

Abstract
The National Comprehensive Cancer Network guidelines recommend radiotherapy as a standard treatment for patients with a high risk of recurrence in gastric cancer. Because radiation is harmful to the surrounding organs, a radiation sensitizer might therefore be useful to decrease the side effects of patients with advanced gastric carcinoma. The aim of the current study was to clarify the effect of a DNA methyltransferase inhibitor, 5‐aza‐2′‐deoxycytidine (CdR), on radiation sensitivity in gastric cancer cells. Five gastric cancer cell lines, OCUM‐2M, OCUM‐12, KATO‐III, MKN‐45, and MKN‐74, were used. The effects of 5‐aza‐CdR with irradiation on the growth activity, cell‐cycle distribution, apoptosis, and apoptosis‐associated gene expression were examined. 5‐aza‐CdR sensitized three of five gastric cancer cell lines to radiation. A combination of irradiation and 5‐aza‐CdR significantly (P < 0.05) decreased the growth activity compared with irradiation alone in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in KATO‐III and MKN‐74 cells. The percentage of cells in G2–M phase and the apoptotic rate with irradiation in combination with 5‐aza‐CdR were increased in OCUM‐2M, OCUM‐12, and MKN‐45 cells compared with irradiation alone, but not in KATO‐III and MKN‐74 cells. 5‐aza‐CdR increased the expression of p53, RASSF1, and death‐associated protein kinases (DAPK) genes compared with the control or irradiation alone. These findings suggest that 5‐aza‐CdR might therefore be useful as a radiation sensitizer to treat some types of gastric carcinoma. The arrest at G2–M phase and increased apoptotic rate might be partly mediated by enhanced expression of the p53, RASSF1, or DAPK gene families by 5‐aza‐CdR. (Cancer Sci 2009; 100: 181–188)
Gastric cancer remains a threat around the world. Patients with gastric cancer frequently experience recurrent tumors even after a curative surgical resection because gastric cancer is frequently diagnosed at an advanced stage. More than 50% of recurrent lesions are local in patients with recurrent gastric cancer.( 1 ) Surgical treatment alone is not useful for patients with local and distal recurrences. Various types of therapies have been tested for effectiveness in recurrent gastric carcinoma, including chemotherapy, hyperthermia, and immunotherapy, but none has been satisfactory.( 2 ) Accordingly, new therapies against the recurrence of advanced gastric cancer are urgently sought.
A clinical trial, INT0116,( 3 ) reported that chemoradiotherapy after surgery prolonged patients’ survival time in comparison to surgery alone, and concluded that postoperative chemoradiotherapy might be useful for patients with a high risk for recurrence of gastric cancer. The National Comprehensive Cancer Network guidelines on gastric cancer treatment include radiotherapy as a standard treatment for patients with a high risk of recurrence.( 4 , 5 , 6 ) Complications of radiotherapy affecting the surrounding organs are severe, although recent irradiation technologies support the role of radiotherapy in gastric cancer.( 7 ) A radiation sensitizer could therefore be useful when using radiotherapy to treat gastric cancer.
Hypermethylation of CpG islands at gene promoter regions on DNA results in gene inactivation of some tumor‐suppressor genes.( 8 ) These include apoptosis‐related genes, such as death‐associated protein kinases (DAPK), Ras association domain family‐1 (RASSF‐1), p16, and thrombospondin (THBS)‐1, and the DNA mismatch repair genes hMLH1 and O(6)‐methylguanine‐DNA methyltransferase (MGMT).( 9 ) Promoter hypermethylation plays a prominent role in genes involved in cell‐cycle regulation, DNA repair, cell signaling, transcription, and apoptosis.( 8 , 9 , 10 ) A DNA methyltransferase inhibitor, 5‐aza‐2′‐CdR, is a prodrug that requires activation via phosphorylation by deoxcytidine kinase, and the nucleotide analog is then incorporated into DNA where it produces irreversible inactivation of DNA methyltransferase.( 9 ) Preliminary clinical trials of 5‐aza‐CdR showed antineoplastic activity in patients with leukemia and myelodysplastic syndrome,( 11 ) whereas it showed little activity on solid tumors as a single agent.( 12 , 13 ) The potential to increase the 5‐aza‐CdR chemosensitivity of cancer cell lines has been proven in vitro;( 9 , 14 ) however, there are few reports of the radiosensitivity associated with exposure to 5‐aza‐CdR. The present study therefore examined the effect of 5‐aza‐CdR on the radiation sensitivity of gastric carcinomas.
Materials and Methods
Cell lines. Five gastric cancer cell lines (OCUM‐2M,( 15 ) OCUM‐12, KATO‐III, MKN‐45,( 16 ) and MKN‐74( 16 )) and two gastric fibroblast cell lines (NF‐24 and NF‐25) were used in the present study. OCUM‐2M, OCUM‐12, KATO‐III, and MKN‐45 were derived from poorly differentiated adenocarcinomas, whereas MKN‐74 was from differentiated adenocarcinomas. These cell lines were cultured in Dulbecco's modified Eagle's medium (Nikken Biomedical Laboratory, Kyoto, Japan) supplemented with 10% fetal bovine serum, 100 IU/mL penicillin (ICN Biomedical, Costa Mesa, CA, USA), 100 mg/mL streptomycin (ICN Biomedical), and 0.5 mmol/L sodium pyruvate (Cambrex, Walkersville, MD, USA). The cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in air.
Chemicals. A stock solution of 5‐aza‐CdR (Sigma, St Louis, MO, USA) at 10 mmol/L in 99% ethanol was stored at –20°C, and used at a concentration of 0.5 or 1 µmol/L.
Irradiation. X‐ray irradiation was administered using an X‐ray machine (MBR‐1520R; Hitachi Medical Corporation, Tokyo, Japan), which operated at 150 kV and 20 mA with 0.5 mmol/L Al and 0.1 mmol/L Cu filtration. X‐ray irradiation was carried out between 2 and 9 Gy at a dose rate of approximately 1.4 Gy/min.
Colony‐forming assay. Cancer cells were trypsinized to generate a single‐cell suspension and a specified number of cells were seeded into each well of six‐well tissue culture plates according to the respective irradiation dose. After allowing the cells time to attach, 5‐aza‐CdR (0.5 or 1 µmol/L) or a vehicle control was added at the specified concentrations, and the plates were irradiated 2 h later. Ten to 12 days after seeding, colonies were stained with crystal violet, the number of colonies containing at least 50 cells was determined, and the surviving fractions were calculated. Survival curves were generated after normalizing for cytotoxicity as follows. The plating efficiency was defined as the total number of colonies in the vehicle control/total number of cells inoculated in the vehicle control. The surviving fraction of each irradiation dose was calculated as the total no. colonies/(total cells inoculated × plating efficiency).
Proliferation assay. Cancer cells were seeded into two 96‐well plates at a concentration of 1000 cells/well with complete culture medium with irradiation (9 Gy) and/or 5‐aza‐CdR (1 µmol/L). After incubation for 72 h, 20 µL 3‐(4,5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetrazoliumbromide (MTT; Sigma) at a concentration of 2 mg/mL (diluted with phosphate‐buffered saline) was added into each well and incubation was continued at 37°C for a further 3 h to allow the MTT to be metabolized. The crystals formed were dissolved by adding 100 µL dimethyl sulfoxide. The formazan product of MTT was measured as absorbance at 570 nm using a microtiter plate reader (PM2004; Wako, Osaka, Japan). The percentage of cell viability was determined as the ratio of the absorbance of the sample to that of the control. The proliferation of each column was calculated as the percentage of cell viability divided by that of the vehicle control.
Cell cycle test. The cell cycle phase distribution was evaluated using flow cytometry. Cancer cells (2 × 104 cells) were seeded into a 100‐mm dish with vehicle or 5‐aza‐CdR (1 µmol/L) for the cell cycle test. After incubation for 72 h, the cells were harvested and managed according to the instructions of the Cycle Test Plus DNA reagent kit protocol (Becton Dickinson, Mountain View, CA, USA), then incubated with ribonuclease A for 10 min at room temperature, and with propidium iodide for 30 min in the dark on ice. The sub‐G0–G1, S, and G2/M fractions of 2 × 104 cells were determined by flow cytometry using a FACScaliber (Becton Dickinson). The results were analyzed using the Modofit software program (Becton Dickinson).
Apoptosis assay. Apoptosis in response to irradiation with or without a combination of 5‐aza‐CdR was tested using flow cytometry by staining the cells with annexin V–fluorescein isothiocyanate (FITC) and propidium iodide labeling (Medical and Biological Laboratories, Nagoya, Japan). The treatment protocols were essentially the same as in the proliferation experiments. Exponentially growing cells were inoculated into 100‐mm dishes at a concentration of 1.0 × 105 cells/mL before irradiation with 0 and 9 Gy, the cells were incubated with or without 5‐aza‐CdR for 2 h before irradiation. After irradiation and further incubation for 72 h with or without 5‐aza‐CdR, cells were harvested and stained with annexin V–FITC and propidium iodide according to the manufacturer's instructions, incubated for 15 min at room temperature in the dark, and analyzed immediately by FACScan flow cytometry. Viable cells do not stain with either dye, apoptotic cells only stain with annexin V–FITC, and secondary necrotic cells stain with both annexin V–FITC and propidium iodide. Three independent experiments were carried out.
Real‐time reverse transcription–polymerase chain reaction. To examine at the mRNA level the expression of genes including p53, RASSF1, caspase‐6, DAPK‐1, DAPK‐2, and DAPK‐3, exponentially growing cells with or without 1 µmol/L 5‐aza‐CdR for 48 h were seeded into 100‐mm dishes at a concentration of 1.0 × 105 cells/mL. The total cellular RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. After the genomic DNA was removed by DNAse, cDNA was prepared from 20 µg RNA with Maloney mouse leukemia virus reverse transcriptase (Invitrogen) using random primers (Invitrogen). To determine fold changes in each gene, real‐time reverse transcription‐polymerase chain reaction (PCR) was done on the ABI Prism 7000 (Applied Biosystems, Foster City, CA, USA) using a commercially available gene expression assay for p53, RASSF1, caspase‐6, DAPK‐1, DAPK‐2, and DAPK‐3 (Hs01034249, Hs00200394, Hs00154250, Hs00234489, Hs00204888, and Hs00154676, respectively). PCR was carried out at 95°C for 15 s and 60°C for 60 s for 40 cycles. As an internal standard to normalize mRNA levels for differences in sample concentration and loading, amplification of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used. The threshold cycle values were used to calculate the relative expression ratios between control and treated cells using the formula described by Pfaffl.( 17 ) Quantitative PCR were done in triplicate.
Statistical analysis. The quantitative ratios of different groups were compared using Student's t‐test. Probability values of P < 0.05 were regarded as statistically significant. All statistical tests were two sided.
Results
Effects of 5‐aza‐CdR and irradiation on the colony‐forming rate of gastric cancer cells. Figure 1 shows the effects of 5‐aza‐CdR in combination with irradiation on the colony‐forming rate of gastric cancer cells. Because KATO‐III cells float, colony formation was not observed in KATO‐III cells. Therefore, the colony‐forming assay was carried out using OCUM‐2M, OCUM‐12, MKN‐45, and MKN‐74. 5‐Aza‐CdR at 0.5 or 1.0 µmol/L suppressed the colony‐forming ability of cancer cells with irradiation in OCUM‐2M, OCUM‐12, and MKN‐45, but not in MKN‐74. A significant difference in the colony‐forming rate was found in combination with irradiation and 5‐Aza‐CdR at 1.0 µmol/L in OCUM‐2M (P < 0.05), OCUM‐12 (P < 0.01), and MKN‐45 (P ≤ 0.01) compared with irradiation alone.
Figure 1.
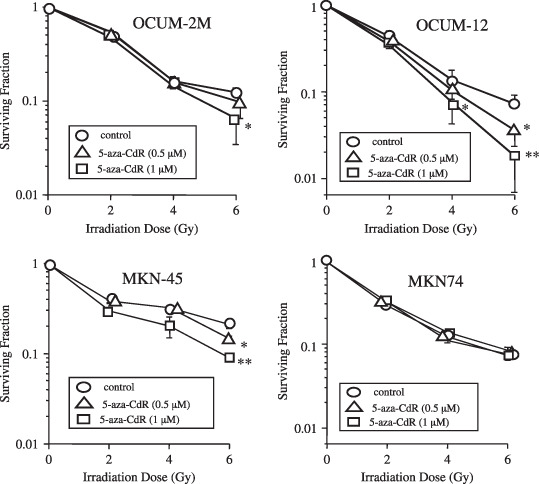
Effects of 5‐aza‐CdR and irradiation on colony formation. In OCUM‐2M cells, the colony‐forming rate after irradiation at 6 Gy was 12.2, 9.7, and 6.4% in combination with vehicle control ( ), 0.5 µmol/L 5‐aza‐CdR (
), 0.5 µmol/L 5‐aza‐CdR ( ), and 1 µmol/L 5‐aza‐CdR (
), and 1 µmol/L 5‐aza‐CdR ( ), respectively. In OCUM‐12 cells, the colony‐forming rate of irradiation at 6 Gy was 9.8, 3.6, and 1.8% in combination with vehicle control, 0.5 µmol/L 5‐aza‐CdR, and 1 µmol/L 5‐aza‐CdR, respectively. In MKN‐45, the colony‐forming rate after irradiation at 6 Gy was 21.6, 15.2, and 9.3% in combination with vehicle control, 0.5 µmol/L 5‐aza‐CdR, and 1 µmol/L 5‐aza‐CdR, respectively. In MKN‐74, the colony‐forming rate after irradiation at 6 Gy was 8.3, 7.0, and 7.4% in combination with vehicle control, 0.5 µmol/L 5‐aza‐CdR, and 1 µmol/L 5‐aza‐CdR, respectively. The plating efficiency was defined as the total no. colonies of 0 Gy/total no. cells inoculated at 0 Gy. The survival fraction of each irradiation dose was calculated as the total no. colonies/(total cells inoculated × plating efficiency). Three independent experiments were carried out. The results are presented as the mean of three independent experiments, and the bar indicates SD. *P < 0.05; **P < 0.01, versus control.
), respectively. In OCUM‐12 cells, the colony‐forming rate of irradiation at 6 Gy was 9.8, 3.6, and 1.8% in combination with vehicle control, 0.5 µmol/L 5‐aza‐CdR, and 1 µmol/L 5‐aza‐CdR, respectively. In MKN‐45, the colony‐forming rate after irradiation at 6 Gy was 21.6, 15.2, and 9.3% in combination with vehicle control, 0.5 µmol/L 5‐aza‐CdR, and 1 µmol/L 5‐aza‐CdR, respectively. In MKN‐74, the colony‐forming rate after irradiation at 6 Gy was 8.3, 7.0, and 7.4% in combination with vehicle control, 0.5 µmol/L 5‐aza‐CdR, and 1 µmol/L 5‐aza‐CdR, respectively. The plating efficiency was defined as the total no. colonies of 0 Gy/total no. cells inoculated at 0 Gy. The survival fraction of each irradiation dose was calculated as the total no. colonies/(total cells inoculated × plating efficiency). Three independent experiments were carried out. The results are presented as the mean of three independent experiments, and the bar indicates SD. *P < 0.05; **P < 0.01, versus control.
Effects of 5‐aza‐CdR and irradiation on the proliferation of gastric cancer cells and gastric fibroblasts. In OCUM‐2M, OCUM‐12, and MKN‐45 cells, the proliferation rate after irradiation with 5‐aza‐CdR decreased significantly (P < 0.05) to 31.8, 43.2, and 55.8%, respectively, compared with irradiation alone (46.8, 64.4, and 66.2%). In contrast, no significant effect was observed in response to irradiation with 5‐aza‐CdR in KATO‐III, MKN‐74, NF‐24, and NF‐25 cells compared with irradiation alone. 5‐aza‐CdR (1 µmol/L) alone did not significantly decrease the proliferation of any cancer cells or normal fibroblasts. Synergistic interactions were found in KATO‐III, MKN‐74, NF‐24, and NF‐25 cells, because the proliferation rate associated with the combination of irradiation and 5‐aza‐CdR (31.8, 43.2, and 55.8%) was less than the expected value (43.1, 60.3, and 59.7%) in OCUM‐2M, OCUM‐12, and MKN‐45 cells (Fig. 2).
Figure 2.
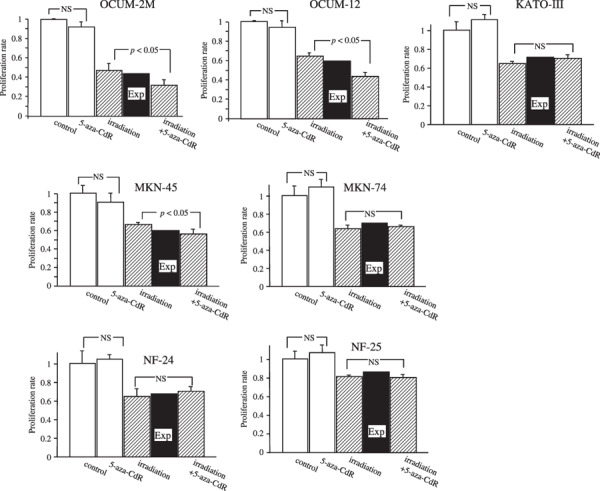
Effects of 5‐aza‐CdR on the proliferation of cancer cells in combination with irradiation. The proliferation effect for cancer cells in response to the combination of 5‐aza‐CdR with irradiation was lower than the expected additive effect in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in KATO‐III, MKN‐74, NF‐24, and NF‐25 cells, thus demonstrating that the combination of 1 µmol/L 5‐aza‐CdR and irradiation shows a synergistic antiproliferative effect in OCUM‐2M, OCUM‐12, and MKN‐45 by 3‐(4,5‐dimethylthiazol‐2,5‐diphenylterazolium bromide (MTT) assay. In contrast, no significant effect was observed in response to 5‐aza‐CdR alone in any cell lines compared with the control. The expected additive value of the combined effects (%) = effects of interaction alone/control × effects of 5‐aza‐CdR alone/control × 100% was calculated. The synergistic interactions were determined when the value was less than the expected value. The results are presented as the mean of three independent experiments, and the bars indicate SD. Exp, an expected additive value; NS, not significant.
5‐aza‐CdR triggers cell‐cycle arrest at G2–M. Figure 3 shows DNA histograms for analysis at 72 h after treatment with 5‐aza‐CdR (1 µmol/L). 5‐aza‐CdR increased the percentage of cells in G2–M phase in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in KATO‐III or MKN‐74 cells. The percentage of cells in G2–M phase in OCUM‐2M, OCUM‐12, and MKN‐45 cells was 50.2, 25.1, and 59.0% after 5‐aza‐CdR treatment, whereas that of the control was 19.7, 9.3, and 33.9%, respectively. In contrast, no effect of 5‐aza‐CdR was observed on G2–M phase in either KATO‐III or MKN‐74 cells.
Figure 3.
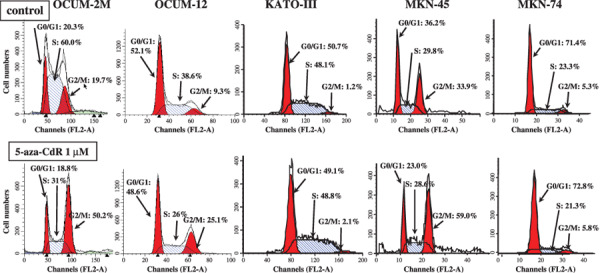
Effects of 5‐aza‐CdR on the cell‐cycle distribution. Gastric cancer cells were exposed to 1 µmol/L 5‐aza‐CdR for 72 h and were subsequently examined by flow cytometry. In OCUM‐2 M cells, 5‐aza‐CdR increased the percentage of cells in G2–M phase (50.2%) and decreased that in S phase (31%), compared with the control (G2–M 19.7%, S phase 60%). In OCUM‐12 cells, 5‐aza‐CdR increased the percentage of cells in G2–M phase (25.1%) and decreased that in S phase (26%), compared with the control (G2–M 9.3%, S phase 38.6%). In MKN‐45 cells, 5‐aza‐CdR increased the percentage of cells in G2–M phase (59%), compared with the control (G2–M 33.9%). In KATO‐III and MKN‐74 cells, no effect of 5‐aza‐CdR was observed on G2–M phase.
5‐Aza‐CdR increased apoptosis induced by irradiation. To clarify the induction of apoptosis by irradiation combined with 5‐aza‐CdR in gastric cancer cell lines, double staining of cells with annexin V (FITC) and PI was carried out. Cells staining annexin V‐positive, PI‐negative were considered to be apoptotic (Fig. 4a). Figure 4b shows the apoptosis rate induced by irradiation with or without 5‐aza‐CdR in five gastric cancer cell lines. In OCUM‐2M, OCUM‐12, and MKN‐45 cells, the apoptosis rate induced by irradiation in combination with 5‐aza‐CdR significantly increased in comparison to that by irradiation alone, but not in either KATO‐III or MKN‐74 cells. The apoptosis rate induced by 5‐aza‐CdR significantly increased in MKN‐45 cells compared with the control.
Figure 4.
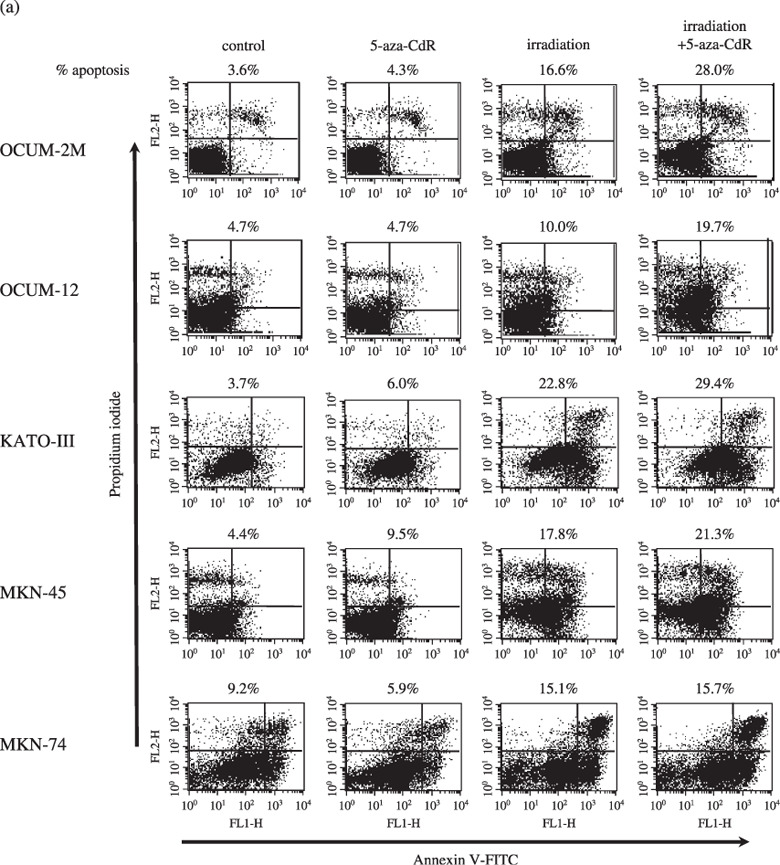
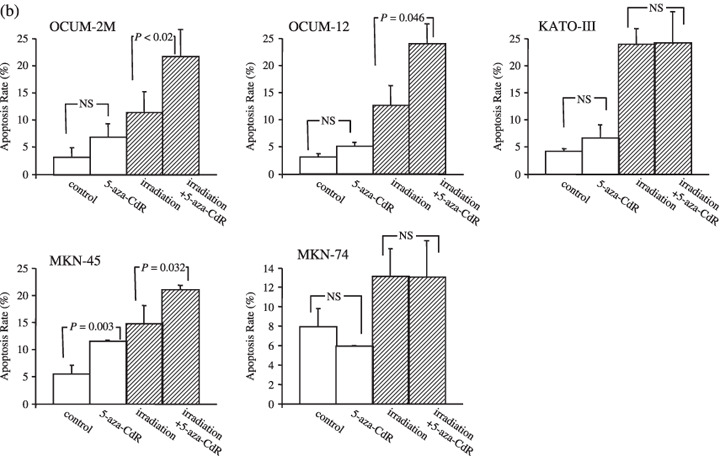
Apoptosis induction by 5‐aza‐CdR in combination with irradiation (a), representative examples of flow cytometric analysis (b). In OCUM‐2M cells, 5‐aza‐CdR alone induced apoptosis at a rate of 5.0% in comparison to the control rate of 3.0%. The apoptosis rate induced by irradiation in combination with 5‐aza‐CdR was 23.9%, and the apoptosis rate induced by irradiation alone was 12.6%. A significant difference was found (P = 0.02) between the two groups. In OCUM‐12 cells, 5‐aza‐CdR alone induced apoptosis at a rate of 6.9%, compared with the control group (3.2%). The apoptosis rate induced by the combination of irradiation and 5‐aza‐CdR was 21.7%, and that induced by irradiation alone was 11.3%, and the difference was significant (P = 0.046). In MKN‐45 cells, 5‐aza‐CdR alone significantly (P = 0.003) increased apoptosis at a rate of 11.4% compared with the control group (5.4%). The apoptosis rate induced by the combination of irradiation and 5‐aza‐CdR was 21%, and that induced by irradiation alone was 14.7%, and the difference was significant (P = 0.032). In contrast, no significant effect of 5‐aza‐CdR was observed in combination with irradiation on the apoptosis in KATO‐III and MKN‐74 cells. The results shown in the isometric chart represent the mean of three independent experiments and the bars show SD.
Effects of 5‐aza‐CdR on gene expression in gastric cancer cells. 5‐aza‐CdR increased the expression of p53, RASSF1, and DAPK genes in OCUM‐2M, OCUM‐12, and MKN‐45 cells compared with the control. p53 mRNA expression was increased by 5‐aza‐CdR with or without irradiation in OCUM‐2M, OCUM‐12, and MKN‐45 cells compared with the control, but not in either KATO‐III or MKN‐74 cells. The DAPK‐1 expression level of MKN‐45 cells was increased by 5‐aza‐CdR compared with the control. The DAPK‐2 expression levels of OCUM‐2M cells treated with 5‐aza‐CdR with or without irradiation were higher than the control. DAPK‐3 expression was increased by 5‐aza‐CdR with or without irradiation in OCUM‐2M, OCUM‐12, and MKN‐45 cells (Fig. 5). No differences in the level of expression were found in any other genes, including THBS1, hMLH1, p16, excision repair cross‐complementation group 1 (ERCC1), and X‐ray repair cross‐complementation group 1 (XRCC1) (data not shown).
Figure 5.
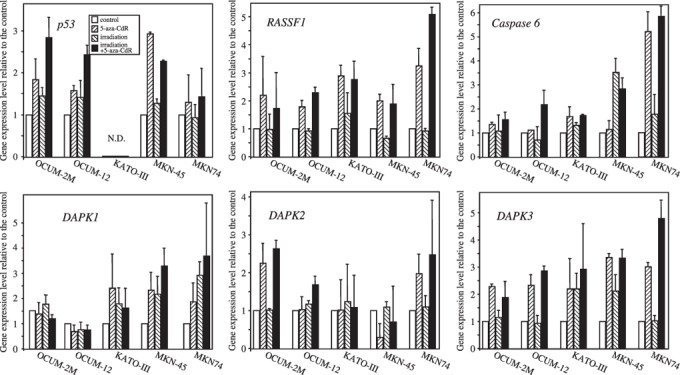
Effects of 5‐aza‐CdR on gene expression with or without irradiation. The expression of p53 mRNA increased in response to 5‐aza‐CdR in OCUM‐2M, OCUM‐12, and MKN‐45 cells. p53 mRNA was not amplified in KATO‐III cells. RASSF1 mRNA expression increased in response to 5‐aza‐CdR with or without irradiation in OCUM‐2M, KATO‐III, MKN‐45, and MKN‐74 cells. Caspase‐6 mRNA expression increased in response to 5‐aza‐CdR in MKN‐74 cells. DAPK‐1 mRNA expression increased was by 5‐aza‐CdR in KATO‐III, MKN‐45, and MKN‐74 cells. DAPK‐2 mRNA expression increased was by 5‐aza‐CdR in OCUM‐2M and MKN‐74 cells. DAPK‐3 mRNA expression was increased by 5‐aza‐CdR in all five gastric cancer cell lines. ND, not determined.
Discussion
The DNA methyltransferase inhibitor 5‐aza‐CdR has been demonstrated to have little activity on solid tumors as a single agent;( 12 , 13 ) however, its effects in combination with irradiation are still unclear. The efficacy of 5‐aza‐CdR in combination with irradiation was investigated in five gastric cancer cell lines in the present study. Based on colony‐forming assays, 5‐aza‐CdR and radiation cooperated to yield fewer and smaller colonies, and there were significant radiosensitization effects in three of four cancer cell lines: OCUM‐2M, OCUM‐12, and MKN‐45, but not MKN‐74. In addition, 5‐aza‐CdR showed synergism of growth inhibition in combination with irradiation, as the proliferation rate in response to a combination of irradiation and 5‐aza‐CdR was less than the expected value in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in KATO‐III and MKN‐74 cells. These results suggest that 5‐aza‐CdR might be a useful radiation sensitizer in some types of gastric carcinoma. Because in vivo experiments are important to clarify the possible activity of 5‐aza‐CdR in radiation therapy, an in vivo study is therefore necessary in the future. Previous study suggested that 5‐aza‐CdR increases the chemosensitivity of gastric cancer cells.( 9 ) Although it might be difficult to compare the advantages between radiotherapy and chemotherapy in combination with 5‐aza‐CdR, the indications of gastric cancer patients might thus be different between radiotherapy and chemotherapy. Radiotherapy might show some advantage for patients with a risk of local recurrence, whereas chemotherapy might show an advantage for distant recurrence. This may therefore be important for determining the adequate indications for either chemotherapy or radiotherapy. Taken together, chemoradiotherapy in combination with 5‐aza‐CdR might be more useful for patients with a high risk of recurrence in gastric cancer.
In the normal fibroblast cells, 5‐aza‐CdR did not show any synergism in growth inhibition in combination with irradiation, whereas irradiation alone decreased the proliferation of normal cells. These findings suggest that 5‐aza‐CdR shows a different synergistic effect of proliferation between normal and gastric cancer cells. Because complications of the surrounding organs are severe with radiotherapy, the administration of drugs that can increase radiosensitivity might thus be helpful to avoid these side effects in the normal cells adjacent to the advanced gastric cancer.
A low dose of 5‐aza‐CdR (1 µmol/L) was used in combination with irradiation in the present study. Yang et al.( 18 ) reported that 5 µmol/L 5‐aza‐CdR is sufficient to reactivate the expression of silenced genes. Their low‐dose prolonged‐exposure 5‐aza‐CdR schedule was well tolerated and delivered on an outpatient basis, and low doses are more effective than higher doses.( 19 ) In a preliminary study, different concentrations of 5‐aza‐CdR, ranging from 0.5 to 10 µmol/L, were applied for the proliferation assay and it was found that 0.5–2 µmol/L 5‐aza‐CdR did not show significant anticancer effects, whereas 5 and 10 µmol/L significantly decreased the proliferation of gastric cancer cells (data not shown). In contrast, 1 µmol/L 5‐aza‐CdR showed synergistic effects in combination with radiation in three of five gastric cancer cell lines, but 0.5 µmol/L 5‐aza‐CdR did not. Therefore, 1 µmol/L 5‐aza‐CdR was used in subsequent experiments. The epigenetic alteration of mRNA by DNA methyltransferase inhibitor is reversible.( 20 ) 5‐aza‐CdR has a short half‐life of 15–25 min due to its rapid inactivation by liver cytidine deaminase in humans.( 21 ) 5‐aza‐CdR exposure for 2 h increased the radiation sensitivity and enhanced the expression of genes associated with radiosensitivity. These findings suggest that a low‐dose administration (1 µmol/L) of 5‐aza‐CdR for 2 h might be effective and safe for radiation therapy. 5‐aza‐CdR has been studied in several clinical trials for solid tumors as well as in different types of leukemia. 5‐aza‐CdR has been shown to have limited efficacy against solid tumors,( 22 , 23 ) although it has shown response rates ranging from 20 to 40% in myelodysplastic syndrome and acute and chronic myelogenous leukemia in multiple phase II and III studies.( 24 , 25 ) 5‐aza‐CdR induces moderate toxicities but not severe toxicities.( 12 ) Therefore, the next approach utilizing 5‐aza‐CdR clinically should be in combination with radiotherapy or chemotherapy at the same dose and schedule as described in previous trials.
It has been reported that 5‐aza‐CdR causes cell‐cycle arrest at G1 or G2–M phase in different cancer cells.( 19 , 20 , 26 ) In the present study, 5‐aza‐CdR resulted in an accumulation of cancer cells in G2–M phase of the cell cycle in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in either KATO‐III or MKN‐74 cells. As cells in G2–M phase are the most sensitive to irradiation,( 27 ) the arrest at G2–M phase caused by 5‐aza‐CdR might contribute to strong antiproliferation effects of irradiation in some types of gastric cancer cells. The rates of apoptosis were significantly increased by irradiation in combination with 5‐aza‐CdR in OCUM‐2M, OCUM‐12 and MKN‐45 cell lines. The mechanisms responsible for the synergistic antiproliferation effect of 5‐aza‐CdR might be partly explained by the induction of apoptosis. These findings provide the rationale for antitumor and cell‐cycle effects of a combination of 5‐aza‐CdR and radiation.
An increase in p53 mRNA expression was observed in response to 5‐aza‐CdR with or without irradiation in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in either KATO‐III or MKN‐74 cells. The p53 gene is wild type in OCUM‐2M, and MKN‐45, mutated in MKN‐74 and OCUM‐12, and deleted in KATO‐III cells. Taken together, the p53 protein level also increased in response to 5‐aza‐CdR with or without irradiation in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in either KATO‐III or MKN‐74 cells (Supporting Fig. S1). Neither the p53 gene nor the p53 protein were detected in KATO‐III cells as KATO‐III cells are p53 deficient. Gastric tumor cell lines expressing wild‐type p53 (OCUM‐2M and MKN‐45) are more sensitive to combination therapy with radiation and 5‐aza‐CdR than those expressing mutant p53 (KATO‐III and MKN‐74). A previous report found that 5‐aza‐CdR induces the p53 expression associated with inhibition of cell proliferation in p53 wild‐type cells, but not in p53 mutant cells in prostate cancer.( 21 , 28 ) We also found that 5‐aza‐CdR induces p53 expression associated with the inhibition of cell proliferation in OCUM‐2M, OCUM‐12, and MKN‐45 cells, but not in KATO‐III and MKN‐74 cells. p53 has been classically described as a mediator of the cytotoxicity of irradiation by promoting either cell‐cycle arrest or apoptosis.( 29 , 30 ) In the case of p53‐proficient cells, treatment with 5‐aza‐CdR results in G2–M arrest. The arrest at G2–M phase and increased apoptotic rate might be partly mediated by enhanced expression of p53 in response to 5‐aza‐CdR, resulting in G2–M arrest. We herein address the role of p53 in regulating cellular responses to 5‐aza‐CdR treatment in human gastric cancer cells with wild‐type p53, although DNA promoter methylation is infrequent in the p53 gene. These findings suggest that p53 might therefore be activated by 5‐Aza‐CdR, and not from direct inhibition of promoter methylation.( 31 )
Inactivation of the tumor suppressor RASSF1 has been implicated in the development of many human cancers.( 32 ) RASSF1 modulates multiple apoptotic and cell‐cycle checkpoint pathways.( 33 ) Its expression is lost in a wide variety of human tumors as a result of silencing, primarily from promoter hypermethylation, leading to cell‐cycle arrest. RASSF1 may therefore also explain the mechanism of synergism of 5‐aza‐CdR in apoptotic signaling. Expression of RASSF1 mRNA was increased in response to 5‐aza‐CdR with or without irradiation in OCUM‐2M, KATO‐III, MKN‐45, and MKN‐74 cells. The pro‐apoptotic DAPK genes, which are also associated with apoptotic pathways, are regulated epigenetically by DNA methyltransferase inhibitors.( 34 ) The DAPK‐1 level of OCUM‐2M and MKN‐45, and the DAPK‐2 level of OCUM‐2M, DAPK‐3, OCUM‐12, and MKN‐45 were increased in response to 5‐aza‐CdR compared with the control. The alterations observed in the DAPK genes might therefore be associated with irradiation sensitivity. These findings suggest that the induction of apoptosis might be associated not only with p53 but also with the RASSF1 and DAPK genes.
In conclusion, 5‐aza‐CdR regulates G2 checkpoint control and sensitizes gastric cancer cells to radiation. The arrest at G2–M phase and increased apoptotic rate might be partly mediated by enhanced expression of the p53, RASSF1, or DAPK families of genes by 5‐aza‐CdR. A DNA methyltransferase inhibitor, 5‐aza‐CdR, could be used as a radiation sensitizer to treat some types of gastric carcinoma.
Conflict of interest statement
None of the authors have a conflict of interest to disclose.
Supporting information
Fig. S1. p53 protein level was increased by 5‐aza‐CdR in OCUM‐2M, OCUM‐12 and MKN‐45, but not in KATO‐III and MKN‐74.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This study was partially founded by a Japan–China Sasagawa Medical Scholarship, Grants‐in Aid for Scientific Research (no. 18591475, 20591073, and 18390369) from the Ministry of Education, Science, Sports, Culture, and Technology of Japan, a Japanese Society of Gastroenterology Grant‐in Aid for Scientific Research, a Grant‐in Aid from the Kobayashi Foundation for Innovative Cancer Chemotherapy, a Grant‐in Aid from the Sagawa Foundation for Cancer Research, and a Grant‐in Aid from the Osaka Medical Research Foundation for Incurable Diseases.
References
- 1. D’Angelica M, Gonen M, Brennan MF, Turnbull AD, Bains M, Karpeh MS. Patterns of initial recurrence in completely resected gastric adenocarcinoma. Ann Surg 2004; 240: 808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kunisaki C, Shimada H, Nomura M et al . Therapeutic strategy for scirrhous type gastric cancer. Hepatogastroenterology 2005; 52: 314–18. [PubMed] [Google Scholar]
- 3. Macdonald JS, Smalley SR, Benedetti J et al . Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001; 345: 725–30. [DOI] [PubMed] [Google Scholar]
- 4. Smalley SR, Gunderson L, Tepper J et al . Gastric surgical adjuvant radiotherapy consensus report: rationale and treatment implementation. Int J Radiat Oncol Biol Phys 2002; 52: 283–93. [DOI] [PubMed] [Google Scholar]
- 5. Zhang ZX, Gu XZ, Yin WB, Huang GJ, Zhang DW, Zhang RG. Randomized clinical trial on the combination of preoperative irradiation and surgery in the treatment of adenocarcinoma of gastric cardia (AGC) – report on 370 patients. Int J Radiat Oncol Biol Phys 1998; 42: 929–34. [DOI] [PubMed] [Google Scholar]
- 6. National Comprehensive Cancer Network . Practice guidelines for upper gastrointestinal carcinomas. Oncology (Williston Park) 1998; 12: 179–223. [PubMed] [Google Scholar]
- 7. Valentini V, Cellini F. Radiotherapy in gastric cancer: a systematic review of literature and new perspectives. Expert Rev Anticancer Ther 2007; 7: 1379–93. [DOI] [PubMed] [Google Scholar]
- 8. Jones PA. DNA methylation and cancer. Oncogene 2002; 21: 5358–60. [DOI] [PubMed] [Google Scholar]
- 9. Zhang X, Yashiro M, Ohira M, Ren J, Hirakawa K. Synergic antiproliferative effect of DNA methyltransferase inhibitor in combination with anticancer drugs in gastric carcinoma. Cancer Sci 2006; 97: 938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lettini AA, Guidoboni M, Fonsatti E, Anzalone L, Cortini E, Maio M. Epigenetic remodelling of DNA in cancer. Histol Histopathol 2007; 22: 1413–24. [DOI] [PubMed] [Google Scholar]
- 11. Momparler RL. Epigenetic therapy of cancer with 5‐aza‐2′‐deoxycytidine (decitabine). Semin Oncol 2005; 32: 443–51. [DOI] [PubMed] [Google Scholar]
- 12. Abele R, Clavel M, Dodion P et al . The EORTC Early Clinical Trials Cooperative Group experience with 5‐aza‐2′‐deoxycytidine (NSC 127716) in patients with colo‐rectal, head and neck, renal carcinomas and malignant melanomas. European J Cancer Clin Oncology 1987; 23: 1921–4. [DOI] [PubMed] [Google Scholar]
- 13. Van Groeningen CJ, Leyva A, O’Brien AM, Gall HE, Pinedo HM. Phase I and pharmacokinetic study of 5‐aza‐2′‐deoxycytidine (NSC 127716) in cancer patients. Cancer Res 1986; 46: 4831–6. [PubMed] [Google Scholar]
- 14. Morita S, Iida S, Kato K, Takagi Y, Uetake H, Sugihara K. The synergistic effect of 5‐aza‐2′‐deoxycytidine and 5‐fluorouracil on drug‐resistant tumors. Oncology 2006; 71: 437–45. [DOI] [PubMed] [Google Scholar]
- 15. Yashiro M, Chung YS, Nishimura S, Inoue T, Sowa M. Establishment of two new scirrhous gastric cancer cell lines: analysis of factors associated with disseminated metastasis. Br J Cancer 1995; 72: 1200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niigata HHJ. Establishment of cultured cell lines of human stomach cancer origin and their morphological characteristics. Exp Med 1977; 91: 737–63. [Google Scholar]
- 17. Pfaffl MW. A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res 2001; 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang QW, Liu S, Tian Y et al . Methylation‐associated silencing of the thrombospondin‐1 gene in human neuroblastoma. Cancer Res 2003; 63: 6299–310. [PubMed] [Google Scholar]
- 19. Zhang B, Huang T, Liu K, Chen J, Wang G. Effects of 5‐Aza‐CdR on cell proliferation of breast cancer cell line MDA‐MB‐435S and expression of maspin gene. J Huazhong Univ Sci Technol Med Sci 2007; 27: 543–6. [DOI] [PubMed] [Google Scholar]
- 20. Yang Q, Shan L, Yoshimura G et al . 5‐aza‐2′‐deoxycytidine induces retinoic acid receptor beta 2 demethylation, cell cycle arrest and growth inhibition in breast carcinoma cells. Anticancer Res 2002; 22: 2753–6. [PubMed] [Google Scholar]
- 21. Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5‐aza‐2′‐deoxycytidine. Mol Pharmacol 2001; 59: 751–7. [PubMed] [Google Scholar]
- 22. Samuels BL, Herndon JE 2nd, Harmon DC et al . Dihydro‐5‐azacytidine and cisplatin in the treatment of malignant mesothelioma: a phase II study by the Cancer and Leukemia Group B. Cancer 1998; 82: 1578–84. [PubMed] [Google Scholar]
- 23. Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J. Pilot phase I–II study on 5‐aza‐2′‐deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anti-Cancer Drugs 1997; 8: 358–68. [DOI] [PubMed] [Google Scholar]
- 24. Oki Y, Issa JP. Review: recent clinical trials in epigenetic therapy. Rev Recent Clin Trials 2006; 1: 169–82. [DOI] [PubMed] [Google Scholar]
- 25. Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Seminars Hematol 2008; 45: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nieto M, Samper E, Fraga MF, Gonzalez de Buitrago G, Esteller M, Serrano M. The absence of p53 is critical for the induction of apoptosis by 5‐aza‐2′‐deoxycytidine. Oncogene 2004; 23: 735–43. [DOI] [PubMed] [Google Scholar]
- 27. Wilson GD. Cell kinetics. Clin Oncol (R Coll Radiol) 2007; 19: 370–84. [DOI] [PubMed] [Google Scholar]
- 28. Pulukuri SM, Rao JS. Activation of p53/p21Waf1/Cip1 pathway by 5‐aza‐2′‐deoxycytidine inhibits cell proliferation, induces pro‐apoptotic genes and mitogen‐activated protein kinases in human prostate cancer cells. Int J Oncol 2005; 26: 863–71. [PubMed] [Google Scholar]
- 29. Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild‐type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA 1992; 89: 7491–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation‐induced apoptosis in mouse thymocytes. Nature 1993; 362: 847–9. [DOI] [PubMed] [Google Scholar]
- 31. Liang G, Gonzales FA, Jones PA, Orntoft TF, Thykjaer T. Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5‐aza‐2′‐deoxycytidine. Cancer Res 2002; 62: 961–6. [PubMed] [Google Scholar]
- 32. Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000; 25: 315–19. [DOI] [PubMed] [Google Scholar]
- 33. Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci 2007; 120: 3163–72. [DOI] [PubMed] [Google Scholar]
- 34. Schildhaus HU, Krockel I, Lippert H, Malfertheiner P, Roessner A, Schneider‐Stock R. Promoter hypermethylation of p16INK4a, E‐cadherin, O6‐MGMT, DAPK and FHIT in adenocarcinomas of the esophagus, esophagogastric junction and proximal stomach. Int J Oncol 2005; 26: 1493–500. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. p53 protein level was increased by 5‐aza‐CdR in OCUM‐2M, OCUM‐12 and MKN‐45, but not in KATO‐III and MKN‐74.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item