

Abstract
The effects of MUC1 DNA vaccination on the orthotopic growth and liver metastasis of colon carcinoma cells were investigated in mice. Vaccination with MUC1 DNA resulted in immune responses that were effective in suppressing mouse colon carcinoma cells transfected with MUC1 cDNA. CD4+ T cells but not CD8+ T cells mediated this antitumor response as shown by the in vivo depletion of lymphocyte subpopulations with the use of anti‐CD4 or anti‐CD8 antibody. The effects of neutralizing antibodies in vivo revealed that the predominant effector molecule in preventing orthotopic tumor growth was FasL, whereas the effector molecule effective in preventing liver metastasis was tumor necrosis factor‐α. Colon carcinoma cells isolated from tumors growing in the ceca, spleens, and livers were shown to be equally sensitive to FasL and tumor necrosis factor‐α. The results strongly suggest that elimination of tumor cells initiated by DNA vaccination in the present protocol is mediated by antigen‐specific CD4+ T cells and the effector mechanisms in the cecum and in the liver are distinct due to a unique organ microenvironment. (Cancer Sci 2008; 99: 2477–2484)
The effector mechanisms in cancer immunotherapy of colon carcinoma were investigated in a unique experimental system. This system was unique because orthotopic and metastatic growth of tumors was examined. A new protocol for cancer immunotherapy is necessary to target residual diseases and micrometastases left after surgical and other treatments. However, it is not known whether the immunological effector mechanisms responsible for the elimination of tumor cells growing at different sites, such as primary sites and metastases, are the same. It is well known that the malignant behavior of tumor cells is affected by the organs where the tumor cells grow.( 1 ) The metastatic potentials of tumor cells are also known to be influenced by the organ microenvironment.( 1 ) Such effects are likely to be mediated, at least in part, by the interactions of tumor cells with host immune systems. Therefore, it is important to use experimental models that mimic the actual consequences of tumor cell–host interactions when the effects of cancer immunotherapy and the effector mechanism in a particular protocol is evaluated for an animal model. Orthotopic transplantations of murine tumor cells are often used to investigate tumor cell behaviors in vivo, as reported previously with lung carcinoma cell growth in the trachea or lung, gastric carcinoma cell growth in the gastric wall, renal cell carcinoma cell growth in the kidney, and colon carcinoma cell growth in the cecum.( 2 ) However, few studies were reported based on the use of these models to evaluate the efficacy of cancer immunotherapy.
The predominant site of colon carcinoma metastasis is the liver. The formation of metastatic foci from micrometastases in the liver is considered to be the most critical event in determining the survival of patients. Therefore, two tumor transplantation models, orthotopic transplantation and liver metastasis, were used to evaluate the effect of the vaccine.
In the present study, we chose the tumor antigen MUC1 as the target of immunotherapy because the level of MUC1 in colon carcinoma correlates with the stage of disease.( 3 ) Carcinoma‐associated MUC1 is known to be recognized by the immune system in cancer patients due at least in part to aberrant glycosylation.( 4 ) MUC1‐specific cyototoxic T lymphocytes (CTL) have been detected in colon, pancreatic, and breast carcinoma patients.( 5 , 6 ) There are reports showing that breast and pancreatic carcinoma patients who have antibodies specific for MUC1 have a better survival rate than those without antibodies.( 7 , 8 ) Therefore, MUC1 is considered to be an effective target of cancer immunotherapy. DNA vaccination with MUC1 was used in our previous studies and experimental lung metastasis of MUC1‐transfected B16‐F10 melanoma cells was inhibited.( 9 ) Effective immunotherapy of infectious disease and cancer was previously achieved by DNA vaccine,( 10 ) and this method was considered to be safe, inexpensive, and effective because plasmid DNA contains CpG motifs that can act as potent adjuvants.( 11 , 12 )
The MUC1 DNA vaccine we used was effective in suppressing tumor growth both in the orthotopic and liver metastasis models. CD4+ T cells but not CD8+ T cells initiated the antitumor immune responses. Furthermore, our attempt to elucidate the effector mechanism after MUC1 DNA vaccination led us to a novel finding that the effector molecules in the orthotopic and liver metastasis sites were distinct. The different effector mechanisms between these two sites were not likely to be due to the properties of tumor cells growing under these organ microenvironments because tumor cells collected from the ceca, spleens, and livers showed equal sensitivity to the effector molecules examined.
Materials and Methods
Mice. Specific pathogen‐free 5‐week‐old female C57BL/6 mice were obtained from Charles River Japan (Yokohama, Japan). B6 perforin‐deficient (Pfp−/– ) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). All experimental animals were housed under specific pathogen‐free conditions and handled according to the guidelines of the Bioscience Committee of the University of Tokyo.
Construction of MUC1 cDNA. The full‐length human MUC1 cDNA that contains 22 tandem repeats was provided originally in the pDKOF vector by Dr O.J. Finn (University of Pittsburgh, Pittsburgh, PA, US). The cDNA was cut out and recloned into the HindIII site of the pCEP4 vector, which includes the CMV promoter (pCEP4‐MUC1). The plasmid without the MUC1 cDNA (pCEP4) was used as a control.
Establishment of MUC1 cDNA‐transfected murine colon carcinoma cell lines. SL4, a highly liver‐metastatic variant of the colon 38 murine colon carcinoma cell line,( 13 ) was transfected with human MUC1 cDNA (pDKOF‐MUC1) with lipofection using DOTAP Liposomal Transfection Reagent (Roche, Basel, Switzerland). After selection with G418 (Wako, Osaka, Japan), the limiting‐dilution method was used to obtain clonal populations. Although SL4 cells, selected for their high metastatic potential to the liver, grew well in the orthotopic site,( 13 ) the ability of the clones to grow at the orthotopic sites and to metastasize to the livers were diverse. Among 70 mock‐transfected and 24 MUC1‐transfected clones, clones suitable for the liver metastasis model were selected according to their ability to metastasize to the liver following intrasplenic injection. Clone SL4‐M11, which showed high metastatic ability to the liver, was chosen and confirmed to grow at the orthotopic site. However, mice injected intrasplenically with this clone died occasionally from unknown causes within short time periods. Therefore, SL4‐M8, which had metastatic ability to the liver that was similar to the parental SL4 cells, was used for the liver metastasis model. As a control, clones that were transfected with empty plasmid vector (pDKOF) and showed similar growth to SL4‐M11 or SL4‐M8 were used (SL4‐P36 for the orthtopic model and SL4‐P46 for the liver metastasis model). The expression of MUC1 on these clones was checked by flow cytometric analysis with Epics XL (Beckman Coulter, Fullerton, CA, USA) using the anti‐MUC1 monoclonal antibody MY.1E12.( 14 )
Immunization. The MUC1 cDNA in the pCEP4 vector (pCEP4‐MUC1) and the pCEP4 plasmid alone were amplified in Escherichia coli strain JM109 and purified using Qiagen Mega‐Plasmid columns (Qiagen, Hilden, Germany). Mice were immunized intradermally three times at weekly intervals in the forelimb with 25 µg DNA dissolved in 50 µL Hanks’ Balanced Salt Solution.
Orthotopic transplantation and experimental liver metastasis models. One week after the final immunization, SL4‐M11 cells (1 × 106 cells/50 µL) were injected into the space under the cecal serosa of mice in the orthotopic model. Twenty‐one days after the challenge, mice were killed and the increase in weight of the cecum due to tumor growth was measured. In the liver metastasis model, SL4‐M8 cells (5 × 105 cells/50 µL) were injected into the spleen. Nineteen days after the challenge, the weights of the spleen and liver were measured.
Cell depletion and neutralization of effector molecules in vivo. For the depletion of lymphocyte subsets, mice were injected with monoclonal antibodies specific for CD4 and CD8. Hybridoma clones 53‐6.72 (anti‐mouse CD8) and GK1.5 (anti‐mouse CD4) were purchased from the American Type Culture Collection (Manassas, VA, USA). Hybridoma culture supernatants were purified on a protein G‐Sepharose 4B (GE Healthcare, Amersham, UK) column. Rat IgG (Sigma, St Louis, MO, USA) was used as a control. One week after the final immunization, mice were injected intravenously with 200 µg monoclonal antibody three times at 4‐day intervals. MUC1‐transfected SL4 cells were injected one day after the final injection of monoclonal antibody.
For the neutralization of effector molecules, mice were injected intraperitoneally with anti‐interferon (IFN)‐γ monoclonal antibody (R4‐6A2, 1 mg), anti‐TNF‐related apoptosis including ligand (TRAIL) monoclonal antibody (N2B2, 300 µg), anti‐tumor necrosis factor (TNF)‐α monoclonal antibody (MP6‐XT22, 500 µg), or anti‐FasL monoclonal antibody (MFL4, 500 µg) three times at 4‐day intervals, starting 6 days after the final immunization. MUC1‐transfected SL4 cells were injected 1 day after the first injection of monoclonal antibody.
Preparation of MUC1‐transfected SL4 cells growing in vivo from mouse organs. Naive C57BL/6 mice were injected with SL4‐M8 or SL4‐M11 cells. One week after the injection, mice were killed and the ceca, spleens, and livers were resected, minced, and washed with phosphate‐buffered saline. The fragments of tumor tissue were digested in RPMI‐1640 containing 10% fetal calf serum, 200 U/mL collagenase (Wako), and 0.02% DNaseI (Roche) and agitated gently at 37°C for 2 h. To detect MHC class I, MHC class II, and Fas expression on the MUC1+ tumor cells, the cells were stained with biotinylated anti‐MUC1 monoclonal antibody (MY.1E12), fluorescein isothiocyanate‐conjugated anti‐MHC I (5F1, kindly provided by Dr Shigeo Koyasu), phycoerythrin (PE)‐conjugated anti‐MHC II (M5/114.15.2; eBioscience, San Diego, CA, US), and PE‐conjugated anti‐Fas monoclonal antibody (Jo‐2; BD Bioscience, Franklin Lakes, NJ, US), respectively, followed by allophycocyanin–streptavidin (eBioscience) and analyzed by FACSAria (BD, Biosciences). To isolate tumor cells growing in vivo, the cell suspensions were sorted by FACSAria for MY.1E12 monoclonal antibody binding. The MUC1‐positive cells were used immediately in the following experiments.
FasL killing assay. To investigate the sensitivity of MUC1‐transfected SL4 cells growing in vivo to FasL, 51Cr release assays were carried out. FasL‐transfected cells (mFasL/L5178Y) used as effector cells in this experiment were established as described previously.( 15 ) 51Cr‐labeled target tumor cells were cultured in triplicate in 96‐well microtiter plates with mFasL/L5178Y or control cells (L5178Y) at different effector : target ratios. After incubation at 37°C for 4 h, 100 µL of the supernatants were harvested and their radioactivity was measured using a gamma counter (Aloka, Tokyo, Japan).
WST‐1 cell proliferation assay. MUC1‐transfected SL4 cells (2 × 104 cells/200 µL) were cultured in 96‐well microtiter plates with different concentrations of recombinant mouse TNF‐α (R&D Systems, Minneapolis, MN, USA). After 3‐day culture, 20 µL WST‐1 reagent (Roche) was added to each well, and incubated at 37°C for 90–120 min. Absorbance values at 450 and 655 nm were measured using a microplate reader and the cell viabilities were calculated. The viability of cells cultured without TNF‐α was regarded as 100%.
Statistical analysis. Statistical analysis was carried out using the Mann–Whitney U‐test for the orthotopic and liver metastasis model. Cytotoxic assays and cell proliferation assays were analyzed using Student's t‐test. P‐values less than 0.05 were considered significant.
Results
MUC1‐specific immune response elicited by MUC1 DNA vaccine suppresses colon carcinoma growth at primary and metastatic sites. To evaluate the efficacy of MUC1 DNA vaccine in the orthotopic and liver metastasis models, the SL4 colon carcinoma cell line was transfected stably with human MUC1 and the clonal populations were obtained (Fig. 1). Female C57BL/6 mice were immunized intradermally three times at weekly intervals with 25 µg of a vector containing full‐length human MUC1 cDNA (pCEP4‐MUC1) or control vector without MUC1 cDNA (pCEP4) each time. One week after the final vaccination, the mice were injected with SL4‐M8 cells into the spleen or SL4‐M11 cells into the space under the cecal serosa. The effect of MUC1 DNA vaccination was evaluated by measuring the increase in weight of the cecum, spleen, and liver due to tumor growth. When the mice were injected with SL4‐P36 or SL4‐P46 cells, the weights of the cecum, spleen, and liver did not change, regardless of immunization. However, when the mice were inoculated with MUC1‐transfected SL4 cells, rejection of tumor cells was observed only in the mice immunized with pCEP4‐MUC1 (Fig. 2). These results indicate that a MUC1‐specific immune response was induced by the MUC1 DNA vaccine as shown previously in the experimental lung metastasis model with B16 melanoma cells,( 9 ) and that MUC1 DNA vaccine was effective in suppressing tumor growth in the orthotopic sites, spleens, and liver metastases.
Figure 1.
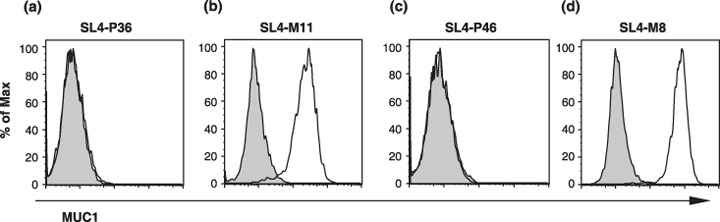
MUC1 was expressed on the cell surfaces of MUC1‐transfected but not mock‐transfected SL4 cells. Mock‐transfected and MUC1‐transfected cells, (a) SL4‐P36 and (b) SL4‐M11 for the orthotopic model and (c) SL4‐P46 and (d) SL4‐M8 for the liver metastasis model, were stained with the anti‐MUC1 monoclonal antibody MY.1E12 and analyzed by flow cytometry. Gray‐filled histograms represent the controls and open histograms represent the binding of anti‐MUC1 monoclonal antibody.
Figure 2.
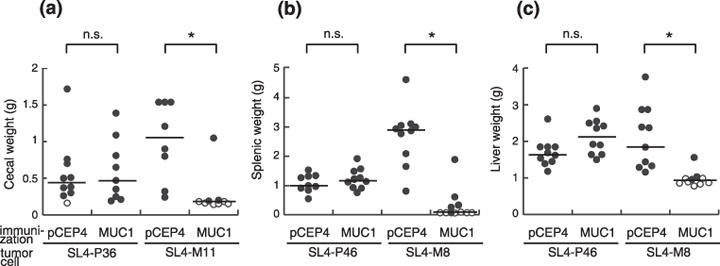
MUC1 DNA vaccine effectively suppressed tumor growth at orthotopic sites and in the liver metastases in a MUC1‐specific manner. One week after the final MUC1 DNA immunization, SL4‐P36 or SL4‐M11 cells (1 × 106 cells/50 µL) were injected into the space under the cecal serosa of mice. (a) Twenty‐one days after the challenge, the mice were killed and the weight of the cecum was examined. In the liver metastasis model, SL4‐P46 or SL4‐M8 cells (5 × 105 cells/50 µL) were injected into the spleen. Nineteen days after the challenge, the weights of the (b) spleen and (c) liver were measured. Open circles represent tumor‐free mice and filled circles represent tumor‐bearing mice. n.s., not significant; *P < 0.005.
Identification of the effector cell population responsible for the tumor rejection. To determine the effector cell population in the suppression of tumor growth and metastasis, mice were injected intravenously with anti‐CD4 or CD8 monoclonal antibody 1 day before the tumor injection. Injection of anti‐CD4 monoclonal antibody completely abolished the vaccine effects in the cecum, spleen, and liver on the growth of SL4‐M8 and M11 cells. In contrast, the anti‐CD8 monoclonal antibody did not influence the therapeutic efficacy of the MUC1 DNA vaccination, as shown in Figure 3. Thus, CD4+ but not CD8+ cells were shown to mediate the tumor rejection induced by MUC1 DNA vaccination in the present study. To confirm that MUC1‐specific CD4+ T cells were induced by the MUC1 DNA vaccination, the Winn assays were carried out (data shown in Suppl. Fig. S1). When CD4+ T cells but not CD8+ T cells isolated from the mice immunized with MUC1 DNA were mixed with MUC1‐transfected B16‐F10 melanoma cells and injected subcutaneously into naive mice, an improved survival rate was observed. The results indicate that MUC1‐specific CD4+ T cells induced by the MUC1 DNA vaccination played a pivotal role in the antitumor immune responses.
Figure 3.
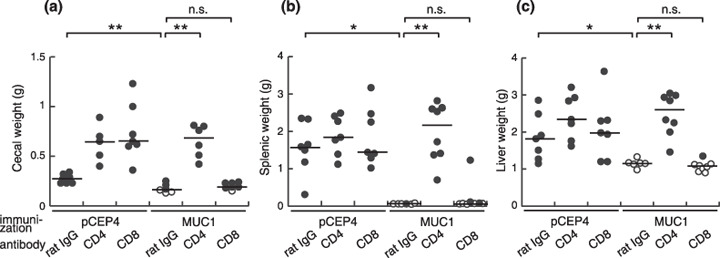
Effector cells were CD4+ in both the orthotopic and liver metastasis models. One week after the final MUC1 DNA immunization, the mice were treated with anti‐CD4 (GK1.5) or CD8 (53–6.72) monoclonal antibody and injected with (a) SL4‐M11 or (b,c) SL4‐M8 as described in Figure 1. Vaccine effects were assessed by tumor growth in the (a) cecum, (b) spleen, and (c) liver. Open circles represent tumor‐free mice and filled circles represent tumor‐bearing mice. n.s., not significant; *P < 0.05; **P < 0.005.
Effector mechanisms induced by MUC1 DNA vaccine. MHC class II expression on tumor cells might be essential for the recognition of MUC1 on the MUC1‐transfected SL4 cells directly by the CD4+ T cells. Thus, MHC molecules expressed on the MUC1‐transfected SL4 cells were examined by the flow cytometric analysis. Although these cells cultured in vitro were shown to be negative for the expression of MHC class I and II (Fig. 4a,c,f,h), cells from tumors growing in the ceca, spleens, and livers obtained by collagenase treatment expressed low but significant levels of MHC class I and II (Fig. 4b,d,e,g,i,j). From these results, it is possible that MUC1‐specific CD4+ T cells recognize MUC1 peptides presented with MHC class II on the MUC1‐transfected SL4 cells in vivo.
Figure 4.
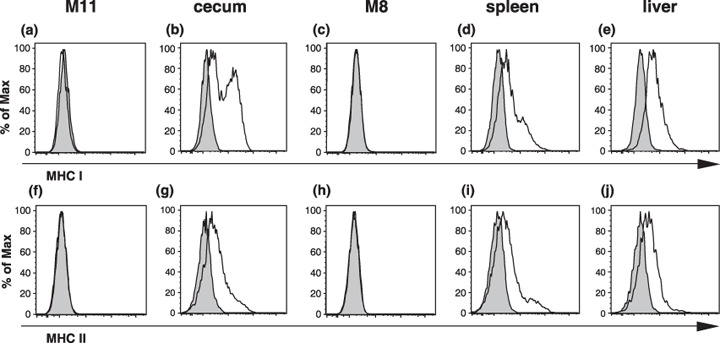
Tumor cells from the ceca, spleens, and livers expressed low but detectable levels of MHC class I and II molecules on their surfaces. The cells grown (a,c,f,h) in vitro in cultures and obtained from tumors growing in (a,f) ceca, (d,i) spleens, and (e,j) livers were stained with the anti‐MUC1 monoclonal antibody MY.1E12 and anti‐MHC class I or II monoclonal antibody. Using flow cytometric analysis, MUC1+ cells regarded as tumor cells were gated and analyzed further for MHC expression. The gray‐filled histograms represent the isotype controls and the open histograms represent (a–e) anti‐MHC class I and (f–j) anti‐MHC class II monoclonal antibody binding.
To further investigate the mechanistic basis of antitumor immune responses induced by MUC1 DNA vaccination, we subsequently examined the effector molecules involved in the rejection of MUC1‐transfected SL4 cells. One of the most important effector molecules determining antitumor effects by CD4+ T cells and other immune cells is postulated to be IFN‐γ.( 16 ) Injection of anti‐IFN‐γ monoclonal antibody starting 1 day prior to the tumor injection did not alter the vaccine effects as shown in Figure 5. The dose of antibody used in the present study was equal to previous reports in which the antibody injection reduced or abolished the effect of IFN‐γ.( 17 , 18 , 19 ) Therefore, we concluded that the antitumor immune responses induced by MUC1 DNA vaccination were independent of IFN‐γ.
Figure 5.
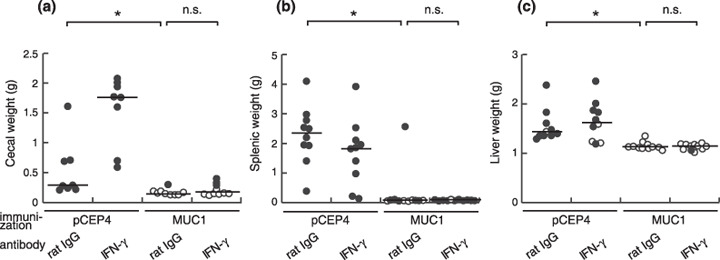
The antitumor effector mechanism induced by the MUC1 DNA vaccine was independent of interferon (IFN)‐γ. After MUC1 DNA immunization, the mice were treated with anti‐IFN‐γ monoclonal antibody (R4‐6A2) and injected with MUC1‐transfected SL4 cells. Vaccine effects were assessed by tumor growth in the (a) cecum, (b) spleen, and (c) liver. Open circles represent tumor‐free mice and filled circles represent tumor‐bearing mice. n.s., not significant; *P < 0.005.
To further investigate other effector molecules involved in the antitumor immune response induced by MUC1 DNA vaccination, we chose perforin, TRAIL, FasL, and TNF‐α as candidates because these molecules are also known to be engaged in antitumor effector mechanisms with CD4+ T cells.( 20 , 21 , 22 , 23 ) To examine the involvement of perforin, B6 perforin‐deficient (Pfp−/– ) mice were used, and neutralizing monoclonal antibodies were used for TRAIL, FasL, and TNF‐α. Immunization with MUC1 DNA vaccine led to rejection of MUC1‐transfected SL4 cells in Pfp−/– mice (n = 8 for the orthotopic model and n = 5 for the liver metastasis model, data not shown). Treatment with anti‐TRAIL monoclonal antibody also did not influence the vaccine effect (n = 10, data not shown). The absence of perforin or TRAIL did not show any influence on MUC1‐specific tumor rejection, therefore perforin and TRAIL were excluded as candidate effector molecules. We found that the neutralization of FasL and TNF‐α resulted in a reduction in the effect of vaccination. In the orthotopic model, the injection of anti‐FasL monoclonal antibody abolished the vaccine effect but anti‐TNF‐α monoclonal antibody did not (Fig. 6a). In contrast, in the liver metastasis model, blocking of TNF‐α but not FasL resulted in a significant decrease in the antitumor immune response both in the spleen and in the liver (Fig. 6b,c).
Figure 6.
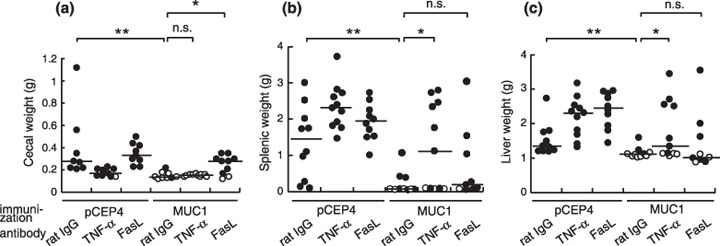
The antitumor effector mechanism at the orthotopic sites was distinct from that in the liver metastasis. After MUC1 DNA immunization, the mice were treated with anti‐tumor necrosis factor (TNF)‐α (MP6‐XT22) or anti‐FasL (MFL4) monoclonal antibody and injected with MUC1‐transfected SL4 cells. Vaccine effects were assessed by tumor growth in the (a) cecum, (b) spleen, and (c) liver. Open circles represent tumor‐free mice and filled circles represent tumor‐bearing mice. n.s., not significant; *P < 0.05; **P < 0.005.
Sensitivity of in vivo‐derived tumor cells to FasL. For further investigation of the differential effector mechanisms in the cecum, spleen, and liver, we next examined whether there were differences in the sensitivities of tumor cells to effector molecules. To investigate the sensitivities of tumor cells to FasL, we first checked whether Fas was expressed on the MUC1‐transfected SL4 cells by flow cytometry. These cells cultured in vitro were shown to be negative for the binding of anti‐Fas monoclonal antibody (Fig. 7a,c). When these cells were tested for their sensitivity as target cells to FasL‐transfected cells in the 51Cr release assay, they were shown to be resistant to FasL‐induced apoptosis (Fig. 7f,h). These results were inconsistent with the in vivo data in which anti‐FasL treatment abolished the vaccine effects on these cells, indicating that FasL was an effector molecule in the cecum. Therefore, MUC1‐transfected SL4 cells were grown in vivo and their sensitivity to FasL‐transfected cells was compared to the cells grown in vitro. The cells obtained from tumors growing in the ceca, spleens, and livers were shown to express Fas (Fig. 7b,d,e), and were sensitive to FasL‐induced apoptosis (Fig. 7g,i,j). SL4‐M8 cells obtained from tumors growing in the spleens and livers and SL4‐M11 cells obtained from tumors growing in the ceca were equally sensitive. Although the effects of the injection of anti‐FasL monoclonal antibody to reverse the DNA vaccine therapy were only observed in tumors growing in the cecum, these data indicate that MUC1‐transfected SL4 cells growing in the spleen and liver could also be killed through Fas–FasL interaction in vitro.
Figure 7.
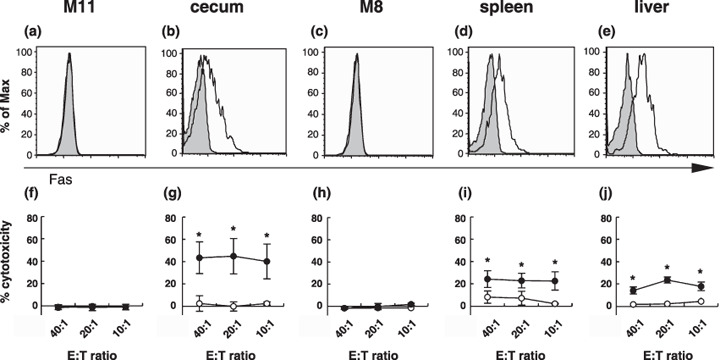
Tumor cells from the ceca, spleens, and livers expressed Fas and were sensitive to FasL‐induced apoptosis. Fas expression and sensitivity to FasL of (c–e,h–j) SL4‐M8 and (a,b,f,g) SL4‐M11 cells were examined. The cells were grown (a,c,f,h) in vitro and in vivo and were collected from (a,f) ceca, (d,i) spleens, and (e,j) livers. (a–e) The cell surface expression of Fas was examined by anti‐Fas monoclonal antibody binding by flow cytometric analysis. MUC1+ cells that were regarded as in vivo‐growing tumor cells were gated and analyzed. The gray‐filled histograms represent the isotype controls and the open histograms represent anti‐Fas monoclonal antibody binding. (f–j) FasL killing assays were carried out. Target tumor cells were collected from ceca, spleens, and livers by cell sorting using anti‐MUC1 monoclonal antibody (MY.1E12) binding. Parental L5178Y cells (open circle) and mFasL/L5178Y cells (filled circle) were used as effector cells. The percentage of specific 51Cr release was calculated according to the following formula: 51Cr release (%) = 100 × ([cpm experiment – cpm spontaneous release]/[cpm maximum – cpm spontaneous release]). Spontaneous release was obtained from target cells incubated with medium alone and the maximum release was obtained from target cells incubated with 1 M HCl instead of effector cells. Data are represented as the mean ± SD (*P < 0.005). E:T, effector:target.
Sensitivity of tumor cells growing in vivo to tumor necrosis factor‐α. The sensitivity of MUC1‐transfected SL4 cells collected from tumors growing in vivo or in vitro to TNF‐α was examined using the cell proliferation assay. As shown in Figure 8a, the viabilities of MUC1‐transfected SL4 cells obtained from tumors growing in the ceca, spleens, and livers were decreased dramatically by the addition of TNF‐α in a dose‐dependent manner, and the sensitivities among MUC1‐transfected SL4 cells from different organs were almost identical. The sensitivity of MUC1‐transfected SL4 cells (SL4‐M8 and SL4‐M11) growing in vitro also did not show any difference in their TNF‐α sensitivity (Fig. 8b). These results indicate that MUC1‐transfected SL4 cells obtained from different organs had similar sensitivities to FasL and TNF‐α, in spite of the organ specificity of effector molecules in vivo as shown in Figure 6.
Figure 8.
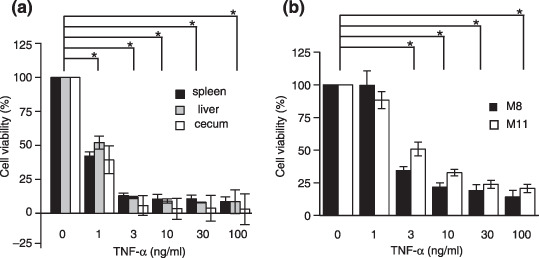
Tumor cells from the ceca, spleens, and livers were sensitive to tumor necrosis factor (TNF)‐α‐induced apoptosis. MUC1‐transfected SL4 cells injected into ceca (SL4‐M11 cells) or spleens (SL4‐M8 cells) were grown in vivo at the injection sites and as liver metastases. One week later, tumors were collected from the ceca, spleens, and livers and MUC1‐positive cells were obtained. Tumor cells grown (a) in vivo and (b) in vitro were incubated with recombinant mouse TNF‐α and cell viability and proliferation were evaluated using WST‐1 cell proliferation assays. The viability of cells cultured without TNF‐α was regarded as 100%. Data are represented as the mean ± SD. *P < 0.005.
Discussion
In the present study, we established models to examine orthotopic growth and experimental liver metastasis in mouse colon carcinoma cells expressing human MUC1. Primary tumor growth and metastasis were prevented by MUC1‐specific antitumor immune responses induced by DNA vaccination. The antitumor effector cells responsible for the elimination of MUC1‐transfected SL4 cells turned out to be CD4+ T cells, not CD8+ T cells. In general, it is believed that CD8+ T cells play important roles in antitumor immune responses and that an immunization protocol to induce strong CD8+ CTL is preferable. However, in the present study CD4+ T cells were able to initiate an antitumor immune response that suppressed murine colon carcinoma cell growth and metastasis. The possibility that natural killer (NK) and NKT cells directly recognize and kill the MUC1‐transfected SL4 cells is not likely because anti‐NK1.1 antibody treatment did not alter the effects of MUC1 DNA vaccination in any organs (data not shown). The reason why only CD4+ T cells act as effector cells is unclear. In general, DNA vaccination is known to be able to induce CD8+ CTL.( 10 ) Furthermore, MUC1‐specific CD8+ T cells were previously reported after various means of immunization in mice.( 24 , 25 , 26 ) Therefore, the dose, timing of vaccination, and experimental model used to determine the vaccine might affect the induction and response of antigen‐specific T cells.
Although activated CD4+ T cells are known to produce a large amount of IFN‐γ, neutralization of IFN‐γ in the effector phase did not affect the antitumor response in our models. IFN‐γ is known to play important roles in tumor surveillance by upregulating MHC and preventing angiogenesis.( 27 , 28 ) In the control (immunized with control vector) mice, IFN‐γ seemed to play a role in such innate antitumor responses at least in the orthotopic model. In contrast, the IFN‐γ‐dependent antitumor effect seemed to be negligible in the immunized group. The roles of perforin, TRAIL, FasL, and TNF‐α were also tested as these molecules were identified as effector molecules involved in tumoricidal activity of CD8+ T cells, NK cells, and CD4+ T cells.( 20 , 21 , 22 , 23 ) The results indicate that different effector molecules are involved in the suppression of orthotopic growth and eradication of metastasis; FasL is an effector molecule in the cecum and TNF‐α is an effector in the spleen and liver.
The differential effector mechanisms in the cecum, spleen, and liver are possibly explained by different susceptibilities of the tumor cells to the effector mechanisms, which are dependent on the organ microenvironments. Another possibility is that the different tumor cell clones (clonal populations) used in the orthotopic and liver metastasis models determined the sensitivity to effector molecules. However, these possibilities were eliminated experimentally (7, 8). We also obtained preliminary data that TNF‐α but not FasL is the effector molecule in the liver even though the clone for the orthotopic model (SL4‐M11 cells) was used in the liver metastasis model. Therefore, the differences in effector molecules revealed by neutralization with the antibodies is likely to be due to the properties of the effector cells and not the properties of the tumor cells in each organ. Our data strongly suggest that the final effector mechanism initiated by the antigen‐specific CD4+ T cells in the cecum was different to that in the liver. This is the first report to show that the same immunization protocol elicits distinct effector mechanisms in an organ‐specific manner.
Whether MUC1‐specific CD4+ T cells directly recognize and killed MUC1‐transfected SL4 cells in vitro and in vivo is presently unknown, although CD4+ T cells were shown to be essential for the rejection of these cells. It is known that CD4+ T cells can also kill target cells through many effector pathways.( 20 , 29 , 30 , 31 ) Because MUC1‐transfected SL4 cells growing in vivo expressed low but detectable levels of MHC class II molecules, it is possible that activated MUC1‐specific CD4+ T cells killed MUC1‐transfected SL4 cells directly. Macrophages should also be considered as candidate cells for killing MUC1‐transfected SL4 cells in a MUC1‐specific manner because there are reports that macrophages expressed TNF‐α and FasL on their surface and kill target cells.( 32 )
FasL and TNF‐α are known to have different impacts on the inflammatory responses of the liver and intestinal tissue. Ogasawara and coworkers reported that mice injected with the anti‐Fas mAb Jo2 died rapidly of fulminant hepatitis.( 33 ) In another report on an acute GvHD model, FasL‐defective T cells showed minimal effects on the liver.( 34 ) The pro‐inflammatory cytokine TNF‐α is known to play a key role in the pathogenesis of inflammatory bowel disease. It has been reported that a pathological concentration of TNF‐α inhibits the proliferation of a mouse intestinal cell line.( 35 ) Anti‐TNF‐α antibody treatment was used effectively for the treatment of Crohn's disease. From these results it is strongly suggested that TNF‐α is harmful to the maintenance of homeostasis in the intestine. Furthermore, pathogenesis after graft versus host diseases (GvHD) was previously shown to be suppressed by anti‐TNF‐α monoclonal antibody in the intestine and by anti‐FasL monoclonal antibody in the liver.( 36 ) Therefore, effector molecules harmful to the organs are apparently suppressed by unknown mechanisms after MUC1 DNA vaccination and recruitment of an effector cell subset might explain such organ specificity. Because tissue‐derived dendritic cells (DC) were reported to imprint the homing of effector T cells,( 37 , 38 ) DC in each organ might generate CD4+ T cells that have distinct effector functions in each draining lymph node. An alternative possibility is that the organ microenvironment itself influences the mode of immune response. The effector function of activated T cells might be regulated under the organ‐specific microenvironment in each organ by different regulatory molecules such as interleukin‐10 or transforming growth factor (TGF)‐β, which are known to play important roles in the maintenance of homeostasis in the intestinal organs.( 39 , 40 , 41 ) Tissue resident cells, such as stromal cells, potentially produce tissue‐specific cytokines and other chemical mediators that may modulate effector functions. Whether the CD4+ T cells in the different organs were derived from the same subpopulation should also be clarified. Because our method of immunization was intradermal, the site where the MUC1‐specific memory T cells were generated was likely to be the skin‐draining lymph nodes and further investigations are necessary to determine the differential characteristics of MUC1‐specific memory T cells migrating from these lymph nodes to the orthotopic and metastatic sites.
In conclusion, experimental orthotopic growth and liver metastasis formation by MUC1‐transfected SL4 cells was suppressed successfully by MUC1 DNA vaccination. The effector mechanism initiated by antigen‐specific CD4+ T cells in the orthotopic site was different from that in the liver metastasis. This is the first report to show that the mode of immune response is organ specific even when the initial immunization is given through the same route. This organ‐dependent immunological regulation should prove to be useful in the development of an effective cancer immunotherapy. Further analysis must be carried out using MUC1 transgenic mice, in which the immune systems recognize MUC1 as a self antigen,( 42 ) considering the clinical application of cancer immunotherapy targeting MUC1.
Supporting information
Fig. S1. One week after MUC1 DNA vaccination, mice were killed. Single‐cell suspensions were obtained from the spleens. After removal of erythrocytes, splenocytes were passed through a nylon wool column to enrich T cells. CD4+ and CD8+ T cells were obtained by negative selection using a mixture of biotinylated monoclonal antibodies (CD11b, Ly6G, DX5, B220, and CD8 or CD4) and streptavidin microbeads on a LS column (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s specifications. These bulk T cells (5 × 106 cells), CD4+ T cells, or CD8+ T cells (2.5 × 106 cells) were mixed with MUC1‐transfected B16‐F10 cells (5 × 104 cells)( 9 ) and injected subcutaneously into naive C57B/6 mice. The survival rates were determined (n = 5).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
Daisuke Sugiura is a recipient of a Japan Society for the Promotion of Science Research Fellowship for Young Scientists. We thank Ms Kyoko Sakai and Ms Miki Noji for their assistance in the preparation of this manuscript. This work was supported by Grants‐in‐Aid from the Ministry of Education, Science, Sports and Culture of Japan. The authors also thank Dr Olivera J. Finn (University of Pittsburgh) for providing pDKOF‐MUC1.
References
- 1. Fidler IJ, Kim SJ, Langley RR. The role of the organ microenvironment in the biology and therapy of cancer metastasis. J Cell Biochem 2007; 101: 927–36. [DOI] [PubMed] [Google Scholar]
- 2. Kubota T. Metastatic models of human cancer xenografted in the nude mouse: the importance of orthotopic transplantation. J Cell Biochem 1994; 56: 4–8. [DOI] [PubMed] [Google Scholar]
- 3. Nakamori S, Ota DM, Cleary KR, Shirotani K, Irimura T. MUC1 mucin expression as a marker of progression and metastasis of human colorectal carcinoma. Gastroenterology 1994; 106: 353–61. [DOI] [PubMed] [Google Scholar]
- 4. Denda‐Nagai K, Irimura T. MUC1 in carcinoma–host interactions. Glycoconj J 2000; 17: 649–58. [DOI] [PubMed] [Google Scholar]
- 5. Barnd DL, Lan MS, Metzgar RS, Finn OJ. Specific, major histocompatibility complex‐unrestricted recognition of tumor‐associated mucins by human cytotoxic T cells. Proc Natl Acad Sci USA 1989; 86: 7159–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jerome KR, Domenech N, Finn OJ. Tumor‐specific cytotoxic T cell clones from patients with breast and pancreatic adenocarcinoma recognize EBV‐immortalized B cells transfected with polymorphic epithelial mucin complementary DNA. J Immunol 1993; 151: 1654–62. [PubMed] [Google Scholar]
- 7. Von Mensdorff‐Pouilly S, Verstraeten AA, Kenemans P et al . Survival in early breast cancer patients is favorably influenced by a natural humoral immune response to polymorphic epithelial mucin. J Clin Oncol 2000; 18: 574–83. [DOI] [PubMed] [Google Scholar]
- 8. Hamanaka Y, Suehiro Y, Fukui M, Shikichi K, Imai K, Hinoda Y. Circulating anti‐MUC1 IgG antibodies as a favorable prognostic factor for pancreatic cancer. Int J Cancer 2003; 103: 97–100. [DOI] [PubMed] [Google Scholar]
- 9. Kamata M, Denda‐Nagai K, Kubota N, Aida S, Takeda K, Irimura T. Vaccination of mice with MUC1 cDNA suppresses the development of lung metastases. Clin Exp Metastasis 2002; 19: 689–96. [DOI] [PubMed] [Google Scholar]
- 10. Donnelly JJ, Ulmer JB, Shiver JW, Liu MA. DNA vaccines. Annu Rev Immunol 1997; 15: 617–48. [DOI] [PubMed] [Google Scholar]
- 11. Hartmann G, Weiner GJ, Krieg AM. CpG DNA: a potent signal for growth, activation, and maturation of human dendritic cells. Proc Natl Acad Sci USA 1999; 96: 9305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hemmi H, Takeuchi O, Kawai T et al . A Toll‐like receptor recognizes bacterial DNA. Nature 2000; 408: 740–5. [DOI] [PubMed] [Google Scholar]
- 13. Morimoto‐Tomita M, Ohashi Y, Matsubara A, Tsuiji M, Irimura T. Mouse colon carcinoma cells established for high incidence of experimental hepatic metastasis exhibit accelerated and anchorage‐independent growth. Clin Exp Metastasis 2005; 22: 513–21. [DOI] [PubMed] [Google Scholar]
- 14. Yamamoto M, Bhavanandan VP, Nakamori S, Irimura T. A novel monoclonal antibody specific for sialylated MUC1 mucin. Jpn J Cancer Res 1996; 87: 488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanabuchi S, Koyanagi M, Kawasaki A et al . Fas and its ligand in a general mechanism of T‐cell‐mediated cytotoxicity. Proc Natl Acad Sci USA 1994; 91: 4930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ikeda H, Old LJ, Schreiber RD. The roles of IFN‐γ in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev 2002; 13: 95–109. [DOI] [PubMed] [Google Scholar]
- 17. Kawakami K, Koguchi Y, Qureshi MH et al . IL‐18 contributes to host resistance against infection with Cryptococcus neoformans in mice with defective IL‐12 synthesis through induction of IFN‐γ production by NK cells. J Immunol 2000; 165: 941–7. [DOI] [PubMed] [Google Scholar]
- 18. Piccotti JR, Li K, Chan SY et al . Alloantigen‐reactive Th1 development in IL‐12‐deficient mice. J Immunol 1998; 160: 1132–8. [PubMed] [Google Scholar]
- 19. Sojka DK, Felnerova D, Mokyr MB. Anti‐metastatic activity of hapten‐modified autologous tumor cell vaccine in an animal tumor model. Cancer Immunol Immunother 2002; 51: 200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Appay V, Zaunders JJ, Papagno L et al . Characterization of CD4+ CTLs ex vivo . J Immunol 2002; 168: 5954–8. [DOI] [PubMed] [Google Scholar]
- 21. Aruga E, Tanigawa K, Aruga A et al . CD95‐mediated tumor recognition by CD4+ effector cells in a murine mammary model. J Immunother 2000; 23: 225–34. [DOI] [PubMed] [Google Scholar]
- 22. Ju ST, Ruddle NH, Strack P, Dorf ME, DeKruyff RH. Expression of two distinct cytolytic mechanisms among murine CD4 subsets. J Immunol 1990; 144: 23–31. [PubMed] [Google Scholar]
- 23. Smyth MJ, Norihisa Y, Ortaldo JR. Multiple cytolytic mechanisms displayed by activated human peripheral blood T cell subsets. J Immunol 1992; 148: 55–62. [PubMed] [Google Scholar]
- 24. Apostolopoulos V, Pietersz GA, Loveland BE, Sandrin MS, McKenzie IF. Oxidative/reductive conjugation of mannan to antigen selects for T1 or T2 immune responses. Proc Natl Acad Sci USA 1995; 92: 10 128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gong J, Chen D, Kashiwaba M, Kufe D. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nat Med 1997; 3: 558–61. [DOI] [PubMed] [Google Scholar]
- 26. Plunkett T, Graham R, Correa I et al . Protection against MUC1 expressing mouse tumours by intra‐muscular injection of MUC1 cDNA requires functional CD8+ and CD4+ T cells but does not require the MUC1 tandem repeat domain. Int J Cancer 2004; 109: 691–7. [DOI] [PubMed] [Google Scholar]
- 27. Mumberg D, Monach PA, Wanderling S et al . CD4+ T cells eliminate MHC class II‐negative cancer cells in vivo by indirect effects of IFN‐γ. Proc Natl Acad Sci USA 1999; 96: 8633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin Z, Blankenstein T. CD4+ T cell‐mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN‐γ receptor expression by nonhematopoietic cells. Immunity 2000; 12: 677–86. [DOI] [PubMed] [Google Scholar]
- 29. Krensky AM, Reiss CS, Mier JW, Strominger JL, Burakoff SJ. Long‐term human cytolytic T‐cell lines allospecific for HLA‐DR6 antigen are OKT4+ . Proc Natl Acad Sci USA 1982; 79: 2365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lukacher AE, Morrison LA, Braciale VL, Malissen B, Braciale TJ. Expression of specific cytolytic activity by H‐2I region‐restricted, influenza virus‐specific T lymphocyte clones. J Exp Med 1985; 162: 171–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCarthy SA, Singer A. Recognition of MHC class I allodeterminants regulates the generation of MHC class II‐specific CTL. J Immunol 1986; 137: 3087–92. [PubMed] [Google Scholar]
- 32. Badley AD, Dockrell D, Simpson M et al . Macrophage‐dependent apoptosis of CD4+ T lymphocytes from HIV‐infected individuals is mediated by FasL and tumor necrosis factor. J Exp Med 1997; 185: 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ogasawara J, Watanabe‐Fukunaga R, Adachi M et al . Lethal effect of the anti‐Fas antibody in mice. Nature 1993; 364: 806–9. [DOI] [PubMed] [Google Scholar]
- 34. Baker MB, Altman NH, Podack ER, Levy RB. The role of cell‐mediated cytotoxicity in acute GVHD after MHC‐matched allogeneic bone marrow transplantation in mice. J Exp Med 1996; 183: 2645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kaiser GC, Polk DB. Tumor necrosis factor α regulates proliferation in a mouse intestinal cell line. Gastroenterology 1997; 112: 1231–40. [DOI] [PubMed] [Google Scholar]
- 36. Hattori K, Hirano T, Miyajima H et al . Differential effects of anti‐Fas ligand and anti‐tumor necrosis factor α antibodies on acute graft‐versus‐host disease pathologies. Blood 1998; 91: 4051–5. [PubMed] [Google Scholar]
- 37. Mora JR, Bono MR, Manjunath N et al . Selective imprinting of gut‐homing T cells by Peyer's patch dendritic cells. Nature 2003; 424: 88–93. [DOI] [PubMed] [Google Scholar]
- 38. Mora JR, Cheng G, Picarella D, Briskin M, Buchanan N, Von Andrian UH. Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin‐ and gut‐associated lymphoid tissues. J Exp Med 2005; 201: 303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berg DJ, Davidson N, Kuhn R et al . Enterocolitis and colon cancer in interleukin‐10‐deficient mice are associated with aberrant cytokine production and CD4+ TH1‐like responses. J Clin Invest 1996; 98: 1010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin‐10‐deficient mice develop chronic enterocolitis. Cell 1993; 75: 263–74. [DOI] [PubMed] [Google Scholar]
- 41. Davidson NJ, Leach MW, Fort MM et al . T helper cell 1‐type CD4+ T cells, but not B cells, mediate colitis in interleukin 10‐deficient mice. J Exp Med 1996; 184: 241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA, Gendler SJ. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res 1998; 58: 315–21. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. One week after MUC1 DNA vaccination, mice were killed. Single‐cell suspensions were obtained from the spleens. After removal of erythrocytes, splenocytes were passed through a nylon wool column to enrich T cells. CD4+ and CD8+ T cells were obtained by negative selection using a mixture of biotinylated monoclonal antibodies (CD11b, Ly6G, DX5, B220, and CD8 or CD4) and streptavidin microbeads on a LS column (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s specifications. These bulk T cells (5 × 106 cells), CD4+ T cells, or CD8+ T cells (2.5 × 106 cells) were mixed with MUC1‐transfected B16‐F10 cells (5 × 104 cells)( 9 ) and injected subcutaneously into naive C57B/6 mice. The survival rates were determined (n = 5).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item