

Abstract
Interferon regulatory factors (IRFs) are transcription factors known to play key roles in innate and adaptive immune responses, cell growth, apoptosis, and development. Their function in tumorigenesis of gastric cancer remains to be determined, however. In the present study, therefore, we examined epigenetic inactivation of IRF1–9 in a panel of gastric cancer cell lines. We found that expression of IRF4, IRF5, and IRF8 was frequently suppressed in gastric cancer cell lines; that methylation of the three genes correlated with their silencing; and that treating the cells with the demethylating agent 5‐aza‐2′‐deoxycytidine (DAC) restored their expression. Expression of IRF5 in cancer cells was enhanced by the combination of DAC treatment and adenoviral vector‐mediated expression of p53, p63, or p73. Interferon‐γ‐induced expression of IRF8 was also enhanced by DAC. Moreover, treating gastric cancer cells with DAC enhanced the suppressive effects of interferon‐α, interferon‐β, and interferon‐γ on cell growth. Among a cohort of 455 gastric cancer and noncancerous gastric tissue samples, methylation of IRF4 was frequently observed in both gastric cancer specimens and noncancerous specimens of gastric mucosa from patients with multiple gastric cancers, which suggests IRF4 methylation could be a useful molecular marker for diagnosing recurrence of gastric cancers. Our findings indicate that epigenetic IRF inactivation plays a key role in tumorigenesis of gastric cancer, and that inhibition of DNA methylation may restore the antitumor activity of interferons through up‐regulation of IRFs. (Cancer Sci 2010)
Gastric cancer arises through the accumulation of multiple genetic changes, including mutation of adenomatous polyposis coli (APC), K‐ras, and p53. ( 1 ) But recent studies have also shown that epigenetic changes such as DNA methylation are also importantly involved in the gene silencing seen in cancer.( 2 ) For instance, genes involved in regulation of the cell cycle and apoptosis are now known to be inactivated by DNA methylation.( 3 , 4 , 5 ) In addition we previously showed that a number of genes involved in signal transduction are epigenetically silenced in cancer. The affected genes include secreted frizzled‐related protein 1 (SFRP1), SFRP2, dickkopf 1 (DKK1), and DKK2, which are negative regulators of WNT signaling,( 6 , 7 ) Ras association domain family member 2 (RASSF2), a negative regulator of Ras;( 8 ) and 14‐3‐3σ and deafness, autosomal dominant 5 (DFNA5), two transcriptional targets of p53.( 9 , 10 ) Because DNA methylation is an epigenetic change, which does not affect gene sequences, the silenced genes can be reactivated by demethylation, making DNA methylation a useful target of cancer therapy.( 11 , 12 )
DNA methylation could also be used as a molecular marker for cancer detection. For instance, methylation of genes such as SFRP2 and GATA binding protein‐4 (GATA‐4) has been detected in stool DNA from colorectal cancer patients.( 13 , 14 ) In gastric cancer, infection by Helicobacter pylori (H. pylori) induces DNA methylation even in noncancerous tissues.( 15 ) In addition, higher levels of methylation are detected in gastric mucosae from cancer patients than in samples from patients without cancer.( 15 , 16 ) Thus, DNA methylation in noncancerous tissues could be a potentially useful marker predicting development or recurrence of gastric cancer.
The interferon regulatory factor gene (IRF) family encodes a group transcription factors induced by interferon. To date, nine IRFs (IRF1–9) have been identified (reviewed in ref. 17), and their products have been shown to be involved in variety of processes, including innate and adaptive immune responses, cell growth, apoptosis, and development.( 17 ) Interferon regulatory factor 1 (IRF1) was the first to be identified as a regulatory factor in the interferon system,( 18 ) and several lines of evidence suggest IRF1 acts as a tumor suppressor in human neoplasias. For instance, IRF1 and p53 cooperate via two parallel but independent pathways leading to the induction of cell cycle arrest and p21 gene transcription.( 19 ) In addition, IRF5 is induced by p53 and is involved in growth suppression,( 20 , 21 ) while both IRF5 and IRF7 are involved in the induction of senescence.( 22 ) And down‐regulation of IRF8 expression contributes to resistance to apoptosis and to the metastatic phenotype in metastatic tumor cells.( 23 ) These findings prompted us to speculate that epigenetic inactivation of IRF expression may play a key role in tumorigenesis.
Epigenetic inactivation of IRF8 has recently been observed in colorectal, nasopharyngeal, esophageal, breast, and cervical cancers,( 23 , 24 ) and inactivation of IRF4 was shown to be silenced by DNA methylation in chronic myeloid leukemia.( 25 ) Thus epigenetic inactivation of IRFs appears to be centrally involved in the development of human neoplasias. However, there has been no comprehensive analysis of the epigenetic alterations of IRFs in gastric cancer. In the present study, therefore, we examined epigenetic inactivation of IRF1–9 in gastric cancer.
Materials and Methods
Cell lines and specimens. Sixteen gastric cancer cell lines (MKN1, MKN7, MKN28, MKN45, MKN74, KatoIII, AZ521, JRST, SNU1, SNU16, NUGC3, NUGC4, AGS, NCI‐N87, SNU16) were obtained from the American Type Culture Collection (Manassas, VA, USA) or the Japanese Collection of Research Bioresources (Tokyo, Japan). In addition, SH101 cells were kindly provided by Dr K. Yanagihara( 26 ) at the National Cancer Center Research Institute and have been described previously. In some cases cancer cell lines were treated with 2 μM 5‐aza‐2′‐deoxycytidine (DAC) (Sigma, St. Louis, MO, USA) for 72 h, replacing the drug and medium every 24 h. When cells were exposed to DAC and either IFN‐α, IFN‐β, or IFN‐γ, 1000 U/mL IFN‐α or IFN‐β or 100 U/mL IFN‐γ was added to the culture for 48 h following incubation with 0.2 μM DAC. The generation and purification of replication‐deficient recombinant adenoviruses encoding p53 (Ad‐p53), p63 (Ad‐p63), p73 (Ad‐p73), or LacZ (Ad‐LacZ), as well as the infection procedure, were all described previously.( 27 , 28 ) At a multiplicity of infection (MOI) of 100, 90–100% of the cells were infected.
Two sets of specimens were used in this study. One set contained a total of 68 primary gastric cancers and 22 corresponding gastric mucosa specimens described previously.( 29 ) The second set contained 35 gastric cancer specimens and 330 noncancerous specimens of gastric mucosa from 165 patients, which were obtained through biopsy during the course of endoscopy. Informed consent was obtained from all patients before collection of the specimens. Genomic DNA was extracted using the standard phenol‐chloroform procedure. Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and then treated with a DNA‐free kit (Ambion, Austin, TX, USA). Total RNA extracted from normal stomach, colon, breast, and pancreas from a healthy individual was purchased from BioChain (Hayward, CA, USA). RNA was also obtained from normal stomach glands using the crypt isolation technique as described previously.( 30 )
Gene expression analysis. Real‐time PCR was carried out using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) and a 7900HT Fast Real‐Time PCR System (Applied Biosystems) according to the manufacturer’s instructions. SDS2.2.2 software (Applied Biosystems) was used for comparative delta Ct analysis, and GAPDH served as an endogenous control. The primers used in this study are shown in Supplementary Table S1. The TaqMan primers/probes used in this study were: IRF1, Hs00971960_m1; IRF2, Hs01082884_m1; IRF3, Hs00155574_m1; IRF4, Hs00180031_m1; IRF5, Hs00158114_m1; IRF6, Hs00608402_m1; IRF7, Hs00242190_g1; IRF8, Hs00175238_m1; and IRF9, Hs00196051_m1.
Methylation analysis. Samples of genomic DNA (2 μg) were modified with sodium bisulfite using an EpiTect Bisulfite Kit (Qiagen, Hilden, Germany). Methylation was determined by methylation specific PCR, bisulfite‐sequencing, and bisulfite‐pyrosequencing, and details of methods are shown in the Supporting Information. The primer sequences are listed in Supplementary Tables S1 and S2.
Statistics. Statistical analyses were carried out using SPSSJ 15.0 (SPSS Japan, Tokyo, Japan). For comparison of methylation levels between cancerous and normal tissues, and for other continuous data, t‐tests or paired t‐tests were performed, as appropriate. Fisher’s exact test and the Mann–Whitney U‐test were used to evaluate the association between IRF methylation, clinicopathological features, and other genetic and epigenetic alterations. Receiver–operator curves (ROC) were constructed based on IRF methylation levels, and P‐values were calculated by comparing the areas under the curves (AUC) with a reference curve. Values of P < 0.05 were considered significant.
Mutation of p53 and KRAS and detection of the presence of CpG island methylator phenotype (CIMP) or Epstein–Barr virus (EBV) were described previously.( 31 ) To determine CIMP status, methylation status of five loci (MINT1, MINT2, MINT12, MINT25, and MINT31) was assessed using combined bisulfite restriction analysis (COBRA). Cases with methylation of four or five loci were defined as CIMP‐H. Cases with methylation of one to three loci were defined as CIMP‐L. Cases with no methylation were defined as CIMP‐N.
Results
Expression of IRF1–9 in gastric cancer cell lines. To determine whether expression of IRF1–9 is altered in gastric cancers, we carried out a real‐time PCR analysis using a panel of gastric cancer cell lines (Fig. 1a). We found that expression of IRF4, IRF5, and IRF8 was frequently down‐regulated in these cell lines. Expression of IRF7 was not detected in normal tissues or in the gastric cancer cell lines, but the remaining IRFs were expressed at various levels in normal tissues (Fig. 1a,b). We also examined expression of IRF1‐9 using cDNA prepared using the gastric gland isolation technique, and similar levels of IRF1–9 expression were observed (Fig. 1b). To determine whether the down‐regulation of the affected IRFs reflected epigenetic modification, we next assessed IRF expression following treatment with the demethylating agent DAC. We found that DAC restored IRF expression in most gastric cancer cell lines showing IRF4, IRF5, and/or IRF8 methylation (Fig. S1). On the other hand, DAC had little effect on several cell lines (i.e. AZ521, AGS, for IRF5; NUGC3 for IRF8), suggesting other stimuli may be required for full reactivation of IRFs.
Figure 1.
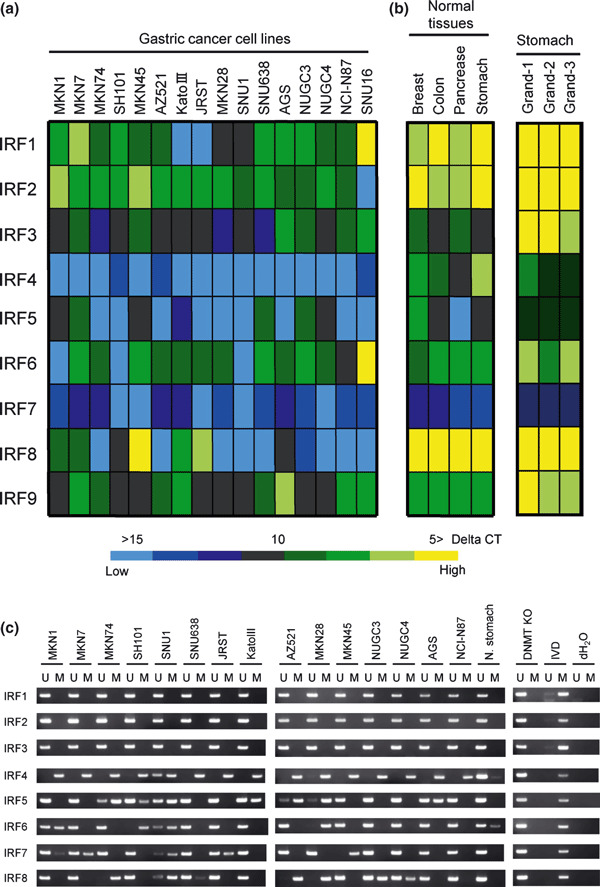
Down‐regulation of interferon regulatory factors (IRFs) in gastric cancer cell lines. The heat map shows the expression profiles in 16 gastric cancer cell lines (a) and four normal tissue specimens (b). Levels of expression are normalized to GAPDH, and delta‐CT values are shown. (c) Analysis of IRF1–8 methylation in gastric cancer cell lines. Methylation of 5′ CpG islands was examined using methylation‐specific PCR. The cell lines examined are shown on the top. DNMT KO: DNMT1−/−, DNMT3B−/− HCT116 cell. IVD, in vitro methylated DNA; M, methylated; N, stomach: normal stomach; U, unmethylated.
Treating cancer cells with DAC restored induction of IRF5 by p53 and of IRF8 by IFN‐γ. Interferon regulatory factor 5 (IRF5) and IRF8 are known to be transcriptional targets of p53( 21 ) and interferon‐γ,( 32 ) respectively. We therefore tested whether DAC would enhance the induction of IRF5 by p53 family members in two gastric cancer cell lines showing IRF5 methylation. When we infected MKN74 and SNU1 cells with Ad‐lacZ, Ad‐p53, Ad‐p73, or Ad‐p63, DAC acted synergistically with the expressed p53 family member to induce IRF5 expression in the cells (Fig. S2a). In similar fashion, we found that treating MKN28 cells with DAC enhanced the induction of IRF8 by interferon‐γ (Fig. S2b).
Methylation of IRF4, IRF5, and IRF8 in gastric cancer cell lines. Database analysis of nine IRF genes showed that all except IRF9 contained CpG islands at their 5′ ends. We therefore used methylation‐specific PCR to examine the methylation status of IRF1‐8 (Fig. 1c). We found that IRF4 was the most frequently methylated in gastric cancer cell lines. In addition, methylation of IRF5, IRF6, IRF7, and IRF8 was detected in subsets of gastric cancer cell lines. No methylation of IRF1, IRF2, or IRF3 was detected in any of the gastric cancer cell lines tested.
We next carried out bisulfite‐pyrosequencing to further examine the role of DNA methylation in the down‐regulation of IRF expression (2, 3, 4). Gastric cancer cell lines that exhibited low or negligible IRF4 expression showed high levels of methylation. Similarly, methylation was well correlated with the down‐regulation of IRF5 and IRF8 in gastric cancer cell lines.
Figure 2.
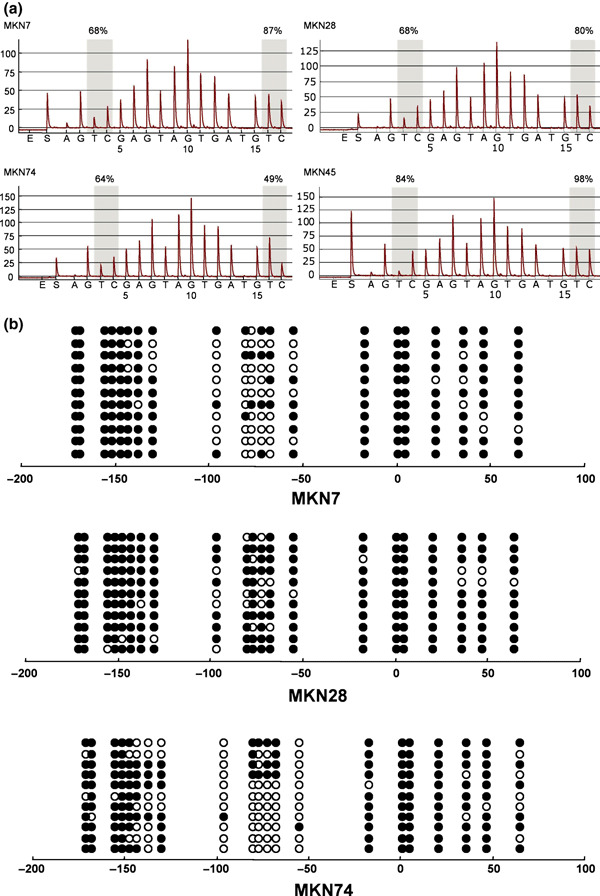
Methylation analysis of interferon regulatory factor (IRF)‐4 in gastric cancer cell lines. (a) Representative pyrosequencing results. Gray columns depict regions of CpG sites, and the percentage methylation at each CpG site is shown on the top. (b) Representative bisulfite‐sequencing results. Each circle represents a CpG dinucleotide. Methylation status: open circles, unmethylated; black circles, methylated. The cell lines examined are shown below the columns.
Figure 3.
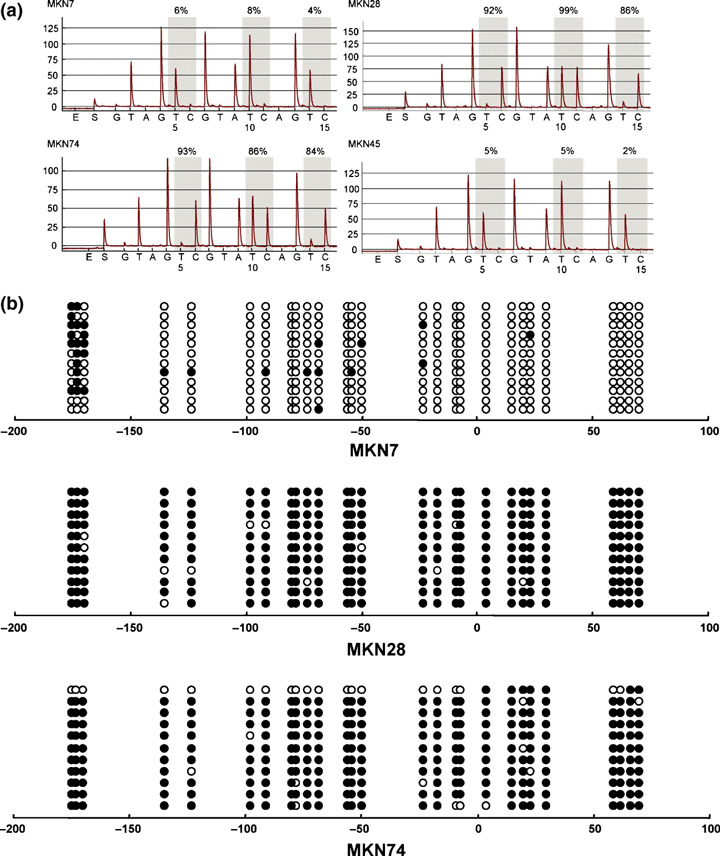
Methylation analysis of interferon regulatory factor (IRF)‐5 in gastric cancer cell lines. (a) Representative pyrosequencing results. (b) Representative bisulfite‐sequencing results.
Figure 4.
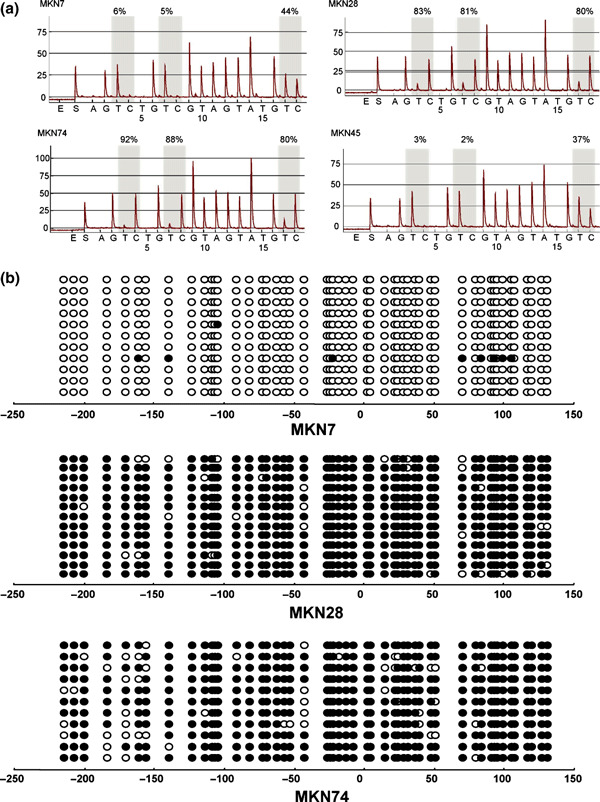
Methylation analysis of interferon regulatory factor (IRF)‐8 in gastric cancer cell lines. (a) Representative pyrosequencing results. (b) Representative bisulfite‐sequencing results.
We then confirmed the methylation status of IRF4, IRF5, and IRF8 using bisulfite‐sequencing (2, 3, 4). High levels of IRF4 methylation were detected in all of the cancer cell lines tested. In the gastric cancer cell lines, for example, heterogeneous methylation was observed in the region spanning positions −50 to −100 from the transcription start site. High levels of IRF5 methylation were detected in two (MKN28 and MKN74) of the cancer cell lines showing low or negligible expression, but only sparse methylation was detected in a third (MKN7). MKN7 cells expressed IRF8 and did not show methylation of that gene. By contrast, MKN28 and MKN74 cells did not express IRF8 and showed dense methylation of the gene.
Suppression of cell growth by DAC + IFN. Given that DAC induces IRFs in gastric cancer cells, we tested whether DAC treatment would enhance the growth suppressive effect of interferon on cancer cells. When we treated four gastric cancer cell lines (SNU1, MKN28, KatoIII, and MKN74) first with DAC for 72 h and then with IFN‐α, ‐β, or ‐γ for 48 h, we found that DAC enhanced the growth suppressive effects of all three interferons (Fig. S3). This prompted us to test the effect of IFN on DNA methylation. Using bisulfite‐pyrosequencing with DNA from cells treated with DAC and/or IFN (Fig. S4), we found that although treatment with DAC induced partial demethylation of IRF4, IRF5, and IRF8, treatment with IFN‐α/β/γ, alone or in combination with DAC, did not induce further demethylation in MKN74 cells.
Methylation of IRF4, IRF5, and IRF8 in primary gastric cancers. To assess IRF methylation in primary tumors, we used bisulfite‐pyrosequencing to examine primary specimens from 68 gastric cancers and 22 noncancerous gastric tissues (Fig. 5a,b). We found that IRF4 was frequently methylated in gastric cancer. In addition, we detected high levels of IRF5 methylation in several gastric cancers, but the average methylation levels did not significantly differ between the cancerous and normal tissues. We did not detect significant methylation of IRF8 in primary gastric cancers.
Figure 5.
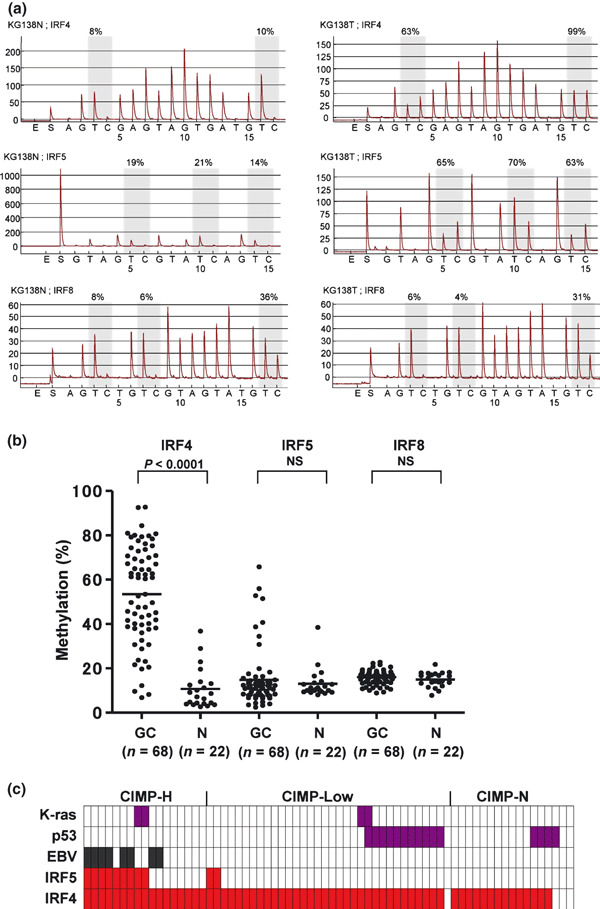
Methylation of interferon regulatory factor (IRF)‐4, IRF5, and IRF8 in primary gastric cancers. (a) Representative bisulfite‐pyrosequencing results. (b) Scatter plot of IRF methylation. GC, gastric cancer; horizontal bars, average methylation levels in total cases; N, normal stomach; NS, not significant. (c) Genetic and epigenetic alterations in gastric cancer. Each row represents the separate gene locus shown on the left. Each column is a primary gastric cancer: red rectangles, methylated tumors; purple rectangles, mutated tumors, grey rectangles, Epstein–Barr virus‐positive tumors.
We next evaluated the correlation between IRF methylation and the clinicopathological features of gastric cancers and between IRF methylation and other genetic and epigenetic alterations in gastric cancer. We selected a 13.9% cut‐off for IRF4 and a 16.6% cut‐off for IRF5 methylation based on our finding that these levels represent the 75th percentile among the control samples. With those thresholds, 64 of 68 cases showed methylation of IRF4, and 11 of 68 cases showed methylation of IRF5 (Fig. 5c). Methylation of IRF4 was detected significantly more frequently in diffuse type and CIMP‐H gastric cancers than in CIMP‐L or CIMP‐N cancers. Methylation of IRF5 was detected significantly more frequently in gastric cancers positive for EBV and in CIMP‐H cancers than in CIMP‐L or CIMP‐N cancers (Table 1).
Table 1.
Correlation between methylation of IRF4/IRF5 and the clinicopathological features of the patients
| Characteristics | IRF4 | IRF5 | |||||
|---|---|---|---|---|---|---|---|
| Total | U | M | P‐value | U | M | P‐value | |
| n | 68 | 4 | 64 | 57 | 11 | ||
| Age | |||||||
| Mean | 64.2 | 66.3 | 64.0 | 0.727 | 64.5 | 62.7 | 0.668 |
| SD | 12.1 | 6.7 | 12.4 | 11.7 | 14.9 | ||
| Sex | |||||||
| Male | 45 | 4 | 41 | 0.292 | 19 | 4 | 1.000 |
| Female | 23 | 0 | 23 | 38 | 7 | ||
| Location | |||||||
| Lower | 30 | 2 | 28 | 0.929 | 26 | 4 | 0.458 |
| Middle | 23 | 1 | 22 | 20 | 3 | ||
| Upper | 15 | 1 | 14 | 11 | 4 | ||
| Type | |||||||
| 0 | 4 | 0 | 4 | 0.605 | 3 | 1 | 0.547 |
| 1 | 5 | 0 | 5 | 4 | 1 | ||
| 2 | 26 | 3 | 23 | 20 | 6 | ||
| 3 | 25 | 1 | 24 | 22 | 3 | ||
| 4 | 8 | 0 | 8 | 8 | 0 | ||
| Histology | |||||||
| D | 38 | 0 | 38 | 0.034 | 29 | 9 | 0.096 |
| I | 30 | 4 | 26 | 28 | 2 | ||
| ly | |||||||
| − | 14 | 0 | 16 | 0.566 | 12 | 4 | 0.272 |
| + | 44 | 4 | 48 | 45 | 7 | ||
| v | |||||||
| − | 16 | 0 | 33 | 0.115 | 27 | 6 | 0.749 |
| + | 52 | 4 | 31 | 30 | 5 | ||
| pT | |||||||
| pT1 | 5 | 0 | 5 | 0.225 | 4 | 1 | 0.352 |
| pT2 | 36 | 1 | 35 | 29 | 7 | ||
| pT3 | 25 | 3 | 22 | 22 | 3 | ||
| pT4 | 2 | 0 | 2 | 2 | 0 | ||
| pN | |||||||
| pN0 | 18 | 2 | 16 | 0.145 | 16 | 2 | 0.855 |
| pN1 | 25 | 2 | 23 | 19 | 6 | ||
| pN2 | 14 | 0 | 14 | 12 | 2 | ||
| pN3 | 11 | 0 | 11 | 10 | 1 | ||
| pM | |||||||
| M0 | 57 | 4 | 53 | 1.000 | 47 | 10 | 0.677 |
| M1 | 11 | 0 | 11 | 10 | 1 | ||
| Stage (pTNM, 1997, 5th ed) | |||||||
| 1A | 3 | 0 | 3 | 0.342 | 2 | 1 | 0.511 |
| 1B | 12 | 0 | 12 | 11 | 1 | ||
| 2 | 13 | 3 | 10 | 10 | 3 | ||
| 3A | 12 | 1 | 11 | 9 | 3 | ||
| 3B | 7 | 0 | 7 | 6 | 1 | ||
| 4 | 21 | 0 | 21 | 19 | 2 | ||
| KRAS | |||||||
| − | 64 | 4 | 60 | 1.000 | 55 | 9 | 0.120 |
| + | 4 | 0 | 4 | 2 | 2 | ||
| p53 | |||||||
| − | 53 | 3 | 50 | 1.000 | 42 | 11 | 0.105 |
| + | 15 | 1 | 14 | 15 | 0 | ||
| EBV | |||||||
| − | 60 | 4 | 56 | 1.000 | 55 | 5 | <0.001 |
| + | 8 | 0 | 8 | 2 | 6 | ||
| CIMP | |||||||
| H | 17 | 0 | 14 | 0.035 | 8 | 9 | <0.001 |
| L | 34 | 1 | 33 | 32 | 2 | ||
| N | 17 | 3 | 14 | 17 | 0 | ||
CIMP, CpG island methylator phenotype; EBV, Epstein–Barr virus; IRF4, interferon regulatory factor 4. ly, lymphatic vessels invasion; pN, pathological node stage; pT, pathological tumor stage; pM, pathological metastasis.
Methylation of IRF4 in noncancerous gastric mucosa is a potential molecular marker for gastric cancer. Several of the cases studied showed high levels of IRF4 methylation, even in noncancerous gastric mucosa (Fig. 5b). We therefore wondered whether levels of IRF4 methylation in noncancerous tissues are associated with the presence of gastric cancer. To address that issue, we examined tissue specimens obtained from 165 patients through endoscopic biopsy, including 35 gastric cancer specimens and 330 noncancerous specimens of gastric mucosa (Fig. 6a, Table S3). We found that methylation of IRF4 in noncancerous gastric tissues was significantly higher in patients with cancer than in those without cancer (P < 0.001). In addition, patients with multiple gastric cancers showed significantly higher levels of IRF4 methylation than patients with a single cancer (P < 0.05). Levels of IRF4 methylation tended to be higher in patients infected with H. pylori than in those without H. pylori, though the difference was not statistically significant.
Figure 6.
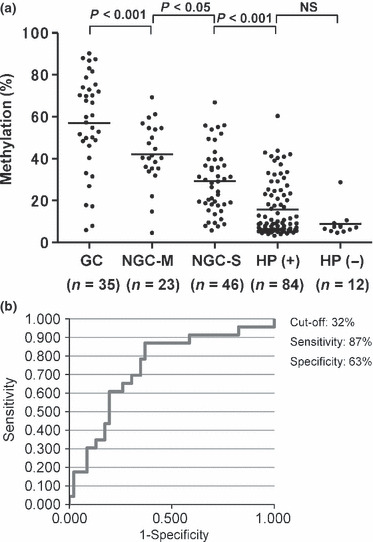
Methylation levels of interferon regulatory factor (IRF)‐4 in gastric cancers and noncancerous gastric mucosae. (a) Distribution of IRF4 methylation in gastric cancer and noncancerous gastric mucosae. GC, gastric cancer; HP(+), gastric mucosae from Helicobacter pylori (H. pylori)‐positive chronic gastritis patients without cancer; HP(−), gastric mucosae from H. pylori‐negative chronic gastritis patients without cancer; NGC‐M, noncancerous gastric mucosae from multiple gastric cancer patients; NGC‐S, noncancerous gastric mucosae from single gastric cancer patients. For noncancerous gastric mucosae, specimens were obtained from the antrum and body, and average methylation levels are shown. Horizontal bars, average methylation levels in total cases. The numbers of cases examined in the study are shown below the column. (b) Receiver–operator curve (ROC) for IRF4 methylation to discriminate patients with multiple gastric cancers from patients with a single gastric cancer.
The clinical usefulness of DNA methylation for distinguishing cancer patients from noncancer patients was confirmed by ROC analysis. Methylation of IRF4 gave highly discriminative ROC profiles, which clearly distinguished patients with a single gastric cancer from H. pylori‐positive gastritis patients without cancer (AUC: 0.77, P < 0.001) (Fig. S5, Table S3). They also distinguished patients with a single or multiple gastric cancers from H. pylori‐positive gastritis patients without cancer (AUC: 0.81, P < 0.001) (Fig. S6, Table S3). More interestingly, when 32%IRF4 methylation in noncancerous gastric mucosae was used as the cut‐off, patients with multiple gastric cancers could be discriminated from patients with a single gastric cancer with a sensitivity of 87% and a specificity of 63% (AUC: 0.74, P < 0.05) (Fig. 6b, Table S3). This suggests methylation of IRF4 in noncancerous gastric mucosae could be used as a molecular marker to predict recurrence of gastric cancer.
Discussion
Interferons play critical roles in regulating immune system function, cell growth, and apoptosis. It is therefore noteworthy that expression of interferon target genes is suppressed in a variety of cancers.( 33 ) For instance, signaling pathways mediated by expression of signal transducer and activator of transcription 1 (STAT1),( 34 ) class II major histocompatibility complex transactivator (CIITA),( 35 ) and XIAP associated factor 1 (XAF1),( 36 ) three genes downstream of interferon, are silenced by epigenetic inactivation in various cancers, which suggests impairment of interferon signaling by epigenetic mechanisms may play an important role in tumorigenesis. Consistent with that idea, a number of earlier studies have shown that IRFs are silenced by DNA methylation in human neoplasias.( 23 , 24 , 25 , 37 , 38 ) Here, we found that DNA methylation of IRF4, IRF5, and/or IRF8 is a frequent event in gastric cancer cell lines and that treatment with a demethylating agent (DAC) restores induction of IRF5 by p53, p63, or p73 and induction of IRF8 by IFN‐γ, which confirms the role played by DNA methylation in silencing the genes. Moreover, when applied together, interferon and DAC acted synergistically to suppress cell growth. Thus inhibition of DNA methylation could be a useful strategy for enhancing the tumor suppressor activity of interferon.
It was previously shown that IRF4 is silenced by DNA methylation in chronic myeloid leukemia.( 25 ) In the present study, we found that IRF4 is frequently silenced by DNA methylation in both gastric cancers and noncancerous gastric mucosae from cancer patients. Such methylation can be readily detected in serum samples and gastric washing solution,( 39 , 40 ) and the high frequency of IRF4 methylation in gastric cancer could be useful for establishing a diagnostic system with DNA methylation as the target. The precise role of IRF4 methylation in the development and progression of gastric cancer remains unknown. It has been suggested that weakly expressed genes are especially susceptible to methylation changes in cancer.( 41 ) In fact, we found that IRF4 expression was minimally expressed in gastric epithelium, which consistent with the report that IRF4 is exclusively expressed in lymphocytic tissues.( 17 ) If that is the case, methylation of IRF4 may not provide a growth advantage to cells, but may reflect epigenetic defects in the gastric mucosa caused by inflammation. Here we showed that levels of IRF4 methylation were high in noncancerous gastric mucosae from gastric cancer patients, especially in those with multiple cancers. Although further prospective study may be necessary, it would appear that methylation of IRF4 could be a molecular marker with which to predict the development or recurrence of gastric cancer.
Several lines of evidence have suggest that IRF5 has tumor suppressor activity, and that in response to DNA damage IRF5 is induced by p53 to promote cell cycle arrest and apoptosis.( 20 , 21 , 42 ) Kulaeva et al. ( 43 )showed that treating spontaneously immortal Li–Fraumeni fibroblasts with DAC induces a senescence‐like state, and that IRF5 is silenced by DNA methylation in the same cells, suggesting IRF5 is involved in mediating cellular senescence.( 22 ) Here we showed that DAC enhanced p53‐induced IRF5 expression, and that IRF5 expression was also induced by p63 and p73, suggesting IRF5 is a target of the p53 gene family. Although, on average, IRF5 methylation was not significantly higher in primary cancers than in noncancerous tissues, several cases did show high levels of IRF5 methylation.
We found that IRF8 expression was down‐regulated in gastric cancer cell lines; that DNA methylation was well correlated with gene silencing; and that treating cells with DAC restored IRF8 expression. This is consistent with earlier reports showing that IRF8 is silenced in colorectal cancer cell lines in a DNA methylation‐dependent manner.( 23 ) In contrast to the data obtained with cell lines, we did not find an increase in IRF8 methylation in primary gastric cancers, as compared to noncancerous tissues. This is in contrast to earlier studies showing that IRF8 is methylated in cancers of the colon, esophagus, and nasopharyngus.( 24 , 37 ) This discrepancy may reflect the different methods used to detect methylation: methylation‐specific PCR was used in those earlier studies, whereas we used bisulfate‐pyrosequencing. Alternatively, methylation of IRF8 may be an early event in tumorigenesis, which starts in subsets of gastric epithelial cells. Consistent with that idea, Lee et al. reported that IRF8 is methylated only in some esophageal tissues from esophageal cancer patients. Further study will be necessary to clarify the significance of IRF8 methylation in primary gastric cancers.
In conclusion, we have shown that IRF4, IRF5, and IRF8 are epigenetically silenced in gastric cancer cells. Methylation of IRF5 was associated with CIMP and EBV infection. Moreover, the high degree of IRF4 methylation in gastric mucosae from cancer patients suggests that DNA methylation of IRF4 could be a useful molecular marker for gastric cancer diagnosis and risk assessment.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Real‐time PCR analysis of interferon regulatory factor (IRF)‐4, IRF5, and IRF8 expression in gastric cancer cell lines.
Fig. S2. Induction of interferon regulatory factor (IRF)‐5 expression by p53 (a) and of IRF8 expression by interferon (IFN)‐ γ (b).
Fig. S3. 5‐Aza‐2′‐deoxycytidine (DAC) enhances suppression of cell growth by interferon.
Fig. S4. Methylation analysis of interferon regulatory factor (IRF)‐4, IRF5, and IRF8 after treatment with 5‐aza‐2′‐deoxycytidine (DAC) and/or interferon (IFN).
Fig. S5. Receiver–operator curve (ROC) for interferon regulatory factor (IRF)‐4 methylation to discriminate patients with a single gastric cancer from patients with Helicobacter pylori‐positive chronic gastritis.
Fig. S6. Receiver–operator curve (ROC) curve for interferon regulatory factor (IRF)‐4 methylation to discriminate patients with a single or multiple gastric cancers from patients with Helicobacter pylori‐positive chronic gastritis.
Table S1. Primers used for methylation‐specific PCR (MSP) used in this study.
Table S2. Primer sequences used for bisulfite‐pyrosequencing and bisulfite‐sequencing.
Table S3. High levels of interferon regulatory factor (IRF)‐4 methylation are associated with multiple gastric cancers.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
The authors thank Dr William F. Goldman for editing the manuscript. This study was supported in part by Grants‐in‐Aid for Scientific Research on Priority Areas (M.T., K.I., and T.T.); the Program for Developing Supporting Systems for Upgrading Education and Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Y.S., M.T.); Grants‐in‐Aid for Scientific Research (S) from the Japan Society for Promotion of Science (K.I.); a Grant‐in‐Aid for the Third‐term Comprehensive 10‐year Strategy for Cancer Control (F.I., M.T.); and Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labor, and Welfare of Japan (M.T.).
References
- 1. Wright PA, Williams GT. Molecular biology and gastric carcinoma. Gut 1993; 34: 145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128: 683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sato F, Meltzer SJ. CpG island hypermethylation in progression of esophageal and gastric cancer. Cancer 2006; 106: 483–93. [DOI] [PubMed] [Google Scholar]
- 4. Suzuki H, Tokino T, Shinomura Y, Imai K, Toyota M. DNA methylation and cancer pathways in gastrointestinal tumors. Pharmacogenomics 2008; 9: 1917–28. [DOI] [PubMed] [Google Scholar]
- 5. Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer 2005; 5: 223–31. [DOI] [PubMed] [Google Scholar]
- 6. Nojima M, Suzuki H, Toyota M et al. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 2007; 26: 4699–713. [DOI] [PubMed] [Google Scholar]
- 7. Sato H, Suzuki H, Toyota M et al. Frequent epigenetic inactivation of DICKKOPF family genes in human gastrointestinal tumors. Carcinogenesis 2007; 28: 2459–66. [DOI] [PubMed] [Google Scholar]
- 8. Maruyama R, Akino K, Toyota M et al. Cytoplasmic RASSF2A is a proapoptotic mediator whose expression is epigenetically silenced in gastric cancer. Carcinogenesis 2008; 29: 1312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki H, Itoh F, Toyota M, Kikuchi T, Kakiuchi H, Imai K. Inactivation of the 14‐3‐3 sigma gene is associated with 5′ CpG island hypermethylation in human cancers. Cancer Res 2000; 60: 4353–7. [PubMed] [Google Scholar]
- 10. Akino K, Toyota M, Suzuki H et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci 2007; 98: 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Issa JP. DNA methylation as a therapeutic target in cancer. Clin Cancer Res 2007; 13: 1634–7. [DOI] [PubMed] [Google Scholar]
- 12. Smits KM, Cleven AH, Weijenberg MP et al. Pharmacoepigenomics in colorectal cancer: a step forward in predicting prognosis and treatment response. Pharmacogenomics 2008; 9: 1903–16. [DOI] [PubMed] [Google Scholar]
- 13. Hellebrekers DM, Lentjes MH, Van Den Bosch SM et al. GATA4 and GATA5 are potential tumor suppressors and biomarkers in colorectal cancer. Clin Cancer Res 2009; 15: 3990–7. [DOI] [PubMed] [Google Scholar]
- 14. Muller HM, Oberwalder M, Fiegl H et al. Methylation changes in faecal DNA: a marker for colorectal cancer screening? Lancet 2004; 363: 1283–5. [DOI] [PubMed] [Google Scholar]
- 15. Maekita T, Nakazawa K, Mihara M et al. High levels of aberrant DNA methylation in Helicobacter pylori‐infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 2006; 12: 989–95. [DOI] [PubMed] [Google Scholar]
- 16. Nakajima T, Maekita T, Oda I et al. Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev 2006; 15: 2317–21. [DOI] [PubMed] [Google Scholar]
- 17. Takaoka A, Tamura T, Taniguchi T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci 2008; 99: 467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miyamoto M, Fujita T, Kimura Y et al. Regulated expression of a gene encoding a nuclear factor, IRF‐1, that specifically binds to IFN‐beta gene regulatory elements. Cell 1988; 54: 903–13. [DOI] [PubMed] [Google Scholar]
- 19. Tanaka N, Ishihara M, Lamphier MS et al. Cooperation of the tumour suppressors IRF‐1 and p53 in response to DNA damage. Nature 1996; 382: 816–8. [DOI] [PubMed] [Google Scholar]
- 20. Barnes BJ, Kellum MJ, Pinder KE, Frisancho JA, Pitha PM. Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res 2003; 63: 6424–31. [PubMed] [Google Scholar]
- 21. Mori T, Anazawa Y, Iiizumi M, Fukuda S, Nakamura Y, Arakawa H. Identification of the interferon regulatory factor 5 gene (IRF‐5) as a direct target for p53. Oncogene 2002; 21: 2914–8. [DOI] [PubMed] [Google Scholar]
- 22. Li Q, Tang L, Roberts PC et al. Interferon regulatory factors IRF5 and IRF7 inhibit growth and induce senescence in immortal Li‐Fraumeni fibroblasts. Mol Cancer Res 2008; 6: 770–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang D, Thangaraju M, Greeneltch K et al. Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res 2007; 67: 3301–9. [DOI] [PubMed] [Google Scholar]
- 24. Lee KY, Geng H, Ng KM et al. Epigenetic disruption of interferon‐gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene 2008; 27: 5267–76. [DOI] [PubMed] [Google Scholar]
- 25. Ortmann CA, Burchert A, Holzle K et al. Down‐regulation of interferon regulatory factor 4 gene expression in leukemic cells due to hypermethylation of CpG motifs in the promoter region. Nucleic Acids Res 2005; 33: 6895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yanagihara K, Ito A, Toge T, Numoto M. Antiproliferative effects of isoflavones on human cancer cell lines established from the gastrointestinal tract. Cancer Res 1993; 53: 5815–21. [PubMed] [Google Scholar]
- 27. Ishida S, Yamashita T, Nakaya U, Tokino T. Adenovirus‐mediated transfer of p53‐related genes induces apoptosis of human cancer cells. Jpn J Cancer Res 2000; 91: 174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maruyama R, Aoki F, Toyota M et al. Comparative genome analysis identifies the vitamin D receptor gene as a direct target of p53‐mediated transcriptional activation. Cancer Res 2006; 66: 4574–83. [DOI] [PubMed] [Google Scholar]
- 29. Watanabe Y, Toyota M, Kondo Y et al. PRDM5 identified as a target of epigenetic silencing in colorectal and gastric cancer. Clin Cancer Res 2007; 13: 4786–94. [DOI] [PubMed] [Google Scholar]
- 30. Sugai T, Habano W, Jiao YF et al. Analysis of allelic imbalances at multiple cancer‐related chromosomal loci and microsatellite instability within the same tumor using a single tumor gland from colorectal carcinomas. Int J Cancer 2005; 114: 337–45. [DOI] [PubMed] [Google Scholar]
- 31. Kusano M, Toyota M, Suzuki H et al. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein‐Barr virus. Cancer 2006; 106: 1467–79. [DOI] [PubMed] [Google Scholar]
- 32. Driggers PH, Ennist DL, Gleason SL et al. An interferon gamma‐regulated protein that binds the interferon‐inducible enhancer element of major histocompatibility complex class I genes. Proc Natl Acad Sci U S A 1990; 87: 3743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shou J, Soriano R, Hayward SW, Cunha GR, Williams PM, Gao WQ. Expression profiling of a human cell line model of prostatic cancer reveals a direct involvement of interferon signaling in prostate tumor progression. Proc Natl Acad Sci U S A 2002; 99: 2830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karpf AR, Peterson PW, Rawlins JT et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natl Acad Sci U S A 1999; 96: 14007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Satoh A, Toyota M, Ikeda H et al. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon‐gamma‐induced HLA‐DR expression in colorectal and gastric cancer cells. Oncogene 2004; 23: 8876–86. [DOI] [PubMed] [Google Scholar]
- 36. Reu FJ, Bae SI, Cherkassky L et al. Overcoming resistance to interferon‐induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J Clin Oncol 2006; 24: 3771–9. [DOI] [PubMed] [Google Scholar]
- 37. McGough JM, Yang D, Huang S et al. DNA methylation represses IFN‐gamma‐induced and signal transducer and activator of transcription 1‐mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol Cancer Res 2008; 6: 1841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tshuikina M, Jernberg‐Wiklund H, Nilsson K, Oberg F. Epigenetic silencing of the interferon regulatory factor ICSBP/IRF8 in human multiple myeloma. Exp Hematol 2008; 36: 1673–81. [DOI] [PubMed] [Google Scholar]
- 39. Ahlquist DA. Next generation stool DNA testing: expanding the scope. Gastroenterology 2009; 136: 2068–73. [DOI] [PubMed] [Google Scholar]
- 40. Watanabe Y, Kim HS, Castoro RJ et al. Sensitive and specific detection of early gastric cancer using DNA methylation analysis of gastric washes. Gastroenterology 2009; 136: 2149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takeshima H, Yamashita S, Shimazu T, Niwa T, Ushijima T. The presence of RNA polymerase II, active or stalled, predicts epigenetic fate of promoter CpG islands. Genome Res 2009; 19: 1974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barnes BJ, Field AE, Pitha‐Rowe PM. Virus‐induced heterodimer formation between IRF‐5 and IRF‐7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J Biol Chem 2003; 278: 16630–41. [DOI] [PubMed] [Google Scholar]
- 43. Kulaeva OI, Draghici S, Tang L, Kraniak JM, Land SJ, Tainsky MA. Epigenetic silencing of multiple interferon pathway genes after cellular immortalization. Oncogene 2003; 22: 4118–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Real‐time PCR analysis of interferon regulatory factor (IRF)‐4, IRF5, and IRF8 expression in gastric cancer cell lines.
Fig. S2. Induction of interferon regulatory factor (IRF)‐5 expression by p53 (a) and of IRF8 expression by interferon (IFN)‐ γ (b).
Fig. S3. 5‐Aza‐2′‐deoxycytidine (DAC) enhances suppression of cell growth by interferon.
Fig. S4. Methylation analysis of interferon regulatory factor (IRF)‐4, IRF5, and IRF8 after treatment with 5‐aza‐2′‐deoxycytidine (DAC) and/or interferon (IFN).
Fig. S5. Receiver–operator curve (ROC) for interferon regulatory factor (IRF)‐4 methylation to discriminate patients with a single gastric cancer from patients with Helicobacter pylori‐positive chronic gastritis.
Fig. S6. Receiver–operator curve (ROC) curve for interferon regulatory factor (IRF)‐4 methylation to discriminate patients with a single or multiple gastric cancers from patients with Helicobacter pylori‐positive chronic gastritis.
Table S1. Primers used for methylation‐specific PCR (MSP) used in this study.
Table S2. Primer sequences used for bisulfite‐pyrosequencing and bisulfite‐sequencing.
Table S3. High levels of interferon regulatory factor (IRF)‐4 methylation are associated with multiple gastric cancers.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item