

Abstract
The association of hepatocyte growth factor (HGF) with its high‐affinity receptor (c‐Met) has been shown to induce mitogenesis, motogenesis and morphogenesis in a variety of cell types. Various point mutations in c‐Met have been identified in hereditary and sporadic papillary renal carcinomas as well as in other carcinomas. In the present study, we examined the effects of c‐Met point mutations on the morphology of a porcine aortic endothelial (PAE) cell line. When cultured in three‐dimensional collagen gel, PAE cells formed branching tubule structures, and HGF treatment caused breakdown of the structures and induced a scattered morphology. The exogenous expression of c‐Met point mutants inhibited the formation of tubules. HGF treatment induced the formation of tubules by PAE cells expressing some c‐Met mutants, but it induced the scattering of PAE cells expressing other c‐Met mutants. The presence of a low concentration of a mitogen‐activated protein kinase/extracellular signal‐regulated kinase kinase (MEK) inhibitor cancelled the inhibitory effect of the c‐Met point mutations on the formation of tubules. These results suggest that c‐Met point mutations affect the extracellular signal‐regulated kinase (ERK) signaling required for the formation of tubules by PAE cells, and HGF binding changes the conformation of c‐Met mutants, leading to the different signals required for formation of tubules and cell scattering. (Cancer Sci 2006; 97: 1343–1350)
Hepatocyte growth factor (HGF) is a mesenchymal cell‐derived glycoprotein described originally as a potent mitogen for primary hepatocytes.( 1 , 2 ) Subsequent studies revealed that HGF is also mitogenic for other cell types including epithelial cells, melanocytes and endothelial cells.( 3 ) Furthermore, HGF is identical to scatter factor, which dissociates epithelial cell colonies by causing a breakdown of intercellular junctions and induces a transition from an epithelial to a scattered fibroblastic morphology.( 4 , 5 ) HGF also induces branching tubule formation of epithelial cells grown in three‐dimensional collagen gel.( 6 , 7 ) In vivo, HGF is a potent angiogenic factor,( 8 , 9 ) and is involved in organ regeneration( 10 ) and tumorigenesis.( 11 ) Mice lacking HGF show placental defects and a reduced liver size and die in utero, indicating that HGF is a growth factor essential for organogenesis during normal embryonic development.( 12 , 13 )
The pleiotropic biological effects of HGF are transduced through the activation of its high‐affinity receptor, the c‐met protooncogene product (c‐Met).( 14 , 15 , 16 , 17 ) c‐Met possesses a tyrosine kinase domain in the cytoplasmic region. Upon binding of HGF to c‐Met, the tyrosine kinase is activated, and several tyrosine residues within the cytoplasmic region are highly phosphorylated. The phosphorylation of two residues (Tyr1234 and Tyr1235) in the kinase domain is essential for the catalytic activity,( 18 , 19 ) whereas the phosphorylation of two residues (Tyr1349 and Tyr1356) in the C‐terminal region mediates the binding of signaling proteins such as the p85 regulatory subunit of phosphoinositide 3‐kinase (PI3K), pp60c–Src, phospholipase Cγ, Shc, Grb2 and Gab1.( 20 , 21 , 22 ) The binding of these proteins stimulates intracellular signaling pathways mediating the biological effects of HGF.
Various point mutations in the c‐met gene, which result in amino acid substitutions in the kinase domain and in the juxtamembrane domain of c‐Met, have been identified in hereditary and sporadic papillary renal carcinoma as well as in other carcinomas.( 23 ) The biochemical and biological properties of these c‐Met mutants have been investigated by exogenously expressing the c‐Met cDNA in a variety of cultured cells such as NIH3T3 cells, MDCK cells and MLP29 cells.( 24 , 25 , 26 ) The c‐Met mutants exhibit increased levels of tyrosine phosphorylation and enhanced kinase activity compared with wild‐type c‐Met. NIH3T3 cells expressing mutant c‐Met molecules form foci in vitro and are tumorigenic in nude mice, and a strong correlation between the kinase activity and the transforming activity is observed.( 24 ) However, other biological properties are not correlated with the levels of kinase activity. MLP29 cells expressing the c‐Met mutants L1195V or Y1230C, which have weaker kinase activity than the c‐Met mutants D1228H and M1250T, are more effective in inducing protection from apoptosis, sustaining anchorage‐independent growth and promoting invasion.( 26 ) These findings indicate that different c‐Met mutants activate distinct signaling pathways, which lead to functional diversity. Thus, it is important to investigate signaling pathways through the c‐Met mutants mediating specific biological consequences.
In the present study, we examined the effects of c‐Met point mutations on the morphology of a porcine aortic endothelial (PAE) cell line. PAE cells formed colonies with intercellular junctions in two‐dimensional culture, and HGF treatment caused the breakdown of intercellular junctions and led to a scattered morphology. When cultured in three‐dimensional collagen gels, PAE cells formed branching tubule structures, and HGF treatment caused breakdown of the structures. The exogenous expression of c‐Met mutants did not affect the morphology of PAE cells in two‐dimensional culture in the absence or presence of HGF. However, it inhibited the formation of tubules by PAE cells in three‐dimensional culture. The inhibitory effects of the c‐Met mutants were examined further.
Materials and Methods
Reagents. Reagents were obtained as follows: antihuman c‐Met antibody (sc‐161) from Santa Cruz Biotechnology; antiphosphotyrosine antibody (PY20) from Transduction Laboratories; anti‐ERK2 antibody from Upstate Biotechnology; anti‐ERK1/2 antibody, antiphospho‐ERK1/2 (Thr202/Tyr204) antibody (E10), anti‐Akt antibody and antiphospho‐Akt (Ser473) antibody from Cell Signaling Technology; horseradish peroxidase (HRP)‐conjugated antimouse Ig and HRP‐conjugated antirabbit Ig from Amersham Pharmacia Biotech; recombinant human HGF from the Research Center of Mitsubishi Chemical Corp.;( 27 ) and PD98059, LY294002 and wortmannin from Calbiochem.
Plasmids. Human c‐Met cDNA was amplified from RNA of the HepG2 human hepatocellular carcinoma cell line by reverse transcription–polymerase chain reaction, and cloned into pME18S. Point mutations were introduced with a QuikChange site‐directed mutagenesis kit (Stratagene). The wild‐type and mutant c‐Met cDNA was excised from pME18S and subcloned into pIRES neo2 (Clonetech).
Cell culture and transfection. Porcine aortic endothelial cells were cultured in Ham's F12 medium containing 10% fetal bovine serum, 100 units/mL penicillin and 100 µg/mL streptomycin in a humidified atmosphere of 5% CO2 at 37°C. Cells were seeded at 3 × 105 cells/dish in a 6‐cm dish. After 24 h, the plasmid (2 µg) was transfected into the cells using the FuGENE‐6 reagent (Roche Molecular Biochemicals), and the transfected cells were cultured for 2 days. The cells were then cultured in selective medium containing G418 (500 µg/mL).
Preparation of cell extracts and immunoprecipitation. Cells were seeded at 5 × 105 cells/dish in a 10‐cm dish. At 72 h after seeding, cells were treated with HGF for 5 min. They were washed with cold phosphate‐buffered saline containing 1% ethylenediaminetetracetic acid (EDTA) and 0.2 mM Na3VO4 before being lysed with cold lysis buffer (137 mM NaCl, 8.1 mM Na2HPO4 · 12H2O, 2.68 mM KCl, 1.47 mM KH2PO4, 1% Nonidet P‐40, 1 mM Na3VO4, 5 mM EDTA and 0.5% sodium deoxycholate) containing 1 µg/mL leupeptin, 1 µg/mL aprotinin, 1 µg/mL pepstain A and 1 mM phenylmethyl sulfonate fluoride. The cell lysates were cleared by centrifugation, and the protein concentration in the lysates was measured using the BCA protein assay reagent (Pierce). Equal amounts of cell lysate were incubated with antibody and 20 µL of a 50% slurry of protein A–Sepharose (Amersham Pharmacia Biotech) for 1 h at 4°C. The immune complexes were precipitated and washed with lysis buffer.
Immunoblotting analysis. Cell lysates and precipitated immune complexes were boiled in Laemmli sample buffer (25 mM Tris‐HCl [pH 6.8], 1% sodium dodecylsulfate [SDS], 5% glycerol, 0.05% bromphenol blue and 1% 2‐mercaptoethanol). The samples were then separated by SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were incubated with the primary antibody for 1 h at room temperature and then with HRP‐conjugated secondary antibody for 1 h at room temperature. Immunoreactive proteins were visualized with an enhanced chemiluminescence western blotting detection system (Amersham Pharmacia Biotech).
Cell proliferation assay. Cells were seeded at 1 × 104 cells/well in 12‐well plates and cultured with Ham's F12 medium containing 10% fetal bovine serum for 24 h. They were subsequently cultured in the presence or absence of HGF. The cells were harvested after trypsinization, and the number of cells was counted using a hemocytometer.
Branching tubule formation assay. A collagen solution was prepared by mixing 0.3% Cell Matrix Type I‐A (Iwaki), 10× Ham's F12, diluted hydrochloric acid (pH 3.0) and reconstitution buffer at a ratio of 6:1:2:1. The collagen solution (500 µL) was solidified in 12‐well plates by incubating at 37°C for 10 min (base layers). The collagen solution (500 µL) containing PAE cells (5 × 104 cells/mL) was put on base layers and solidified by incubating at 37°C for 10 min (cell layers). Ham's F12 containing 10% fetal bovine serum was overlaid on the gels in the presence or absence of HGF.
Electron microscopy. Samples were fixed in 2.5% glutaraldehyde. Sections for electron microscopy were postfixed with osmium tetroxide for 1 h, dehydrated with ethanol, and embedded in Epon 812. Sections were then stained with uranyl acetate and lead citrate, and analyzed using a JEOL 1200EX electron microscope (JEOL Ltd.).
Results
Morphology of PAE cells in the absence or presence of HGF. We chose the PAE cell line, established from porcine aortic endothelial cells,( 28 ) to investigate the effects of point mutations in c‐Met on cell morphology, because overexpression of c‐Met in this cell line results in a scattering of cells in two‐dimensional culture.( 29 ) Because the effect of HGF on the morphology of PAE cells has not been studied, we first examined whether the morphology of PAE cells changed in two‐dimensional culture in response to HGF. In the absence of HGF, PAE cells formed a tight monolayer with junctional complexes. When added to the culture, HGF caused the breakdown of intercellular junctions and led to a scattered morphology (Fig. 1A). PAE cells expressing vascular endothelial growth factor receptor‐1 plated on growth factor‐reduced Matrigel rearrange into tubule structures.( 30 ) Moreover, HGF induces the formation of branching tubules by MDCK cells cultured in three‐dimensional collagen gel,( 6 ) and HGF stimulates the formation of tubules by bovine brain endothelial cells cultured in dishes coated with Matrigel.( 31 ) Thus, it is assumed that HGF induces PAE cells to form tubules. To test this, we examined the effect of HGF on the morphology of PAE cells cultured in three‐dimensional collagen gel. Unexpectedly, PAE cells formed branching tubule‐like structures in the absence of HGF (Fig. 1B). To examine the tubule‐like structures in more detail, sections were analyzed by electron microscopy. The analysis showed that the structures were hollows surrounded with cells (Fig. 1C), indicating that PAE cells form tubule structures when cultured in three‐dimensional collagen gel in the presence of serum. Addition of HGF to the culture caused the breakdown of the tubule structures and led to a scattered morphology (Fig. 1B). HGF did not significantly affect the proliferation of PAE cells cultured in the presence of serum (data not shown). These results indicate that PAE cells have endogenous c‐Met, and HGF treatment does not enhance the proliferation of PAE cells but induces a scattered morphology both in two‐dimensional culture and in three‐dimensional culture in collagen gel.
Figure 1.
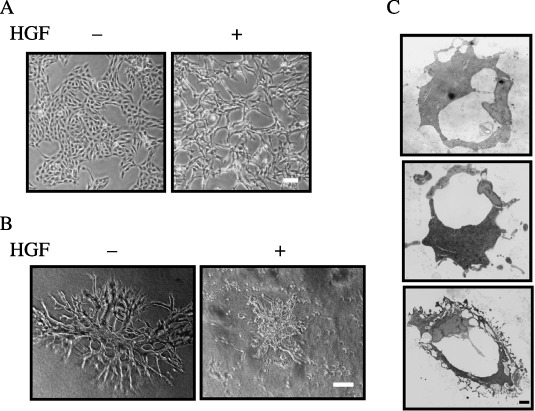
Morphology of porcine aortic endothelial (PAE) cells in the absence or presence of hepatocyte growth factor (HGF). (A) Morphology of PAE cells in two‐dimensional culture. PAE cells were cultured for 24 h in the absence (–) or presence (+) of HGF (50 ng/mL). The morphology of the cells was analyzed by light microscopy. Scale bar = 10 µm. (B) Morphology of PAE cells cultured in three‐dimensional collagen gel. PAE cells were cultured for 8 days in the absence (–) or presence (+) of HGF (50 ng/mL). The morphology of the cells was analyzed by light microscopy. Scale bar = 10 µm. (C) Electron microscopic analysis of sections of the tubule structure formed by PAE cells in three‐dimensional culture in the absence of HGF. Scale bar = 1 µm.
Isolation of PAE cell clones expressing mutant forms of c‐Met. Various point mutations have been identified in the kinase domain of c‐Met.( 23 ) We selected four well‐characterized mutations to examine their effects on the morphology of PAE cells. Two of them (M1131T and V1188L) were located in the hinge region, one (Y1230C) was in the activation loop, and one (M1250T) was in the P + 1 loop. It has been reported that c‐Met mutants M1131T and V1188L show weaker kinase activity and less transforming activity than c‐Met mutants Y1230C and M1250T when expressed in NIH3T3 cells.( 24 )
To obtain PAE cell clones expressing the mutant forms of c‐Met, cDNA encoding each of the mutant forms was inserted into the expression plasmid pIRESneo2. To obtain control cells, cDNA encoding wild‐type c‐Met was also inserted into the same plasmid. The expression plasmids were transfected into PAE cells. Clones resistant to G418 were selected and expanded. They were subjected to an analysis of the expression of c‐Met proteins by immunoblotting. Two clones for each mutant c‐Met and wild‐type c‐Met were selected for further analysis. c‐Met proteins were highly expressed in all clones (Fig. 2B). To examine the tyrosine‐phosphorylation status of c‐Met, each clone was left untreated or treated with HGF and phosphorylation was analyzed by immunoprecipitation with an anti‐c‐Met antibody followed by immunoblotting with an antiphosphotyrosine antibody. The phosphorylation levels were undetectable or low in untreated cells expressing wild‐type and mutant c‐Met. HGF treatment enhanced the phosphorylation of all c‐Met mutants as well as wild‐type c‐Met (Fig. 2A).
Figure 2.
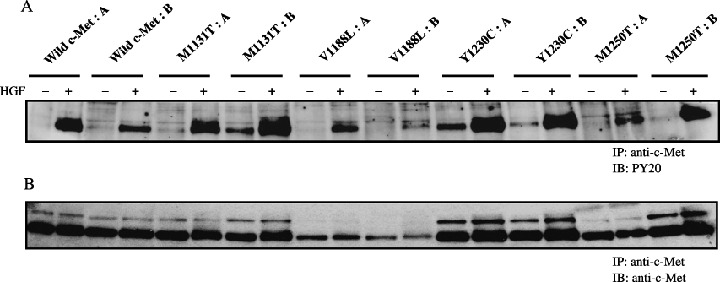
Immunoblotting analysis of the expression and phosphorylation of c‐Met proteins. Porcine aortic endothelial (PAE) cells expressing c‐Met proteins were treated for 5 min with (+) or without (–) hepatocyte growth factor (HGF) (50 ng/mL). Lysates of the cells were immunoprecipitated (IP) with an anti‐c‐Met antibody and the immunoprecipitates were immunoblotted (IB) with an antiphosphotyrosine antibody (PY20) (A) and with an anti‐c‐Met antibody (B).
Phenotypes of PAE cells expressing c‐Met mutants. Phenotypes of PAE cells expressing c‐Met mutants were examined in three assay systems. First, the effect of HGF on the proliferation of PAE cells expressing c‐Met mutants was analyzed. The PAE cell clones were cultured for 3 days in the absence or presence of HGF, and cell numbers were counted. HGF treatment did not significantly affect the proliferation of any of the cell clones (data not shown). Second, the effect of HGF on cell scattering in two‐dimensional culture was analyzed. All clones formed a tight monolayer with junctional complexes in the absence of HGF similar to parental PAE cells. HGF treatment for 24 h induced a scattered morphology (Fig. 3). Finally, the effect of HGF on the formation of tubule structures in three‐dimensional collagen gel was analyzed. PAE cell clones expressing wild‐type c‐Met formed branching tubule structures in the absence of HGF, and HGF treatment caused breakdown of the tubule structures, similar to parental cells (Fig. 4). Interestingly, PAE cell clones expressing c‐Met mutants did not form the tubule structures in the absence of HGF. HGF‐treated cells expressing M1131T or V1188L formed less‐branched tubule structures, whereas cells expressing Y1230C or M1250T did not form tubule structures and showed a scattered morphology (Fig. 4). These results indicate that the point mutations in c‐Met do not affect the proliferation of PAE cells and the morphology of PAE cells cultured in two‐dimensions in the absence or presence of HGF, but they inhibit the formation of tubules by PAE cells cultured in three‐dimensions.
Figure 3.
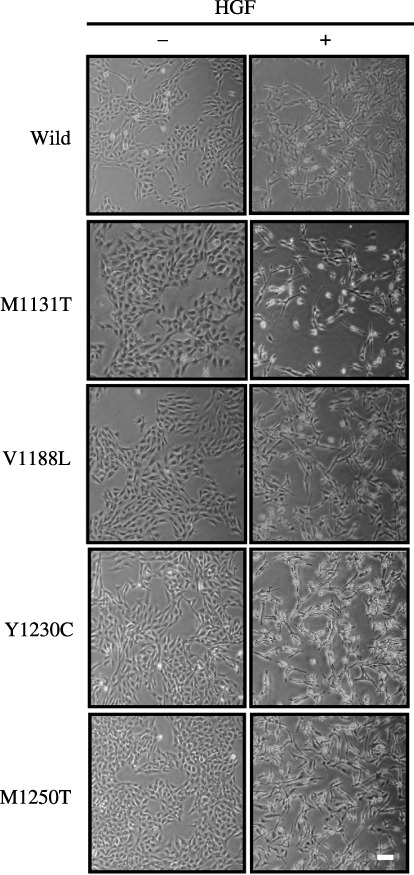
Morphology of porcine aortic endothelial (PAE) cells expressing c‐Met mutants in two‐dimensional culture. PAE cells were cultured for 24 h in the absence (–) or presence (+) of hepatocyte growth factor (HGF) (50 ng/mL). The morphology of the cells was analyzed by light microscopy. Similar results were obtained in two clones of wild‐type and mutant c‐Met. The results for one of them are shown. Scale bar = 10 µm.
Figure 4.
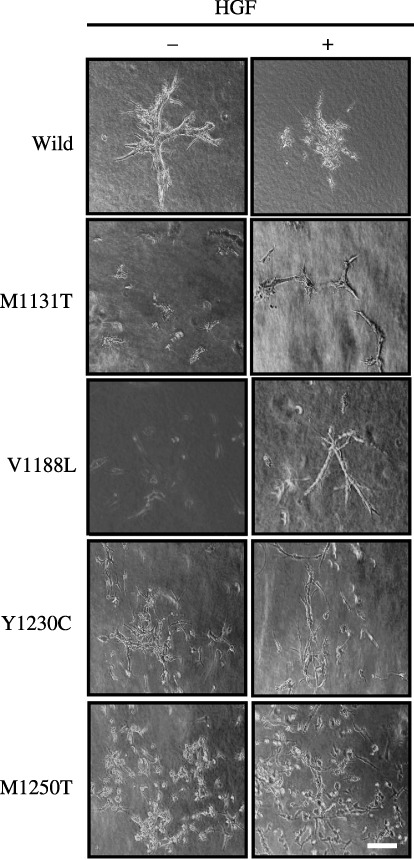
Morphology of porcine aortic endothelial (PAE) cells expressing c‐Met mutants in three‐dimensional culture. PAE cells were cultured for 8 days in the absence (–) or presence (+) of hepatocyte growth factor (HGF) (50 ng/mL). The morphology of the cells was analyzed by light microscopy. Similar results were obtained in two clones of the wild‐type and mutant c‐Met. The results for one of them are shown. Scale bar = 10 µm.
Effects of MEK‐specific and PI3K‐specific inhibitors on the morphology of PAE cells. Because the Ras/ERK and PI3K pathways play a critical role in the formation of tubules by other cell types,( 7 , 32 , 33 , 34 ) they may be involved in the formation of tubules by PAE cells. The effects of a specific inhibitor of MEK (PD98059), an upstream kinase of ERK, and specific inhibitors of PI3K (LY294002 and wortmannin) were tested on the morphology of PAE cells. The concentration of MEK inhibitor that suppressed MEK activity was determined using immunoblotting analysis of ERK1 and 2 phosphorylation. The phosphorylation of ERK2 was detected by a mobility shift of the phosphorylated protein using an ERK2‐specific antibody (Fig. 5A), and also by immunoblotting analysis using an antiphosphorylated ERK1/2 antibody, which also detected the phosphorylation of ERK1 (Fig. 5B). Expression levels of ERK1 and 2 were analyzed by immunoblotting using an anti‐ERK1/2 antibody, which recognized both ERK1 and 2. The expression level of ERK2 was much higher than that of ERK1 (Fig. 5B). The phosphorylation of ERK1 and 2 was undetectable in the absence of HGF, and HGF treatment induced the phosphorylation. This phosphorylation was completely suppressed by pretreatment with 50 µM PD98059 (Fig. 5A,B). The concentrations of PI3K inhibitors that suppressed PI3K activity were determined using immunoblotting analysis of the phosphorylation of Ser473 of Akt, a downstream target of PI3K. Phosphorylation of Akt was undetectable in the absence of HGF, and HGF treatment induced the phosphorylation. This phosphorylation was completely suppressed by pretreatment with 100 nM wortmannin or 50 µM LY294002 (Fig. 5C). Addition of 50 µM PD98059, 100 nM wortmannin or 50 µM LY294002 inhibited the formation of tubules by PAE cells in three‐dimensional culture in the absence of HGF (Fig. 6). These inhibitors also inhibited the HGF‐induced scattering of PAE cells in two‐dimensional culture (data not shown). These results suggest that both the Ras/ERK and PI3K pathways are involved in the formation of tubules by PAE cells and HGF‐induced scattering of PAE cells.
Figure 5.
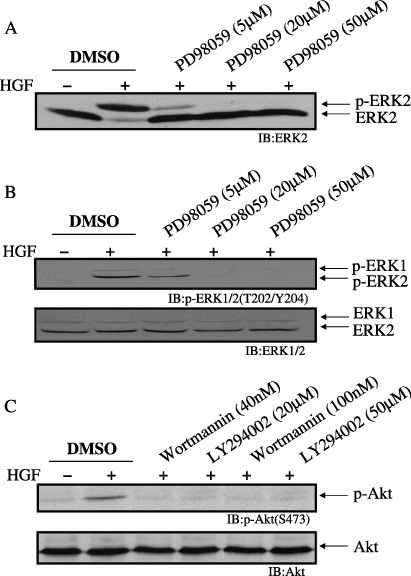
Effect of mitogen‐activated protein kinase/extracellular signal‐regulated kinase kinase (MEK)‐ or phosphoinositide 3‐kinase (PI3K)‐specific inhibitors on extracellular signal‐regulated kinase (ERK) phosphorylation and Akt (Ser473) phosphorylation. Porcine aortic endothelial (PAE) cells were pretreated for 1 h with vehicle (dimethylsulfoxide; DMSO), PD98059 (5 µM, 20 µM, 50 µM) (A and B), wortmannin (40 nM or 100 nM) or LY294002 (20 µM or 50 µM) (C), and treated for 5 min with (+) or without (–) hepatocyte growth factor (HGF) (50 ng/mL). Lysates of the cells were immunoblotted with an anti‐ERK2 antibody (A), an anti‐p‐ERK1/2 (T202/Y204) (B, upper panel) or anti‐ERK1/2 antibody (B, lower panel), an anti‐p‐Akt (S473) (C, upper panel) or anti‐Akt antibody (C, lower panel). (A) Phosphorylation of ERK2 proteins is indicated by a shift to a slower electrophoretic mobility. This mobility shift was not detected in (B), because the sodium dodecylsulfate–polyacrylamide gel electrophoresis system in (B) was different from that in (A).
Figure 6.
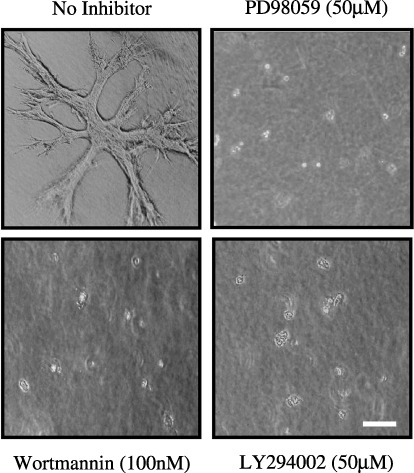
Effect of mitogen‐activated protein kinase/extracellular signal‐regulated kinase kinase (MEK)‐ or phosphoinositide 3‐kinase (PI3K)‐specific inhibitors on the morphology of porcine aortic endothelial (PAE) cells in three‐dimensional culture. PAE cells were cultured for 8 days without (no inhibitor) or with PD98059 (50 µM), wortmannin (100 nM) or LY294002 (50 µM). The morphology of the cells was analyzed by light microscopy. Scale bar = 10 µm.
Effect of the MEK‐specific inhibitor PD98059 on the morphology of PAE cells expressing c‐Met mutants in three‐dimensional culture. To examine the signaling pathways involved in the inhibitory effect of point mutations in c‐Met on the formation of tubules by PAE cells, PI3K and MEK inhibitors were tested. The presence of PI3K inhibitors, at both low and high concentrations, or 50 µM PD98059 inhibited the formation of tubules by PAE cells overexpressing wild‐type c‐Met in the absence of HGF, and also inhibited their scattering in the presence of HGF (data not shown). Similar inhibition was observed in PAE cells expressing the c‐Met mutants (data not shown). The presence of 5 µM PD98059 inhibited the formation of branching tubules by PAE cells overexpressing wild‐type c‐Met in the absence of HGF, but short and less‐branched tubule‐like structures were observed (Fig. 7B). Similar tubule‐like structures were also observed in PAE cells expressing the c‐Met mutants in the presence of 5 µM PD98059 and in the absence of HGF (Fig. 7B). These results suggest that the expression of c‐Met mutants affects the ERK pathway required for the formation of tubules by PAE cells. Treatment of HGF strongly induced the phosphorylation of ERK2 in PAE cells expressing wild‐type c‐Met and c‐Met mutants, and addition of 5 µM PD98059 partially suppressed the phosphorylation (Fig. 7A). Addition of HGF disrupted the short tubule‐like structures in PAE cells expressing wild‐type c‐Met and c‐Met mutants in the presence of 5 µM PD98059 (Fig. 7B).
Figure 7.
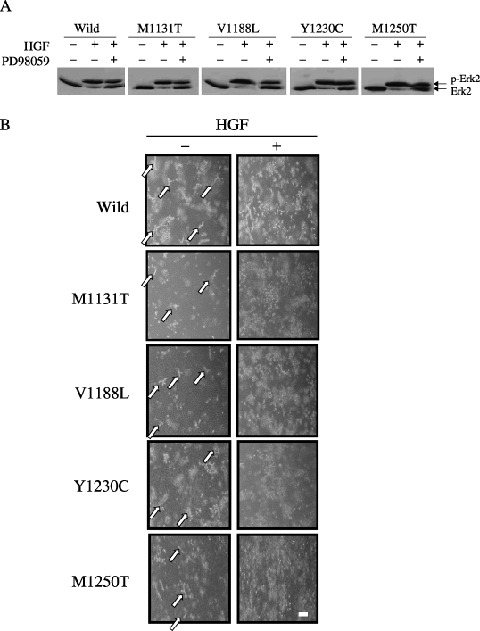
Effect of PD98059 on the morphology of porcine aortic endothelial (PAE) cells expressing c‐Met mutants in three‐dimensional culture. (A) PAE cells cultured in two‐dimensions were pretreated for 1 h with (+) or without (–) 5 µM PD98059, and treated with (+) or without (–) hepatocyte growth factor (HGF) (50 ng/mL). Lysates of the cells were immunoblotted with an anti‐ERK2 antibody. (B) PAE cells were cultured for 8 days in the absence (–) or presence (+) of HGF with 5 µM PD98059. The morphology of the cells was analyzed by light microscopy. Similar results were obtained in two clones of the wild‐type and mutant c‐Met. The results for one of them are shown. The arrows indicate the short tubule‐like structures. Scale bar = 20 µm.
Discussion
Hepatocyte growth factor dissociates epithelial cell colonies into individual cells and induces a scattered fibroblastic morphology in two‐dimensional culture.( 4 , 5 ) In the present study, we demonstrated that HGF also dissociates colonies of PAE cells and induces a scattered morphology in two‐dimensional culture (Fig. 1A), indicating that PAE cells have endogenous c‐Met and respond to HGF. The Ras/ERK and PI3K pathways are involved in the scattering of epithelial cells.( 32 , 33 , 34 , 35 , 36 ) These signaling pathways may mediate the scattering of PAE cells, because the MEK inhibitor and PI3K inhibitors suppressed the phenotype (data not shown). We also demonstrated that PAE cells form branching tubule structures when cultured in three‐dimensional collagen gel in the absence of HGF (Fig. 1B,C), suggesting that a factor present in serum induces PAE cells to form branching tubules. The formation of tubules is likely to be mediated by the ERK and PI3K pathways, because the MEK and PI3K inhibitors blocked the process (Fig. 6). However, we can not rule out the possibility that other pathways, which are suppressed by the MEK and PI3K inhibitors, mediate the formation of tubules, because it has not been shown that the serum factor activates the ERK and PI3K pathways (Fig. 5, HGF‐lanes). Epithelial cells cultured in three‐dimensional collagen gel do not form tubule structures in the presence of serum, and HGF is required for the formation of tubules.( 7 ) Thus, PAE cells possess the receptor for the serum factor, whereas epithelial cells may not. Alternatively, it is possible that an autocrine factor, to which epithelial cells respond, might inhibit the signaling inducing the formation of tubules, and HGF treatment suppresses the production of the inhibitory factor.( 37 ) The presence of HGF caused the breakdown of the tubule structures of PAE cells and led to a scattered morphology (Fig. 1B), suggesting that the signaling by HGF inducing the scattered phenotype is stronger than the signaling by the serum factor inducing the formation of tubules by PAE cells. It is possible that the HGF‐induced breakdown of the tubule structure of PAE cells is caused by induction of apoptosis of the cells. However, extended two‐dimensional culture of PAE cells in the presence of HGF did not induce apoptosis, which was assessed by counting the number of viable cells in the presence or absence of HGF (data not shown). Thus, the breakdown of the tubule structure by HGF treatment may not be caused by induction of apoptosis.
Porcine aortic endothelial cells expressing the c‐Met mutants and wild‐type c‐Met formed colonies with tight intercellular junctions in two‐dimensional culture, and HGF treatment dissociated the colonies and induced a scattered morphology (Fig. 3). These phenotypes were similar to those of parental PAE cells. Thus, the mutations in c‐Met may not affect the morphology of PAE cells in two‐dimensional culture. Nakamura et al. reported that PAE cells overexpressing c‐Met show a scattered morphology in the absence of HGF.( 29 ) The discrepancy between our findings and theirs is probably due to different expression levels of the receptor.
Porcine aortic endothelial cells overexpressing wild‐type c‐Met formed branching tubule structures when cultured in three‐dimensional collagen gel, and HGF treatment caused breakdown of the tubule structures and induced a scattered morphology (Fig. 4). These phenotypes were similar to those of parental PAE cells, indicating that overexpression of wild‐type c‐Met does not affect the formation of tubules by PAE cells in the presence of the serum factor. In contrast, point mutations in c‐Met affect the formation of tubules by PAE cells. PAE cells expressing the c‐Met mutants did not form tubule structures (Fig. 4), indicating that expression of the c‐Met mutants inhibits the signaling required for the formation of tubules. HGF treatment induced the formation of tubules by PAE cells expressing the c‐Met mutants M1131T or V1188L, in spite of these cells exhibiting a scattered morphology in two‐dimensional culture. These results suggest that the binding of HGF to these mutants changes their conformation to that leading to the formation of tubules. In contrast, PAE cells expressing the c‐Met mutants Y1230C or M1250T did not form tubule structures in the presence of HGF, and exhibited a more scattered morphology than the PAE cells expressing wild‐type c‐Met (Fig. 4), suggesting that these mutant receptors induce the signaling mediating the scattered morphology. Analyses of the three‐dimensional structures of the wild‐type and mutant c‐Met catalytic core domains show that mutations M1131T and V1188L increase flexibility at critical points of the tertiary structure and facilitate subdomain movements, and mutations Y1230C and M1250T directly alter contacts between residues in the activation loop and those in the main body of the catalytic domain.( 38 ) These activation mechanisms may lead to the different three‐dimensional morphology of PAE cells expressing the c‐Met mutants.
Short and less‐branched tubule structures, which are likely to be a preceding stage to long and full‐branched tubule structures, were observed in PAE cells overexpressing wild‐type c‐Met treated with a low concentration (5 µM) of the MEK inhibitor PD98059 (Fig. 7B). Similar structures were observed in the PAE cells expressing the c‐Met mutants treated with the same concentration of PD98059 (Fig. 7B). These results suggest that the inhibitory effect of point mutations in c‐Met is cancelled by the presence of the low concentration of PD98059, and thus the ERK pathway downstream of the mutant c‐Met is involved in the inhibitory effect. It has been shown that sustained activation of the ERK pathway is required for MDCK cells to form the branching tubules, and the tyrosine phosphatase SHP‐2 is required for sustained activation.( 39 ) Thus, if a similar mechanism was to be used for the formation of tubules by PAE cells, point mutations in c‐Met might affect the sustained activation of the ERK pathway.
The biological properties of c‐Met point mutants have been investigated by expressing the c‐Met cDNA exogenously in a variety of cultured cells. These mutations cause focus formation, anchorage‐independent growth and cell motility, and are tumorigenic in nude mice.( 24 , 25 , 26 , 40 ) In addition, the influence of the mutations on tumorigenesis and metastasis in vivo has been examined. Mice with targeted mutations in the murine c‐met locus develop sarcomas, lymphomas and carcinomas.( 41 ) Transgenic mice harboring c‐Met mutants develop metastatic mammary carcinoma.( 25 ) These results indicate that the point mutations in c‐Met cause not only tumorigenesis but also metastasis. HGF is a potent angiogenic factor, and thus the c‐Met signaling could be involved in tumor angiogenesis. However, the effect of point mutations in c‐Met on angiogenesis has not been examined. In the present study, we found that the mutations inhibit the formation of branching tubules by PAE cells. Our findings raise the possibility that the mutations influence the signaling in tumor angiogenesis. Further analysis is needed to explore the possibility.
Acknowledgments
We thank Shigeru Iwasaka for technical assistance with electron microscopy. This work was supported by a Grant‐in‐Aid for Cancer Research (No. 17014032) from the Ministry of Education, Science, Sports and Culture of Japan to N. K.
References
- 1. Miyazawa K, Tsubouchi H, Naka D et al. Molecular cloning and sequence analysis of cDNA for human hepatocyte growth factor. Biochem Biophys Res Commun 1989; 163: 967–73. [DOI] [PubMed] [Google Scholar]
- 2. Nakamura T, Nishizawa T, Hagiya M et al. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989; 342: 440–3. [DOI] [PubMed] [Google Scholar]
- 3. Rubin JS, Chan AM‐L, Bottaro DP et al. A broad‐spectrum human lung fibroblast‐derived mitogen is a variant of hepatocyte growth factor. Proc Natl Acad Sci USA 1991; 88: 415–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast‐derived modulator of epithelial cell motility. Nature 1987; 327: 239–42. [DOI] [PubMed] [Google Scholar]
- 5. Weidner KM, Arakaki N, Hartmann G et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci USA 1991; 88: 7001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast‐derived epithelial morphogen as hepatocyte growth factor. Cell 1991; 67: 901–8. [DOI] [PubMed] [Google Scholar]
- 7. Rosario M, Birchmeier W. How to make tubes: signaling by the Met receptor tyrosine kinase. Trends Cell Biol 2003; 13: 328–35. [DOI] [PubMed] [Google Scholar]
- 8. Bussolino F, Di Renzo MF, Ziche M et al. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol 1992; 119: 629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grant DS, Kleinman HK, Goldberg ID et al. Scatter factor induces blood vessel formation in vivo . Proc Natl Acad Sci USA 1993; 90: 1937–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matsumoto K, Nakamura T. Roles of HGF as a pleiotropic factor in organ regeneration. In: Goldberg ID, Rosen EM, eds. Hepatocyte Growth Factor‐Scatter Factor (HGF‐SF) and the c‐Met Receptor. Basel: Birkhauser‐Verlag, 1993; 225–49. [PubMed] [Google Scholar]
- 11. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4: 915–25. [DOI] [PubMed] [Google Scholar]
- 12. Schmidt C, Bladt F, Goedecke S et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995; 373: 699–702. [DOI] [PubMed] [Google Scholar]
- 13. Uehara Y, Minowa O, Mori C et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995; 373: 702–5. [DOI] [PubMed] [Google Scholar]
- 14. Bottaro DP, Rubin JS, Faletto DL et al. Identification of the hepatocyte growth factor receptor as the c‐met proto‐oncogene product. Science 1991; 251: 802–4. [DOI] [PubMed] [Google Scholar]
- 15. Naldini L, Weidner KM, Vigna E et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J 1991; 10: 2867–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weidner KM, Sachs M, Birchmeier W. The Met receptor tyrosine kinase transduces motility, proliferation, and morphogenic signals of scatter factor/hepatocyte growth factor in epithelial cells. J Cell Biol 1993; 121: 145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Komada M, Kitamura N. The cell dissociation and motility triggered by scatter factor/hepatocyte growth factor are mediated through the cytoplasmic domain of the c‐Met receptor. Oncogene 1993; 8: 2381–90. [PubMed] [Google Scholar]
- 18. Komada M, Kitamura N. Regulatory role of major tyrosine auto‐phosphorylation site of kinase domain of c‐Met receptor (scatter factor/hepatocyte growth factor receptor). J Biol Chem 1994; 269: 16131–6. [PubMed] [Google Scholar]
- 19. Longati P, Bardelli A, Ponzetto C, Naldini L, Comoglio PM. Tyrosines 1234–1235 are critical for activation of the tyrosine kinase encoded by the MET proto‐oncogene (HGF receptor). Oncogene 1994; 9: 49–57. [PubMed] [Google Scholar]
- 20. Ponzetto C, Bardeli A, Zhen Z et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994; 77: 261–71. [DOI] [PubMed] [Google Scholar]
- 21. Zhu H, Naujokas MA, Fixman ED, Torossian K, Park M. Tyrosine 1356 in the carboxyl‐terminal tail of the HGF/SF receptor is essential for the transduction of signals for cell motility and morphogenesis. J Biol Chem 1994; 269: 29943–8. [PubMed] [Google Scholar]
- 22. Weidner KM, Di Cesare S, Sachs M, Brinkmann V, Behrens J, Bichmeier W. Interaction between Gab1 and the c‐Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996; 384: 173–6. [DOI] [PubMed] [Google Scholar]
- 23. Danilkovitch‐Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest 2002; 109: 863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeffers M, Schmidt L, Nakaigawa L et al. Activating mutations for the Met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci USA 1997; 94: 11 445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jeffers M, Fiscella M, Webb CP, Anver M, Koochekpour S, Vande Woude GF. The mutationally activated Met receptor mediates motility and metastasis. Proc Natl Acad Sci USA 1998; 95: 14417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giordano S, Mafee A, Williams TA et al. Different point mutations in the met oncogene elicit distinct biological properties. FASEB J 2000; 14: 399–406. [DOI] [PubMed] [Google Scholar]
- 27. Strain AJ, Ismail T, Tsubouchi H et al. Native and recombinant human hepatocyte growth factors are highly potent promoters of DNA synthesis in both human and rat hepatocytes. J Clin Invest 1991; 87: 1853–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyazono K, Okabe T, Urabe A, Yamanaka M, Takaku F. A platelet factor that stimulates the proliferation of vascular endothelial cells. Biochem Biophys Res Commun 1985; 126: 83–8. [DOI] [PubMed] [Google Scholar]
- 29. Nakamura T, Kanda S, Yamamoto K et al. Increase in hepatocyte growth factor receptor tyrosine kinase activity in renal carcinoma cells is associated with increased motility partly through phosphoinositide 3‐kinase activation. Oncogene 2001; 20: 7610–23. [DOI] [PubMed] [Google Scholar]
- 30. Bussolati B, Dunk C, Grohman M, Kontos CD, Mason J, Ahmed A. Vascular endothelial growth factor receptor‐1 modulates vascular endothelial growth factor‐mediated angiogenesis via nitric oxide. Am J Pathol 2001; 159: 993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosen EM, Grant D, Kleinman H et al. Scatter factor stimulates migration of vascular endothelium and capillary‐like tube formation. In: Goldberg ID, ed. Cell Motility Factors. Basel: Birkhauser‐Verlag, 1991; 77–88. [DOI] [PubMed] [Google Scholar]
- 32. Derman MP, Cunha MJ, Barros EJG, Nigam SK, Cantley LG. HGF‐mediated chemotaxis and tubulogenesis require activation of the phosphatidylinositol 3‐kinase. Am J Physiol 1995; 268: F1211–17. [DOI] [PubMed] [Google Scholar]
- 33. Khwaja A, Lehmann K, Marte BM, Downward J. Phosphoinositide 3‐kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J Biol Chem 1998; 273: 18793–801. [DOI] [PubMed] [Google Scholar]
- 34. Terauchi R, Kitamura N. Requirement of regulated activation of Ras for response of MDCK cells to hepatocyte growth factor/scatter factor. Exp Cell Res 2000; 256: 411–22. [DOI] [PubMed] [Google Scholar]
- 35. Royal I, Park M. Hepatocyte growth factor‐induced scatter of Madin–Darby canine kidney cells requires phosphatidylinositol 3‐kinase. J Biol Chem 1995; 270: 27780–7. [DOI] [PubMed] [Google Scholar]
- 36. Potempa S, Ridley AJ. Activation of both MAP kinase and phosphatidylinositide 3‐kinase by Ras is required for hepatocyte growth factor/scatter factor‐induced adherens junction disassembly. Mol Biol Cell 1998; 9: 2185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maeshima A, Zhang Y‐Q, Furukawa M, Naruse T, Kojima I. Hepatocyte growth factor induces branching tubulogenesis in MDCK cells by modulating the activin–follistatin system. Kidney Int 2000; 58: 1511–22. [DOI] [PubMed] [Google Scholar]
- 38. Miller M, Ginalski K, Lesyng B, Nakaigawa N, Schmidt L, Zbar B. Structural basis of oncogenic activation caused by point mutations in the kinase domain of the MET proto‐oncogene: modeling studies. Proteins 2001; 44: 32–43. [DOI] [PubMed] [Google Scholar]
- 39. Maroun CR, Naujokas MA, Holgado‐Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP‐2 required for sustained activation of extracellular signal‐regulated kinase and epithelial morphogenesis downstream from the Met receptor tyrosine kinase. Mol Cell Biol 2000; 20: 8513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma PC, Kijima T, Maulik G et al. c‐Met mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res 2003; 63: 6272–81. [PubMed] [Google Scholar]
- 41. Graveel C, Su Y, Koeman J et al. Activating Met mutations produce unique tumor profiles in mice with selective duplication of the mutant allele. Proc Natl Acad Sci USA 2004; 101: 17198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]