

Abstract
Intraperitoneal (i.p.) administration of paclitaxel nanoparticles (PTX‐30W) prepared by solubulization with the amphiphilic copolymer of 2‐methacryloxyethyl phosphorylcholine and n‐butyl methacrylate can efficiently suppress the growth of peritoneal metastasis. In this study, we characterized the drug distribution of i.p. injected PTX‐30W in peritoneal tumor and liver in a mouse model using MKN45, human gastric cancer cells. Oregon green‐conjugated PTX‐30W showed perivascular accumulation in MKN45 tumor in the peritoneum at 24 h after intravenous (i.v.) injection; however, the amount of PTX in tumor was markedly less than that in liver. In contrast, a larger amount of PTX accumulated in the peripheral area of disseminated nodules at 1 h after i.p. injection and the area gradually enlarged. The depth of PTX infiltration reached 1 mm from the tumor surface at 48 h after i.p. injection, and the fluorescence intensity was markedly greater than that in liver. Interestingly, i.p. injected PTX preferentially accumulated in relatively hypovascular areas, and many tumor cells in the vicinity of PTX accumulation showed apoptosis. This unique accumulation pattern and lesser washout in hypovascular areas are thought to be attributable to the superior penetrating activity of PTX‐30W, and thus, PTX‐30W is considered to be highly suitable for i.p. chemotherapy for peritoneal dissemination. (Cancer Sci 2011; 102: 200–205)
Peritoneal metastasis is one of the commonest patterns of tumor progression in gastrointestinal as well as ovarian cancer.( 1 , 2 ) Almost all previous reports have suggested that systemic perfusion of anti‐cancer drugs has a limited effect on peritoneal lesions, possibly due to the peritoneum‐plasma barrier, which prevents effective drug delivery from the blood stream into the peritoneal cavity.( 3 ) In comparison, intraperitoneal (i.p.) chemotherapy appears to have an advantage for peritoneal dissemination because the anti‐cancer agents are delivered directly into the peritoneal cavity, which enables contact of a high concentration of drug with tumor nodules. In particular, paclitaxel (PTX) is slowly absorbed, and consequently retained in the peritoneal cavity for longer periods due to its high molecular weight and hydrophobicity,( 4 , 5 ) and is thus thought to be suitable for i.p. chemotherapy. In fact, i.p. administration of PTX has been shown to have excellent clinical effects against peritoneal metastases in gastric cancer( 6 ) as well as ovarian cancer.( 7 , 8 , 9 )
For clinical use, hydrophobic PTX is usually dissolved in Cremophor EL, a mixture of polyoxyethylated castor oil and dehydrated ethanol. A water‐soluble and amphiphilic polymer composed of 2‐methacryloxyethyl phosphorylcholine (MPC) and n‐butyl methacrylate (BMA) (PMB30W) forms spontaneous nanoparticles containing PTX approximately 50 nm in diameter with almost 0 mV surface zeta potential( 10 , 11 ) and thus provides a highly water soluble formulation, and the outer MPC units diminish non‐specific capture by the reticuloendothelial system.( 12 ) In our previous study, we demonstrated that i.p. administration of nanoparticles containing PTX (PTX‐30W) significantly inhibited the growth of peritoneal metastases of gastric cancer and prolonged survival compared with conventional PTX dissolved in Cremophor EL in vivo, possibly due to the higher accumulation in peritoneal nodules.( 13 ) This is compatible with the previous study suggesting that intracavitary administration of macromolecular drugs results in slow clearance into the blood.( 14 )
In this study, therefore, we used fluorescence‐labeled PTX‐30W and examined the differences in the specific distribution of PTX‐30W in peritoneal nodules and the liver after i.v. and i.p. administration, and evaluated the clinical usefulness of i.p. chemotherapy from the viewpoint of drug delivery.
Materials and Methods
Materials. Oregon green‐conjugated 488 PTX (OG‐PTX) and a secondary antibody labeled with Alexa Fluors 594 were purchased from Molecular Probes (Portland, OR, USA). Rat monoclonal antibody to mouse platelet/endothelial cell adhesion molecule 1 (PECAM‐1) and DAPI were purchased from BD PharMingen (San Diego, CA, USA) and Wako Pure Chemical Industries Ltd (Osaka Japan), respectively. Hoechst 33342 was purchase from Sigma‐Aldrich (St Louis, MO, USA).
Cell culture. A human gastric cancer variant line producing peritoneal dissemination, MKN45P, established in our department,( 15 ) was routinely cultured in DMEM supplemented with 10% FCS, 100 units/mL penicillin and 100 μg/mL streptomycin (Sigma). After achieving subconfluence, the cells were removed by treatment EDTA and trypsin, and then used for experiments.
Preparation of PTX formulations. PMB30W was synthesized by a conventional radical polymerization reaction reported previously.( 12 , 16 ) The weight‐averaged molecular weight of the polymer was determined to be approximately 50 KDa. Then, 50 mg PMB30W was dissolved in 10 mL distilled water to make a 5.0% solution. Next, 50 mg PTX was dissolved in 1.0 mL ethanol. The PTX solution (1 mL) and the PMB30W aqueous solution (10 mL) were mixed in a sample tube and filtered using a micropore filter with 0.22 μm pore diameter. The ethanol was then removed under reduced pressure. The supernatant of the polymer solution was withdrawn to measure the peak area of PTX by high‐performance liquid chromatography (HPLC) with an ultraviolet detector.
Distribution of PTX in peritoneal nodules and the liver as determined by fluorescence microscopy. Four‐week‐old specific‐pathogen‐free conditioned female BALB/c nude mice were purchased from Charles River Japan, Inc. (Yokohama, Japan), and fed in a temperature‐controlled, light‐cycled room. At 5 weeks after birth, the mice were intraperitoneally inoculated with 3.0 × 106 MKN45P cells suspended in 1 mL PBS. On day 21 after inoculation, Oregon green 488 PTX (OG‐PTX) dissolved in PMB30W was administered via the i.p. or i.v. route. The total amount of administered liquid was fixed at 0.1 mL for the i.v. and 1 mL for the i.p. route, and the dosage of PTX was fixed at 5 μg/g weight in each group. After 3, 12 and 24 h, the peritoneal nodules were excised, fixed for 1 h in 10% neutral buffered formalin at room temperature, washed overnight in PBS containing 10% sucrose at 4°C, embedded in optimal cutting temperature compound (Tissue‐Tek; Sakura Finetek, Torrance, CA, USA) and snap frozen in dry‐iced acetone for immunohistochemical examination.
Then 10‐μm cryostat sections of post‐fixed frozen samples were examined for green fluorescein of OR‐PTX using a fluorescence stereomicroscope (BZ8000; Keyence, Osaka, Japan). The same sections were immunostained with a rat monoclonal antibody to mouse platelet/endothelial cell adhesion molecule 1 (PECAM‐1; 1:200 dilution) to detect blood vessels. Subsequently, specimens were incubated with the corresponding secondary antibody labeled with Alexa Fluor 594 at 1:200 dilution. Cell nuclei were also counterstained with DAPI. OGP‐PTX, PECAM‐1 and DAPI were imaged using a green, red and blue filter, respectively.
Statistical analysis. The results were statistically examined by paired Student’s t test when appropriate. Survival was analyzed by the Kaplan–Meier method. Results are given as mean ± SD, and differences with P < 0.05 were considered significant.
Results
Distribution of i.v. injected PTX in peritoneal nodules and liver. To examine the topical distribution of PTX‐30W in metastatic nodules in the peritoneal cavity, Oregon green‐conjugated PTX (OG‐PTX) diluted in PMB30W solution was injected via the i.v. or i.p. route in mice developing peritoneal metastasis. As shown in Figure 1A–C, OG ‐PTX (here we deliberately consider OG‐PTX as PTX) was rarely detected in peritoneal nodules at 1 h after i.v. injection. However, 24 h later the green signal of PTX was positively detected in tumors, which was significantly reduced in intensity 48 h later.
Figure 1.
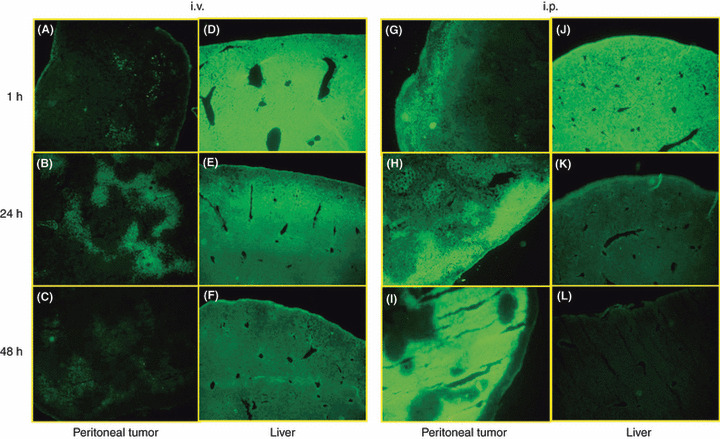
Accumulation pattern of paclitaxel (PTX) in peritoneal tumor (A–C, G–I) and liver (D–F, J–L) after intravenous (i.v.) or intraperitoneal (i.p.) injection of Oregon‐green‐labeled PTX‐30W in mice developing peritoneal metastasis of MKN45. At 1 (A,D,G,J), 24 (B,E,H,K) and 48 (C,F,I,L) h after PTX‐30W administration, peritoneal tumors 2–3 mm in size and the liver were excised, fixed and sections were observed under fluorescence microscopy.
To examine the drug distribution pattern in normal organs in the abdominal cavity, we next evaluated the liver in the same mice. As shown in Figure 1D–F, the liver was brightly and homogenously stained green at 1 h after i.v. injection, indicating that a large amount of PTX is easily delivered and distributed to the liver parenchyma within 1 h. The green PTX gradually reduced in intensity, but was still significantly detected at 48 h after i.v. injection in normal liver.
Distribution of i.p. injected PTX in peritoneal nodules and liver. In contrast to i.v. injection, the PTX signal was detected in the peripheral area of peritoneal nodules at 1 h after i.p. injection of PTX‐30W, and the area and intensity of green fluorescence were markedly increased at 24–48 h (Fig. 1G–I). At 24 h, green fluorescence was restricted to the peripheral area of the tumor, while a strong fluorescent signal was observed in the deeper area at 48 h. The distribution of i.p. injected PTX in the liver was markedly different from that in peritoneal nodules (Fig. 1J–L). After 1 h, green PTX was strongly positive in the liver, similar to that with i.v. injected PTX, although the intensity appeared to be less than in the case of i.v. administration. The PTX signal was markedly decreased at 24 h, and was almost negative at 48 h after i.p. injection. The PTX signal was markedly weaker in the liver than in peritoneal tumor nodules at 24–48 h, and the particular gradation of fluorescence was not observed in the liver at any time point.
When the fluorescence intensity was objectively measured by fluorescence microscopy, the distribution of PTX showed clear contrast between i.v. and i.p. administration (Fig. 2). This clearly indicates that i.p. injected PTX could be delivered into peritoneal nodules mainly through direct infiltration from the tumor surface, and accumulation of PTX in the tumor was markedly greater than that of i.v. administered PTX. Then, we measured the distance of penetration of i.p. injected OG‐labeled PTX‐30W into the peritoneal nodules. As shown in Figure 2, the fluorescence intensity was quantified under a fluorescence microscope, and the distance from the tumor surface to the deepest points, which showed a significantly stronger PTX signal than the background level, was calculated in sections of peritoneal tumors. As shown in Figure 3, the distance of PTX‐30W infiltration reached approximately 500 μm and 1 mm, at 24 and 48 h after i.p. injection, respectively. When examined by the same method, the distance of infiltration was much less for conventional PTX formulated in Cremophol EL.
Figure 2.
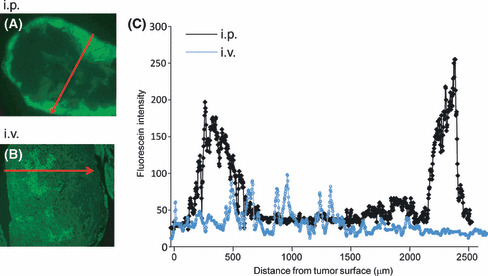
Intratumoral fluorescein intensity in peri‐toneal tumor at 24 h after i.p. and i.v. injection of paclitaxel nanoparticles (PTX‐30W). A representative line, which appears to reflect the deepest infiltration of green PTX, was set as the red arrow in sections of peritoneal tumors (A: i.p., B: i.v. administration), and the fluorescence intensity was measured on the line and plotted. The distance of the deepest infiltration of PTX after i.p. injection in each tumor is shown in Figure 2C.
Figure 3.
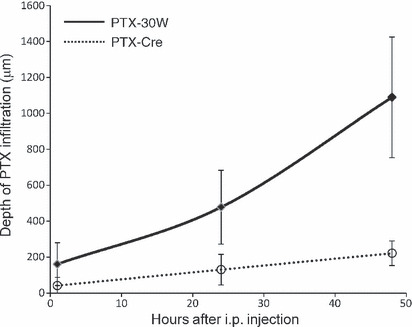
Paclitaxel nanoparticles (PTX‐30W) infiltrate deeper into peritoneal nodules than conventional PTX solubilized with Cremphol EL (PTX‐Cre) after i.p. injection. As shown in Figure 2, the depth of infiltration was determined in five different peritoneal nodules at each time point, and mean ± SD were calculated for PTX‐30W and PTX‐Cre. *P < 0.01.
Intraperitoneally injected PTX preferentially accumulated in avascular areas and induced apoptosis of tumor cells. Multicolor staining showed that the PTX signal was mainly detected around the vessels stained red by PECAM‐1 24 h after i.v. administration (Fig. 4A). Interestingly, however, i.p. administered PMB30W was not evenly accumulated in peritoneal nodules. As shown in Figure 4B, green fluorescence was relatively strong in the PECAM‐1‐negative area at 24 h, and was not detected in the area around PECAM‐1‐positive tumor vessels, even in the tumor periphery at 48 h. This is a marked contrast from i.v. injected PTX‐30W, which showed preferential accumulation in the perivascular area in tumor nodules.
Figure 4.
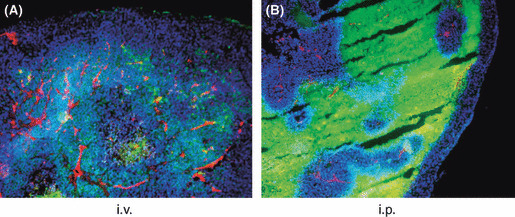
Accumulation pattern of PTX in peritoneal tumors. Peritoneal tumors were excised at 24 h after i.v. (A) or 48 h after i.p. (B) injection of paclitaxel nanoparticles (PTX‐30W), and vessels were stained red with anti‐PECAM‐1 mAb and nuclei were stained with DAPI as described in the Materials and Methods. Most of the green PTX signal was associated with PECAM‐1‐positive vessels in (A), whereas it was strongly detected in the PECAM‐1‐negative area and the area around PECAM‐1‐positive vessels lacked PTX accumulation in (B).
We next excised the tumor nodules from the mesentery and soaked them in PTX‐30W diluted in PBS buffer. In this condition, infiltration of PTX from the tumor surface was mostly even, and lack of PTX accumulation around PECAM‐1‐positive vessels was not observed (Fig. 5), indicating that the unique accumulation pattern was observed only in an in vivo situation.
Figure 5.
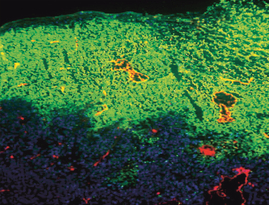
PTX‐30W equally infiltrate into resected tumor irrespective of vascularity. Peritoneal nodules were excised and soaked in PTX‐30W‐containing solution for 24 h and fixed. Similarly, PTX‐30W accumulated in the area of PECAM‐1‐positive tumor vessels.
More importantly, many tumor cells around the PTX‐accumulated area showed condensed nuclei with bright staining when stained by Hoechst 33342 (Fig. 6). The same morphological change could be observed in perivascular tumor cells after i.v. injection, although the number of apoptotic cells was much lower than that after i.p. injection. Moreover, such change was rarely observed in the liver after either i.v. or i.p. injection. This finding suggests that the retention of a high concentration of PTX for a relatively long period efficiently induced apoptosis in tumor cells in the peripheral area of disseminated tumor nodules when a polymeric formulation was used.
Figure 6.
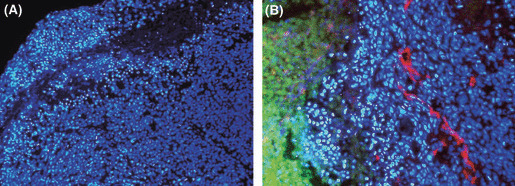
Apoptosis of tumor cells located in the vicinity of a paclitaxel (PTX)‐accumulated area. Peritoneal tumors were excised at 24 h after i.p. injection of PTX‐30W, and the nuclei were stained in blue with Hoechst 33342 (×200) (A). The nuclear staining was overlayed with the green fluorescent signal of OG‐PTX and counterstaining of tumor vessels with anti‐PECAM‐1 in red (×400) (B). Apoptotic nuclei were condensed with brighter staining with Hoechst 33342.
Discussion
Recent studies have suggested that anti‐cancer agents incorporated in polymeric nanomicelle carriers are retained for a long time in the blood stream and are favorably extravasated from vessels into the interstitium of tumor tissue due to their enhanced permeability and retention (EPR) effect.( 17 , 18 , 19 ) In fact, the benefits of PTX‐incorporated nanomicelles have been shown in clinical studies targeting solid tumors such as breast, gastric or lung cancer.( 20 , 21 , 22 ) However, all studies have evaluated systemic administration via the i.v. route, and the clinical effects of such nanomicelle PTX after i.p. administration have not yet been clarified. Furthermore, therapeutic efficacy of peritoneal and pleural carcinomatosis has not improved in the past 30 years using conventional low molecular weight (MW) chemotherapeutic agents.
Under such circumstances, PTX formulated with PMB‐30W (PTX‐30W) is a nanomicellar PTX that shows excellent inhibitory effects on peritoneal metastasis, due to the higher accumulation in tumor nodules after i.p. administration in a mouse model.( 13 ) In this study, we used florescence‐labeled PTX‐30W and compared the specific distribution of PTX‐30W after i.v. and i.p. injection. Our results clearly revealed that i.p. administered PTX‐30W showed a unique distribution in peritoneal nodules, which was markedly different from that with i.v. administration. Under fluorescence microscopy, retention of PTX‐30W was observed mainly in the perivascular area of peritoneal nodules 24 h after i.v. administration, which is consistent with the results of other studies.( 23 , 24 ) However, in our study, we confirmed that the fluorescence intensity in peritoneal nodules was markedly less than that in the liver at any time point. In contrast, PTX highly accumulated in the peripheral area of disseminated tumor even at 1 h after i.p. injection, and the area of PTX accumulation gradually enlarged at least up to 48 h; the intensity was much greater than that in the liver, which is considered to be a big advantage from the viewpoint of drug delivery. We also examined the hepatotoxicity of PTX‐30W and confirmed that i.p. administration of PTX‐30W caused transient alanin aminotrausterase (ALT) elevation in mice, which was tolerable enough (data not shown).
Moreover, the time course of PTX distribution indicates that the intratumoral accumulation of i.p. injected PTX‐30W is largely attributable to direct penetration into peritoneal nodules. The depth of the PTX signal of fluorescence in tumor nodules reached approximately 1 mm from the tumor surface at 48 h, although longer treatment did not increase the depth of PTX infiltration (data not shown). This value is markedly larger than the data of previous studies showing that direct penetration of anti‐cancer drugs into tumor tissue was limited, at least up to 100–200 μm from the surface, using various models,( 25 , 26 , 27 ) and suggests that the present nanomicelle drugs have a higher capacity to penetrate tumor tissue. In fact, we confirmed that PMB‐30W was more infiltrative than conventional PTX solubilized with Cremophor EL in an i.p. model evaluated with a different method using colon26 in Balb/c mice also (data not shown). One can imagine the case of agar gel diffusion of antibody (IgG, 160 KDa), in that the diffusion of antibody can be far greater than a few millimeter in agar gel in overnight.
Whether PTX observed in tumor nodules is encapsulated in the carrier or released from the carrier is another interesting issue. In an in vitro study, we have examined that PTX‐30W is highly permeable through the cell membrane,( 11 ) as well as matrigel, suggesting that PTX‐30W penetrates the tumor tissue in nanomicellar form. The low surface electronic potential of PTX‐30W might be related to the high permeability of this nanoparticle.
An interesting finding in our fluorescence study is that i.p. administered PTX‐30W tended to be strongly detected in relatively hypovascular areas in peripheral tumor sites, which was a marked contrast to the results of i.v. injection. The exact mechanism for the lack of PTX accumulation in perivascular areas after i.p. injection is as yet unclear. In our data, this pattern of accumulation was not observed when the tumor was excised and soaked in PTX‐containing solution, suggesting the possibility that PTX infiltrating the tumor might be washed out via the tumor vessels. In fact, this high accumulation pattern was never observed in the liver, which contains rich vasculature. Therefore, the unique accumulation of PTX is thought to be related to the low vascularity of tumor nodules on the peritoneum and thus have less efficient wash out. Many factors such as the size, charge and water solubility of drugs are known to be related to the distribution and diffusion of anti‐cancer agents in solid tumors.( 28 ) Moreover, drug penetration has been reported to be impeded by severe fibrosis and high interstitial pressure.( 29 , 30 ) These physicochemical features of tumor nodules are thought to be involved in the unique drug distribution of PTX after i.p. injection.
Another important finding in our study is that many tumor cells located around the PTX‐accumulated area showed apoptotic nuclei. In the case of i.v. administration, some apoptotic cells were also observed in the perivascular area, but the number of apoptotic cells was much greater in the peripheral area after i.p. administration. Kuh et al. ( 31 ) used a xenograft model and showed that high tumor cell density is a strong barrier to PTX infiltration, and thus the induction of apoptosis of tumor cells results in enhancement of drug penetration in solid tumors. This suggests that apoptosis of tumor cells induced by a high concentration of PTX in the peripheral area enables drug penetration to a deeper area in peritoneal nodules.
In summary, we demonstrated that i.p. administration of PTX‐30W results in high accumulation in disseminated nodules, especially in hypovascular areas in the peripheral part of peritoneal nodules, presumably due to its superior penetrating activity directly into malignant tissue. In contrast, i.v. injection of PTX‐30W provides a much lower amount of PTX into peritoneal nodules and the range was mostly limited to around tumor vessels. The limited depth of direct penetration of anti‐cancer drugs by diffusion from the free surface is probably the most important problem in regional or intracavitary anti‐cancer therapy. In fact, previous reports of ovarian cancer have suggested that i.p. chemotherapy using free doxorubicin is not so beneficial in bulky tumors because of limited drug penetration.( 32 , 33 ) Based on these facts, i.p. chemotherapy using PTX‐30W is considered to be an ideal therapeutic approach for peritoneal carcinomatosis from the viewpoint of drug delivery.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
The authors thank Chieko Uchikawa and Kahoru Amitani for their excellent technical assistance. This study was funded by the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Ministry of Health, Labor and Welfare of Japan.
References
- 1. D’Angelica M, Gonen M, Brennan MF, Turnbull AD, Bains M, Karpeh MS. Patterns of initial recurrence in completely resected gastric adenocarcinoma. Ann Surg 2004; 240: 808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Legge F, Petrillo M, Adamo V, Pisconti S, Scambia G, Ferrandina G. Epithelial ovarian cancer relapsing as isolated lymph node disease: natural history and clinical outcome. BMC Cancer 2008; 8: 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jacquet P, Sugarbaker PH. Peritoneal‐plasma barrier. Cancer Treat Res 1996; 82: 53–63. [DOI] [PubMed] [Google Scholar]
- 4. Eiseman JL, Eddington ND, Leslie J et al. Plasma pharmacokinetics and tissue distribution of paclitaxel in CD2F1 mice. Cancer Chemother Pharmacol 1994; 34: 465–71. [DOI] [PubMed] [Google Scholar]
- 5. Gelderblom H, Verweij J, van Zomeren DM et al. Influence of Cremophor El on the bioavailability of intraperitoneal paclitaxel. Clin Cancer Res 2002; 8: 1237–41. [PubMed] [Google Scholar]
- 6. Ishigami H, Kitayama J, Kaisaki S et al. Phase II study of weekly intravenous and intraperitoneal paclitaxel combined with S‐1 for advanced gastric cancer with peritoneal metastasis. Ann Oncol 2010; 21: 67–70. [DOI] [PubMed] [Google Scholar]
- 7. Armstrong DK, Bundy B, Wenzel L et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 2006; 354: 34–43. [DOI] [PubMed] [Google Scholar]
- 8. Markman M, Bundy BN, Alberts DS et al. Phase III trial of standard‐dose intravenous cisplatin plus paclitaxel versus moderately high‐dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small‐volume stage III ovarian carcinoma: an intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J Clin Oncol 2001; 19: 1001–7. [DOI] [PubMed] [Google Scholar]
- 9. de Bree E, Rosing H, Michalakis J et al. Intraperitoneal chemotherapy with taxanes for ovarian cancer with peritoneal dissemination. Eur J Surg Oncol 2006; 32: 666–70. [DOI] [PubMed] [Google Scholar]
- 10. Ishihara K, Iwasaki Y, Nakabayashi N. Polymeric lipid nanosphere consisting of water‐soluble poly(2‐methacryloyloxyethyl phosphorylcholine‐co‐n‐butyl methacrylate). Polym J 1999; 31: 1231–6. [Google Scholar]
- 11. Goda T, Goto Y, Ishihara K. Cell‐penetrating macromolecules: direct penetration of amphipathic phospholipid polymers across plasma membrane of living cells. Biomaterials 2010; 31: 2380–7. [DOI] [PubMed] [Google Scholar]
- 12. Konno T, Watanabe J, Ishihara K. Enhanced solubility of paclitaxel using water‐soluble and biocompatible 2‐methacryloyloxyethyl phosphorylcholine polymers. J Biomed Mater Res A 2003; 65: 209–14. [DOI] [PubMed] [Google Scholar]
- 13. Soma D, Kitayama J, Konno T et al. Intraperitoneal administration of paclitaxel solubilized with poly(2‐methacryloxyethyl phosphorylcholine‐co n‐butyl methacrylate) for peritoneal dissemination of gastric cancer. Cancer Sci 2009; 100: 1979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura M, Konno T, Miyamoto Y, Kojima Y, Maeda H. Intracavitary administration: pharmacokinetic advantages of macromolecular anticancer agents against peritoneal and pleural carcinomatoses. Anticancer Res 1998; 18: 2547–50. [PubMed] [Google Scholar]
- 15. Sako A, Kitayama J, Koyama H et al. Transduction of soluble Flt‐1 gene to peritoneal mesothelial cells can effectively suppress peritoneal metastasis of gastric cancer. Cancer Res 2004; 64: 3624–8. [DOI] [PubMed] [Google Scholar]
- 16. Ishihara K, Ueda T, Nakabayashi N. Preparation of phospholipid polymers and their properties as polymer hydrogel membrane. Polym J 1990; 22: 355–60. [Google Scholar]
- 17. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 18. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 2000; 65: 271–84. [DOI] [PubMed] [Google Scholar]
- 19. Vicent MJ, Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol 2006; 24: 39–47. [DOI] [PubMed] [Google Scholar]
- 20. Gradishar WJ, Tjulandin S, Davidson N et al. Phase III trial of nanoparticle albumin‐bound paclitaxel compared with polyethylated castor oil‐based paclitaxel in women with breast cancer. J Clin Oncol 2005; 23: 7794–803. [DOI] [PubMed] [Google Scholar]
- 21. Hamaguchi T, Matsumura Y, Suzuki M et al. NK105, a paclitaxel‐incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer 2005; 92: 1240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rizvi NA, Riely GJ, Azzoli CG et al. Phase I/II trial of weekly intravenous 130‐nm albumin‐bound paclitaxel as initial chemotherapy in patients with stage IV non‐small‐cell lung cancer. J Clin Oncol 2008; 26: 639–43. [DOI] [PubMed] [Google Scholar]
- 23. Lankelma J, Dekker H, Luque FR et al. Doxorubicin gradients in human breast cancer. Clin Cancer Res 1999; 5: 1703–7. [PubMed] [Google Scholar]
- 24. Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res 2005; 11: 8782–8. [DOI] [PubMed] [Google Scholar]
- 25. Los G, Mutsaers PH, van der Vijgh WJ, Baldew GS, de Graaf PW, McVie JG. Direct diffusion of cis‐diamminedichloroplatinum(II) in intraperitoneal rat tumors after intraperitoneal chemotherapy: a comparison with systemic chemotherapy. Cancer Res 1989; 49: 3380–4. [PubMed] [Google Scholar]
- 26. Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res 2002; 8: 878–84. [PubMed] [Google Scholar]
- 27. Kyle AH, Huxham LA, Yeoman DM, Minchinton AI. Limited tissue penetration of taxanes: a mechanism for resistance in solid tumors. Clin Cancer Res 2007; 13: 2804–10. [DOI] [PubMed] [Google Scholar]
- 28. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer 2006; 6: 583–92. [DOI] [PubMed] [Google Scholar]
- 29. Yashiro M, Chung YS, Nishimura S, Inoue T, Sowa M. Fibrosis in the peritoneum induced by scirrhous gastric cancer cells may act as “soil” for peritoneal dissemination. Cancer 1996; 77: 1668–75. [DOI] [PubMed] [Google Scholar]
- 30. Jain RK. Barriers to drug delivery in solid tumors. Sci Am 1994; 271: 58–65. [DOI] [PubMed] [Google Scholar]
- 31. Kuh HJ, Jang SH, Wientjes MG, Weaver JR, Au JL. Determinants of paclitaxel penetration and accumulation in human solid tumor. J Pharmacol Exp Ther 1999; 290: 871–80. [PubMed] [Google Scholar]
- 32. Carney ME, Lancaster JM, Ford C, Tsodikov A, Wiggins CL. A population‐based study of patterns of care for ovarian cancer: who is seen by a gynecologic oncologist and who is not? Gynecol Oncol 2002; 84: 36–42. [DOI] [PubMed] [Google Scholar]
- 33. Bristow RE, Zahurak ML, del Carmen MG et al. Ovarian cancer surgery in Maryland: volume‐based access to care. Gynecol Oncol 2004; 93: 353–60. [DOI] [PubMed] [Google Scholar]