no depletion, right panel; depletion of AsialoGM1+ cells). Original magnification ×200 for each panel. Results are representative of two separate experiments.
Combination therapy resulted in the recruitment of mobilized DC into the tumor bed, although Flt3L–IVE alone recruited mobilized DC into the peri‐tumoral area. It is known that Flt3L mobilizes DC to the local tumor site. To evaluate the distribution of DC by local gene transfer of IL‐18, we examined tumor‐infiltrating DC in the injected tumors with immunohistochemical staining. Mice received an intradermal inoculation of 1 × 106 MCA205 cells in the right flank on day 0, electroporation with expression plasmid on day 5, and the intra‐tumoral injection of adenoviral vector on day 9. On day 12, tumor samples of the mice in each group of tumors were excised for immunostaining with anti‐CD11c antibody (Fig. 3a). We examined both the peri‐tumoral area and the tumor bed and quantified the number of tumor‐infiltrating DC in both fields (Fig. 3b). In control treatment with EGFP–IVE and Ad.EGFP, a few CD11c+ cells were observed in the peri‐tumoral area (a) and no CD11c+ cells were observed in the tumor bed (b). In the mice treated with Flt3L–IVE and Ad.EGFP, marked accumulation of CD11c+ cells were shown in the peri‐tumoral area (c), but very few CD11c+ cells in the tumor bed (d). The mice treated with EGFP–IVE and Ad.IL‐18 showed the same finding as EGFP–IVE and Ad.EGFP in the peri‐tumoral area (e). The number of CD11c+ cells in the tumor bed was marginally increased when compared with those of the mice treated with EGFP–IVE and Ad.EGFP, or Flt3L–IVE and Ad.EGFP (f). In the mice treated with the combination of Flt3L–IVE and Ad.IL‐18, a marked increase of tumor‐infiltrating CD11c+ cells were found not only in the peri‐tumoral area but also in the tumor bed (g, h). As shown in Fig. 3(b), Flt3L–IVE significantly mobilized DC into the peri‐tumoral area. On the other hand, combination therapy with Flt3L–IVE and Ad.IL‐18 synergistically and significantly mobilized tumor‐infiltrating DC into the tumor bed.
Figure 3.
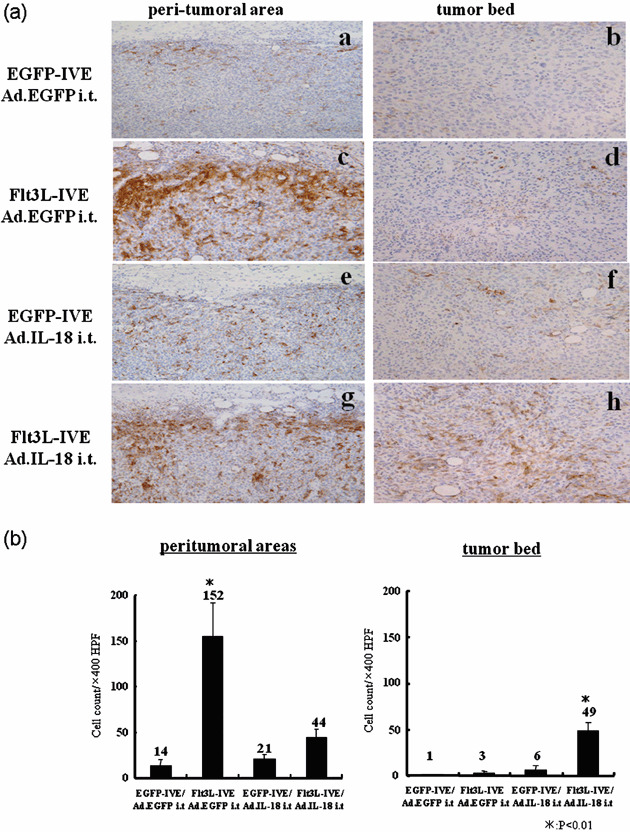
Combination therapy of Fms‐like thyrosine kinase 3 ligand (Flt3L)–in vivo electroporation (IVE) and adenovirus‐vector expressing (Ad.) interferon‐γ‐inducing factor (IL‐18) i.t. resulted in an increased number of dentritic cells (DC) into the tumor bed. (a) Mice were inoculated with MCA205 cells into the right flank of mice on day 0. On day 5, mice were electroporated with expression plasmid into bilateral hind legs, and on day 9, each mouse received 1 × 108 pfu of adenoviral vector. On day 12, DC infiltration at the treated tumor site was analyzed by immunohistochemistry. Serial frozen tumor sections were stained with anti‐CD11c antibody. (left column; peri‐tumoral area, right column; tumor bed). Original magnification ×400 for each panel. Results are representative of two separate experiments. (b) As in (a), quantification of the number of tumor‐infiltrating DC at the tumor site. Results are representative of two separate experiments.
Ad.IL‐18 injection augments CD86 expression on tumor‐infiltrating DC with immuno‐stimulatory capacity. To further examine tumor‐infiltrating dendritic cells in vivo, we separated CD11c‐positive cells from the tumors and examined CD86, a typical maturation marker of DC, and the stimulatory capacity of tumor‐infiltrating DC against allogeneic T‐cells (Fig. 4a,b). The Flt3L–IVE alone did not alter CD86 expression on tumor‐infiltrating CD11c+ DC. Intra‐tumoral treatment of Ad.IL‐18 significantly augmented CD86 expression compared with those with Ad.EGFP as a control (Fig. 4a). In the mice treated with Ad.IL‐18, significantly higher allostimulatory activities were observed when compared with those with the other treatment (Fig. 4b).
Figure 4.

Adenovirus‐vector expressing (Ad.) interferon‐γ‐inducing factor (IL‐18) i.t. up‐regulated CD86 expression on tumor‐infiltrating dendritic cells (DC), and these DC showed stronger allogeneic T‐cell stimulatory capacity. (a) CD86 expression of tumor‐infiltrating CD11c+ DC by fluorescence‐activated cell sorting (FACS) analysis. Results are representative of three separate experiments. (b) Their ability to stimulate allogeneic T‐cell proliferation. *P < 0.05, **P < 0.01 Results are representative of three separate experiments.
Recombinant IL‐18 induces the maturation of myeloid DC propagated by Flt3L in vitro. To confirm the in vivo results of Ad.IL‐18, we generated DC by Flt3L in vitro and exposed them to recombinant IL‐18 (rIL‐18) protein for 48 h. Flow cytometric analysis of IL‐18‐exposed Flt3L DC showed up‐regulated surface expression of CD86, CD80, CD40, MHC class I and MHC class II in a dose‐dependent manner (Fig. 5a). These results suggest that IL‐18 induces the maturation of Flt3L‐propagated myeloid DC. However, rIL‐18 did not alter the expression of CD11c, CD11b and B220 on Flt3L propagated cells (data not shown).
Figure 5.

The BMDC propagated with Fms‐like thyrosine kinase 3 ligand (Flt3L) in vitro showed up‐regulated expression of costimulatory and major histocompatability complex (MHC) molecules with the addition of interferon‐γ‐inducing factor (IL‐18) in a dose‐dependent manner. This effect was partially dependent on interferon (IFN)‐γ. Bone marrow cells (cells) were cultured for 9 days in the presence of human Flt3L. After culture with IL‐18 for the last 48 h, the phenotype was determined by double staining with anti‐CD11c and a panel of Abs to costimulatory molecule (a) IL‐18 alone and (b) IL‐18 with anti‐IFN‐γ antibody; The results shown are the mean of three separate experiments. *P < 0.05, **P < 0.01.
To elucidate whether IFN‐γ was related to DC maturation, we added anti‐IFN‐γ antibody to the culture medium. The effects of up‐regulation of CD86, CD40 and MHC class I molecules by rIL‐18 on DC were significantly but partially suppressed when compared with those with isotype antibody. These results indicated that up‐regulation of costimulatory molecules with IL‐18 treatment was partially dependent on IFN‐γ induced by IL‐18 (Fig. 5b).
Discussion
In the present study, we have shown that local gene transfer of IL‐18 combined with DC mobilization in vivo with Flt3L–IVE may enhance the antitumor effect through the induction of potent systemic antitumor immunity. We have also shown that intra‐tumoral gene transfer of IL‐18 may activate not only NK‐cells but also tumor‐infiltrating DC mobilized by Flt3L in vivo. These results might suggest that IL‐18 activated both DC and NK‐cells, which were mobilized in vivo into the tumor environment by Flt3L–IVE.
DC are potent APC and have a unique capacity to effectively acquire, process and present the antigens to T‐cells, and express costimulatory molecules and pro‐inflammatory cytokines. The clinical application of DC for cancer vaccines, however, has been only moderately successful.( 15 ) In most clinical approaches, DC have been propagated, stimulated to be matured in vitro, pulsed with tumor antigens including peptides and tumor lysate, and they have been infused. However, these cultured DC might not have optimal homing capacity in vivo to lymph nodes where DC most efficiently present tumor antigens to T‐cells and generate tumor‐specific immunity.( 16 ) If DC with appropriate functions could be efficiently propagated in vivo, we could avoid troublesome procedures related to in vitro culture including leukocyte apheresis and concerns about the dysfunction of cultured DC. Furthermore, it would not be necessary to identify tumor‐associated antigens which are readily available from tumors existing in the patients’ nown bodies. Flt3L is a cytokine that mobilizes both DC and NK‐cells in vivo.( 2 , 3 ) Although some studies using Flt3L have shown the induction of effective antitumor immunity as well with Flt3L administration alone, only a marginal antitumor effect was observed in our study using IVE with Flt3L.( 6 ) Several clinical studies have demonstrated that DC mobilized by Flt3L in vivo were only partially activated, and the expected clinical responses were not obtained.( 9 , 17 ) Thus, further investigations appear to be necessary to induce most potent antitumor immune responses using Flt3L. IL‐18 was initially termed IFN‐γ‐inducing factor based its ability to induce IFN‐γ secretion from NK and T‐cells at levels substantially greater than that observed with IL‐12, another potent IFN‐γ inducer.( 18 , 19 )
We have previously reported that DC can capture tumor antigens from tumor cells killed by NK‐cells activated with IL‐18 and efficiently induce tumor‐specific CTL in vitro.( 11 ) These findings strongly suggest that IL‐18 could exert significant in vivo immunoregulatory functions that could modulate the tumor microenvironment where an appropriate number of DC is available.
In the present study, we showed that combination therapy of Flt3L–IVE with Ad.IL‐18 was superior to Flt3L alone or Ad.IL‐18 i.t. alone both in the suppression of the injected and the distant un‐injected tumors. Depletion of asialoGM1 positive cells completely impaired the accumulation of CD8‐positive T‐cells in the tumor. We have previously reported that IL‐18 induces specific immunity but depletion of asialo GM1 positive cells, primarily NK‐cells, which substantially abrogate the IL‐18‐mediated antitumor effects.( 20 ) Furthermore, we have also previously demonstrated that tumor cell destruction by NK‐cells is an important initiating event for inducing tumor‐specific CTL by IL‐18 in vitro.( 11 ) These findings suggest that IL‐18 enhances NK activity, inducing death of viable tumor cells that in turn seem to be acquired and processed by DC to promote CTL generation from näive T‐cells. Thus, DC mobilization into the tumor microenvironment with Flt3L treatment can promote their ability to capture the antigens of tumor cells killed by NK‐cells activated with IL‐18 and Flt3L in vivo. Recently, it has been demonstrated that NK–DC interaction promotes the CTL induction, and NK‐cells can facilitate adaptive antitumor immunity by producing IFN‐γ and other cytokines that recruit and activate DC.( 21 , 22 ) Our results suggest that these synergistic effects induce systemic and tumor specific cellular immune responses. The depletion of asialo GM1 positive cells may involve not only NK‐cells but also NKT‐cells. We previously demonstrated that NK‐cells, but not NKT‐cells, are the major effectors in IL‐18‐induced innate immunity.( 23 ) However, NKT‐cells may participate in induction of CD8 positive CTL mediated by IL‐18 in this study. We will further examine the roles of NKT‐cells in this combination therapy.
We also found a difference in the localization of mobilized DC between combination therapy and Flt3L treatment alone. Vermi et al. reported that tumor‐associated DC are present mostly at the periphery of tumors.( 24 ) Recently, multiple studies have demonstrated that DC in the tumor have functional defects that are affected by the tumor milieu.( 25 ) However, Furumoto et al. suggested that tumor‐infiltrating DC with both penetration and activation capacities into the tumor bed could induce adaptive immunity using a CCL20‐transfected tumor vaccine model.( 26 ) In our study, Flt3L primarily mobilize DC into the peri‐tumoral area of the tumors. Interestingly, Flt3L can mobilize these DC not only into the peri‐tumoral area but also into the tumor bed only when IL‐18 treatment is combined. These tumor‐infiltrating DC showed more potent allostimulatory capacities compared with those with Flt3L alone. Furthermore, our in vitro results may suggest that the maturation of Flt3L‐propagated DC with IL‐18 is primarily mediated by IFN‐γ induced with IL‐18. Thus, this combination therapy may augment the capacity of tumor‐infiltrating DC to induce potent and systemic antitumor immunity.
In conclusion, we found that local administration of IL‐18 combined with DC mobilization in vivo with Flt3L could increase and activate tumor‐infiltrating DC and NK‐cell, resulting in enhancement of the antitumor effect through the induction of potent systemic antitumor immunity. This combination of immunotherapy targeting DC in and NK‐cells in vivo may provide a novel strategy to manipulate the local tumor microenvironment that could be altered to support the initiation and propagation of an effective tumor‐specific immune response.
Acknowledgments
This study was supported by Grant‐in‐aid for Scientific Research from the Japan Society for Promotion of Science (to T.T.). We thank Ms S. Nakayama and Ms. A. Asami (Department of Surgery and Bioengineering, Advanced Clinical Research Center, Institute of Medical Science, University of Tokyo) for their excellent technical assistance.
References
- 1. Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol 2003; 3: 630–41. [DOI] [PubMed] [Google Scholar]
- 2. Maraskovsky E, Brasel K, Teepe M et al . Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand‐treated mice: multiple dendritic cell subpopulations identified. J Exp Med 1996; 184: 1953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shaw SG, Maung AA, Steptoe RJ, Thomson AW, Vujanovic NL. Expansion of functional NK cells in multiple tissue compartments of mice treated with Flt3‐ligand: implications for anti‐cancer and anti‐viral therapy. J Immunol 1998; 161: 2817–24. [PubMed] [Google Scholar]
- 4. Chaudhry UI, Katz SC, Kingham TP et al . In vivo overexpression of Flt3 ligand expands and activates murine spleen natural killer dendritic cells. FASEB J 2006; 20: 982–4. [DOI] [PubMed] [Google Scholar]
- 5. Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo . Nat Med 1997; 3: 625–31. [DOI] [PubMed] [Google Scholar]
- 6. Shimao K, Takayama T, Enomoto K et al . Cancer gene therapy using in vivo electroporation of Flt3‐ligand. Int J Oncol 2005; 27: 457–63. [PubMed] [Google Scholar]
- 7. Favre‐Felix N, Martin M, Maraskovsky E et al . Flt3 ligand lessens the growth of tumors obtained after colon cancer cell injection in rats but does not restore tumor‐suppressed dendritic cell function. Int J Cancer 2000; 86: 827–34. [DOI] [PubMed] [Google Scholar]
- 8. Chen K, Braun S, Lyman S et al . Antitumor activity and immunotherapeutic properties of Flt3‐ligand in a murine breast cancer model. Cancer Res 1997; 57: 3511–6. [PubMed] [Google Scholar]
- 9. Morse MA, Nair S, Fernandez‐Casal M et al . Preoperative mobilization of circulating dendritic cells by Flt3 ligand administration to patients with metastatic colon cancer. J Clin Oncol 2000; 18: 3883–93. [DOI] [PubMed] [Google Scholar]
- 10. Dinarello CA, Novick D, Puren AJ et al . Overview of interleukin‐18: more than an interferon‐gamma inducing factor. J Leukoc Biol 1998; 63: 658–64. [PubMed] [Google Scholar]
- 11. Tanaka F, Hashimoto W, Okamura H, Robbins PD, Lotze MT, Tahara H. Rapid generation of potent and tumor‐specific cytotoxic T lymphocytes by interleukin 18 using dendritic cells and natural killer cells. Cancer Res 2000; 60: 4838–44. [PubMed] [Google Scholar]
- 12. Chaudhry UI, Kingham TP, Plitas G, Katz SC, Raab JR, DeMatteo RP. Combined stimulation with interleukin‐18 and CpG induces murine natural killer dendritic cells to produce IFN‐gamma and inhibit tumor growth. Cancer Res 2006; 66: 10 497–504. [DOI] [PubMed] [Google Scholar]
- 13. Goto H, Osaki T, Nishino K et al . Construction and analysis of new vector systems with improved interleukin‐18 secretion in a xenogeneic human tumor model. J Immunother 2002; 25 (Suppl 1): S35–41. [DOI] [PubMed] [Google Scholar]
- 14. Gilliet MA, Boonstra C, Paturel S et al . The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3‐ligand and granulocyte/macrophage colony‐stimulating factor. J Exp Med 2002; 195: 953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10: 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fong L, Brockstedt D, Benike C, Wu L, Engleman EG. Dendritic cells treated via different routes induce immunity in cancer patients. J Immunol 2001; 166: 4254–9. [DOI] [PubMed] [Google Scholar]
- 17. Marroquin CE, Westwood JA, Lapointe R et al . Mobilization of dendritic cell precursors in patients with cancer by flt3 ligand allows the generation of higher yields of cultured dendritic cells. J Immunother 2002; 25: 278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okamura H, Tsutsi H, Komatsu T et al . Cloning of a new cytokine that induces IFN‐gamma production by T cells. Nature 1995; 378: 88–91. [DOI] [PubMed] [Google Scholar]
- 19. Osaki T, Péron JM, Cai Q et al . IFN‐gamma‐inducing factor/IL‐18 administration mediates IFN‐gamma‐ and IL‐12‐independent antitumor effects. J Immunol 1998; 160: 1742–9. [PubMed] [Google Scholar]
- 20. Osaki T, Hashimoto W, Gambotto A et al . Potent antitumor effects mediated by local expression of the mature form of the interferon‐gamma inducing factor, interleukin‐18 (IL‐18). Gene Ther 1999; 6: 808–15. [DOI] [PubMed] [Google Scholar]
- 21. Kelly JM, Darcy PK, Markby JL et al . Induction of tumor‐specific T cell memory by NK cell‐mediated tumor rejection. Nat Immunol 2002; 3: 83–90. [DOI] [PubMed] [Google Scholar]
- 22. Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural‐killer cells and dendritic cells: ‘l’union fait la force’. Blood 2005; 106: 2252–8. [DOI] [PubMed] [Google Scholar]
- 23. Hashimoto W, Tanaka F, Robbins PD et al . Natural killer, but not natural killer T, cells play a necessary role in the promotion of an innate antitumor response induced by IL‐18. Int J Cancer 2003; 103: 508–13. [DOI] [PubMed] [Google Scholar]
- 24. Vermi W, Bonecchi R, Facchetti F et al . Recruitment of immature plasmacytoid dendritic cells (plasmacytoid monocytes) and myeloid dendritic cells in primary cutaneous melanomas. J Pathol 2003; 200: 255–68. [DOI] [PubMed] [Google Scholar]
- 25. Gabrilovich D. Mechanisms and functional significance of tumour‐induced dendritic‐cell defects. Nat Rev Immunol 2004; 4: 941–52. [DOI] [PubMed] [Google Scholar]
- 26. Furumoto K, Soares L, Engleman EG, Merad M. Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J Clin Invest 2004; 113: 774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]