

Abstract
Neutral endopeptidase 24.11 (NEP), a cell‐surface enzyme expressed by epithelial cells that cleaves and inactivates biologically active small peptides, is downregulated in various cancers. NEP is encoded by a gene that contains a CpG island in the promoter region, whose hypermethylation appears related to decreased expression. Altered expression of NEP has also been reported in human hepatocellular carcinoma (HCC), suggesting its possible role in hepatocarcinogenesis. To elucidate the status of NEP in HCC, methylation in the promoter region of the gene that encodes NEP in male Fischer 344 rats with HCC, induced by a choline‐deficient, l‐amino acid‐defined diet, was investigated by methylation‐specific polymerase chain reaction, combined bisulfite restriction analysis, and bisulfite genomic sequencing. These analyses together showed the promoter to be frequently methylated in HCC in contrast to its unmethylated status in normal liver, the degree of methylation being inversely related to the level of mRNA expression evaluated by reverse transcription–polymerase chain reaction (P = 0.031). In two rat liver cell lines, RLC‐16 and RLC‐27, the promoter was heavily methylated and NEP mRNA expression was negative. However, administration of 5‐aza‐2′‐deoxycytidine caused NEP expression, suggesting that methylation of CpG is a factor regulating transcriptional expression. Together with the data from microarray analyses performed previously using the same animal model, the current results suggest that reduced expression of NEP or other ectopeptidases could impact on molecules involved in signal‐transducing systems, including G‐protein coupled receptors, via modified turnover of extracellularly active small peptides. (Cancer Sci 2006; 97: 611–617)
Abbreviations:
- CDAA
choline‐deficient, l‐amino acid‐defined
- COBRA
combined bisulfite restriction analysis
- GPCR
G‐protein coupled receptor
- HCC
hepatocellular carcinoma
- MSP
methylation‐specific PCR
- NEP
neutral endopeptidase 24.11
- PCR
polymerase chain reaction
- PMA
protein kinase C activator
- RT‐PCR
reverse transcription–polymerase chain reaction
- TE
Tris‐EDTA.
Cell‐surface proteolytic enzymes regulate cell growth by cleaving and inactivating regulatory small peptides and peptide hormones in normal cells.( 1 ) Neutral endopeptidase 24.11 (CD10, enkephalinase, neprilysin, CALLA, EC 3.4.24.11, metallomembrane endopeptidase) is a type II transmembrane zinc‐containing endopeptidase expressed normally in numerous tissues. The enzyme cleaves peptide bonds on the amino side of hydrophobic amino acids and inactivates a variety of physiologically active peptides, including atrial natriuretic factor, substance P, bradykinin, oxytocin, Leu‐ and Met‐enkephalines, neurotensin, bombesin, endothelin‐1 and bombesin‐like peptides. Loss or decreases in NEP expression have been reported in a variety of malignancies, such as lung,( 2 , 3 ) endometrial,( 4 ) prostate,( 5 ) and renal cell( 6 ) cancers. The roles of NEP in regulation of cell proliferation and other functions have also been investigated in various types of cells.( 7 , 8 , 9 , 10 , 11 ) Overexpression of NEP in mutant Jerkat T cells that failed to express NEP resulted in restoration of PMA‐induced growth arrest.( 7 ) Inhibition of cell proliferation was observed in androgen‐independent prostate cancer cells transfected with NEP.( 10 ) Cervical carcinoma cells overexpressing NEP revealed reduced cell proliferation accompanied by a decreased concentration of endothelin‐1 in the conditioned medium.( 8 ) Reduced NEP may promote peptide‐mediated proliferation by allowing accumulation of higher peptide concentrations at the cell surface, and facilitate the development or progression of neoplasia.( 12 ) Possible causes include a change in methylation status. Hypermethylation of promoter‐associated CpG islands is an important mechanism for inactivation of tumor‐suppressor genes in carcinogenesis,( 13 , 14 ) and promoter hypermethylation of NEP has been reported for androgen‐independent prostate cancers( 15 , 16 ) and for adult acute lymphocytic leukemia.( 17 ) In these malignancies, hypermethylation of CpG dinucleotides in the promoter region of the NEP gene has thus been associated with reduced expression.
Hepatocellular carcinoma remains one of the most common malignancies in the world. Hepatocarcinogenesis is a multistep process involving a variety of genetic and epigenetic alterations.( 18 ) In a rat model induced by a CDAA diet in males of the Fischer 344 and Wistar strains, HCC develops on a background of liver cirrhosis, similar to the human case.( 19 ) Several molecular abnormalities have been described for HCC induced by a CDAA diet.( 20 , 21 ) We have recently carried out an oligonucleotide microarray analysis using rat liver HCC induced by a CDAA diet,( 21 ) which indicated that NEP expression in normal liver was 3.1 times higher than that in HCC (see http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE1946). In human HCC, reduced expression has also been reported,( 22 ) along with increased expression of endothelin‐1, a substrate of NEP.( 23 , 24 , 25 ) On the other hand, increased expression of NEP in HCC tissues has been correlated with longer survival.( 26 ) These findings may suggest its possible inhibitory effects on liver cell proliferation. So far, however, no mutations in the human NEP gene have been observed in HCC. Frequencies of allelic or regional losses involving the NEP gene, located in 3q21‐q27, are not particularly high compared to other regions.( 18 ) These observations may suggest that epigenetic mechanisms could be important in the regulation of NEP expression. In the present study we therefore concentrated attention on the methylation status of the CpG island in the promoter region of the rat NEP gene in HCC induced with a CDAA diet and rat liver cell lines by MSP, COBRA, and bisulfite genomic sequencing.
Materials and Methods
Ethical considerations
The experimental protocols were approved by the Animal Experimentation Committee of the Sasaki Institute (Tokyo, Japan) prior to their execution under monitoring by the Committee in accordance with the National Institutes of Health Guideline for the Care and Use of Laboratory Animals, the Japanese Government Animal Protection and Management Law Number 105, and the Japanese Government Notification on Feeding and Safekeeping of Animals Number 6.
Animals, diets and animal treatment
Male Fischer 344 rats (5 weeks old) were purchased from Charles River Japan (Kanagawa, Japan) and housed in plastic cages with white flake bedding in an air‐conditioned room (25 ± 3°C, 55 ± 8% relative humidity, 10–12 times/h ventilation and 12:12 h L:D cycle). They were used for the experimentation after a 1‐week acclimation on basal diet (CRF‐1; Oriental Yeast Corporation, Itabashi, Tokyo) and allowed free access to food and tap water throughout the acclimation and experimental periods. Bodyweight, food consumption and water intake were monitored weekly.
After acclimation, rats were allocated randomly to receive either basal diet or CDAA diet (Dyets, Bethlehem, PA, USA). Five rats on the basal diet and 12 rats on the CDAA diet were killed at 70 weeks after diet initiation. The livers were taken and examined macroscopically. Portions of the macroscopic tumors and background tissue were fixed in 10% neutrally buffered formalin for 24 h, embedded in paraffin, processed for the routine hematoxylin and eosin staining procedure, and examined histologically. Remaining portions were frozen immediately in liquid nitrogen and stored at −80°C.
Cell culture
RLC‐16 is an epithelial cell line from rat liver( 27 ) and RLC‐27 is a liver‐derived cell line, which is tumorigenic in nude mice. Both cell lines were purchased from Riken Bioresourse Center Cell Bank (Tsukuba, Ibaraki) and cultured in Dulbecco's Modified Eagle Medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum in collagen‐coated Petri dishes at 37°C in a humidified 5% CO2 atmosphere.
Genomic DNA and total RNA extraction
Genomic DNA was isolated from liver and liver cell lines using DNeasy Tissue Kit (QIAGEN, Hilden, Germany) and its quality checked by measurement of OD260 and OD280 and 1% agarose gel electrophoresis with ethidium bromide staining. Total RNA was isolated using an RNeasy Midi kit (QIAGEN) and its integrity confirmed by electrophoresis on 1% agarose–formaldehyde gels.
Bisulfite modification
DNA (1 µg) was digested with restriction endonuclease HindIII (Promega, Madison, WI, USA), followed by phenol, phenol/chloroform and chloroform extractions, and subsequent ethanol precipitation. After purification, the DNA was denatured in a volume of 20 µL NaOH (final concentration 0.3 M) for 15 min at 37°C. After denaturation, 8.4 µL of 10 mM hydroquinone (Sigma, St Louis, MO, USA) and 120 µL of 3.6 M sodium hydrogensulfite (Sigma) at pH 5, both prepared freshly, were added and, after mixing, the samples were incubated in a thermal cycler with 15 cycles of 30 s at 50°C and 15 min at 50°C. Modified DNA was purified using the Wizard DNA purification resin (Promega) according to the manufacturer's protocol and eluted into 50 µL of water. Modification was completed with NaOH (final concentration 0.3 M) treatment for 5 min at room temperature, followed by ethanol precipitation. DNA was suspended in TE buffer and used immediately or stored at −20°C. Normal rat liver DNA treated with the CpG methylase, M. SssI (New England Biolabs, Beverly, MA, USA) and rat testis DNA were used as methylated and unmethylated DNA controls, respectively (data not shown). The methylation status of these controls was confirmed by bisulfite genomic sequencing.
MSP analysis
Two sets of primers were used to amplify the region of interest: 5NEP‐U and 3NEP‐U recognize a sequence in which CpG sites are unmethylated (modified to UpG by the bisulfite treatment), and 5NEP‐M and 3NEP‐M recognize a sequence in which CpG sites are methylated (unmodified by the bisulfite treatment). Each set of primers was designed to examine the methylation status of five CpG sites within the CpG island in the second promoter of the NEP gene (designated 1, 2 and 20–22; Fig. 1; Table 1). The cycling conditions were 2 min at 94°C, followed by 35 cycles of 94°C for 1 min, 65°C for 1 min and 72°C for 1 min, with final extension elongated to 7 min, for both reactions. The products were visualized on 10% non‐denaturing polyacrylamide gels with ethidium bromide.
Figure 1.
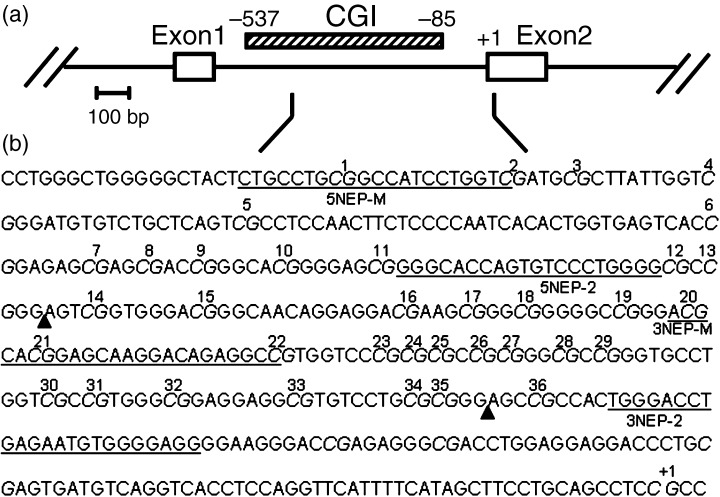
(a) Genomic structure of the rat NEP gene. The open boxes represent the exons and the hatched box the CpG island (CGI). (b) Nucleotide sequence of the rat NEP gene. The sequence is numbered from the exon 2 transcription start site, indicated as +1. Sequences of representative primers are underlined. CpG present in the indicated sequence are in italic and those analyzed by methylation‐specific polymerase chain reaction (MSP), combined bisulfite restriction analysis (COBRA), or bisulfite genomic sequencing are numbered. HinfI recognition sites for methylated CpG are indicated as closed triangles.
Table 1.
Primer sequences designed to examine the methylation status of CpG sites within the CpG island in the second promoter of the NEP gene
| Method | Name | Sequence (5′→3′) | Primer position † | Product size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|---|
| MSP | 5NEP‐M | TTGTTTGCGGTTATTTTGGTC | −409 | 219 | 65 |
| 3NEP‐M | GACCTCTATCCTTACTCCGTACGT | −191 | |||
| 5NEP‐U | TATTTTGTTTGTGGTTATTTTGGTT | −413 | 223 | 65 | |
| 3NEP‐U | AACCTCTATCCTTACTCCATACATC | −191 | |||
| Bisulfite sequencing | 5NEP‐1 | GGGTATTAGTGTTTTTGG | −287 | 197 | 60 |
| 3NEP‐1 | CCTCCCCACATTCTCAAATC | −91 | |||
| 5NEP‐2 | GGGTATTAGTGTTTTTGGGG | −287 | 197 | 68 | |
| 3NEP‐2 | CCTCCCCACATTCTCAAATCCCA | −91 |
Relative to exon 2 transcription start.
COBRA assay
Using 3 µL of sodium bisulfite‐treated DNA, PCR was carried out using a hot start in a 50‐µL reaction with primers 5NEP‐1 and 3NEP‐1 (Table 1). The cycling conditions were 2 min at 94°C, followed by 15 cycles of 94°C for 1 min, 60°C for 1 min and 68°C for 1 min, with final extension elongated to 7 min. One microliter of the PCR product was used for the following nested reaction using a hot start with primers 5NEP‐2 and 3NEP‐2 (Fig. 1; Table 1), which gives a 197‐bp main product. The cycling conditions were 2 min at 94°C, followed by 25 cycles of 94°C for 1 min, 68°C for 1 min and 74°C for 1 min, with final extension elongated to 30 min. After amplification, PCR products were digested with restriction endonuclease HinfI (Promega) that digests alleles methylated before bisulfite treatment at 37°C for 4 h, and separated on 10% non‐denaturing polyacrylamide gels with ethidium bromide staining. This assay was designed to examine the methylation status of two CpG sites in the CpG island (designated 14 and 36; Fig. 1; Table 1). The proportion of methylated versus unmethylated products (digested vs undigested) was quantitated by densitometry using NIH Image software (Bethesda, MD, USA) to determine the extent of methylation.
Bisulfite genomic sequencing of individual alleles
A 197‐bp DNA fragment was amplified by PCR as described above. The products included 25 CpG sites in the second promoter of the rat NEP gene (designated 12–36; Fig. 1). PCR products were cloned into the TA vector pCR2.1‐TOPO (Invitrogen) and transformed into bacteria. Plasmid DNA from isolated clones was purified using the Plasmid Midi kit (QIAGEN) and analyzed by automated DNA sequencing. Complete conversion of cytosines not flanked by guanine was confirmed.
Expression of NEP in liver tissues and cell lines
Reverse transcription was carried out on 3 µg of total RNA using a First‐Strand cDNA Synthesis kit (Amersham Biosciences, Uppsala, Sweden) and oligo(dT)18 primer according to the manufacturer's protocol in a total volume of 15 µL. The entire first‐strand reaction mixture was amplified by adding PCR primers and Taq polymerase in a volume of 50 µL. The primers used were 5′‐AGAGGAGGAAGACGCTGAAT‐3′ (sense) and 5′‐TCTTAACTATCTATCTTGGC‐3′ (antisense) and the cycling conditions were 2 min at 94°C, followed by 23 cycles of 94°C for 1 min, 55°C for 30 s and 72°C for 30 s, which amplified a 460‐bp product. PCR products were separated on 2% agarose gels, visualized with ethidium bromide staining, and subsequently underwent densitometry. The values obtained were then normalized to those for β‐actin.
Re‐expression of NEP by 5‐aza‐2′‐deoxycytidine treatment
Rat cultured cells (5 × 106 cells) were grown for 4 days in the presence of 10 µM of 5‐Aza‐2′‐deoxycytidine (Fluka, St Louis, MO, USA), then harvested for isolation of total RNA and performance of semiquantitative RT‐PCR as described above.
Statistical analyses
The statistical significance of differences of means between two groups was assessed using the unpaired Student's t‐test. A value of P < 0.05 was considered to be statistically significant.
Results
Histological findings
All rats survived until their scheduled killing in relatively healthy conditions. Control rats fed a basal diet showed no particular pathological changes. All of the livers from rats fed the CDAA diet were macroscopically yellowish‐white and appeared cirrhotic with one to three dark‐colored large tumoral nodules. Tumors were diagnosed histologically as relatively well‐differentiated HCC. The surrounding, non‐cancerous areas were cirrhotic, featuring frequent hepatocellular apoptosis and nuclear divisions of hepatocytes. These findings were largely in accordance with our previous reports.( 18 , 20 )
Methylation status of the NEP promoter in HCC, surrounding liver, non‐cancerous liver, normal liver and rat liver cell lines
Two separate regulatory regions have been shown for the NEP gene,( 15 , 28 ) and sequence analysis of the rat NEP promoter has identified a CpG island in the second promoter region located between exons 1 and 2, as defined by the criteria of Gardiner‐Garden and Frommer (G + C content > 50% and observed/expected CpG dinucleotide ratio > 0.6).( 29 ) The CpG island is a 453‐bp segment, ranging from −537 to −85 relative to the exon 2 transcription start (Fig. 1). We first evaluated the methylation status of the CpG island by MSP using unmethylated and methylated primer sets (Fig. 2a). The DNA from age‐matched normal rats (e.g. N1‐N3) demonstrated negative methylation patterns in the region tested, whereas HCC samples exhibited various methylation patterns from negative (e.g. T8) to relatively heavy (e.g. T9) methylation. The two rat liver cell lines RLC‐16 and RLC‐27 showed a complete methylation pattern. Because the degree of methylation evaluated by MSP differed extensively among HCC samples, a more quantitative method was needed, and therefore we also carried out COBRA (Fig. 2b). In this analysis, DNA amplified by PCR was treated with HinfI, which cleaved CpG that were methylated before bisulfite treatment. CpG sites in HCC exhibited positive digestion of the PCR products (e.g. T1, T3, T7 and T9) or negative results (e.g. T8). When a threshold of ≥15% was used to define a sample as methylated,( 17 ) 75% (nine out of 12 samples) of HCC samples were methylated, whereas none of the normal liver samples (five samples) were methylated.
Figure 2.

Methylation status of the NEP gene was assayed by methylation‐specific polymerase chain reaction (MSP), combined bisulfite restriction analysis (COBRA), and bisulfite genomic sequencing. (a) Examples of MSP analysis of hepatocellular carcinoma (HCC) and normal liver samples from control rats. Bisulfite‐modified genomic DNA from HCC (T) and normal control livers (N) was amplified with methylated DNA‐specific primers or unmethylated DNA‐specific primers. 16, RLC‐16; 27, RLC‐27. (b) Examples of COBRA with HinfI digestion. Polymerase chain reaction products from bisulfite‐treated DNA obtained from HCC (T) and normal livers (N) were treated with restriction endonuclease HinfI. Only specimens that had methylated CpG were restricted. The gels were stained with ethidium bromide. Unmethylated (top rows) and methylated (bottom rows, arrows) products were quantitated by densitometry. Methylation densities (percentage of restricted vs unrestricted fragments) are shown below each lane. (c) Results of bisulfite genomic sequencing. Normal liver tissue (N1), three HCC samples (T3, T7 and T9) and a rat liver cell line (RLC‐27) were analyzed. For each sample, eight clones for the region in the CpG island (Fig. 1) were sequenced. Each square indicates a CpG site and each line of squares represents the analysis of a single‐cloned allele. Methylated CpG sites are shown as closed squares and their unmethylated counterparts as open squares. (d) MSP analysis of surrounding, non‐cancerous tissues (NC). Bisulfite‐treated DNA samples NC1, NC3 and NC9 were obtained from rats that had HCC T1, T3 and T9, respectively. In NC3 and NC9, methylated primers also provided positive bands; however, by comparing the intensities of bands, the extent of methylation was considered weaker in surrounding areas than in corresponding HCC themelves (a).
We next examined the methylation status of a DNA segment containing 25 CpG sites (designated 12–36; Fig. 1) in the CpG island using bisulfite genomic sequencing for normal liver tissue (N1), HCC samples (T3, T7 and T9) and a liver cell line (RLC‐27). Eight clones were analyzed for each sample. As shown in Fig. 2c, in the normal liver CpG sites were almost completely unmethylated, whereas in the HCC samples examined they were partially methylated. From the results of sequencing, we have presumed that positive bands detected with unmethylated MSP primers in HCC may be largely due to partially methylated alleles in tumor cells, but the possibility of contamination of surrounding non‐cancerous tissues could not be excluded. Complete methylation of CpG sites within the region tested was detected in the RLC‐27 case.
Methylation status was also investigated by MSP in three surrounding, non‐cancerous liver samples and they gave strong unmethylated bands (Fig. 2d). However, in two of these samples (NC3 and NC9), methylated bands were also detected, although the extent of methylation appeared weaker than in HCC themselves (T3 and T9, respectively, Fig. 2a).
Expression of NEP in HCC, normal liver and rat liver cell lines
Previous studies have shown aberrant methylation of the CpG island located in the second promoter region to be a mechanism of inactivation of the NEP gene in prostate cancer( 15 ) and in adult acute lymphocytic leukemia.( 17 ) Based on this consideration, expression of the rat NEP gene was evaluated by semiquantitative RT‐PCR. Examples of the analysis of HCC and normal liver tissues are illustrated in Fig. 3a. A HCC sample with low‐level methylation had a high expression level similar to control liver (T8), whereas HCC with medium‐to‐high levels of methylation (T1, T7 and T9) demonstrated reduced expression (Fig. 3a). In the two rat liver cell lines, RLC‐16 and RLC‐27, NEP mRNA expression was negative under basal conditions (lanes 1 and 3; Fig. 4). The mRNA level of NEP expressed as a ratio to β‐actin in age‐matched control normal livers was higher than in HCC samples (P < 0.05) (Fig. 3b). In the 12 HCC samples used in this study, NEP methylation estimated by COBRA was inversely correlated with NEP expression at the mRNA level (r = –0.622, P = 0.031).
Figure 3.
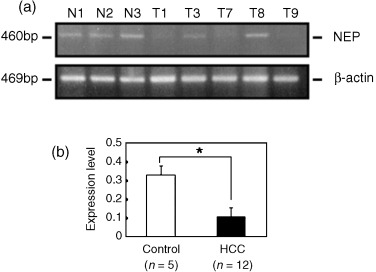
(a) Examples of mRNA expression for NEP (upper panel) and β‐actin (lower panel) in hepatocellular carcinoma (HCC) and normal liver tissues from control rats assayed by semiquantitative reverse transcription–polymerase chain reaction. (b) Expression of NEP at mRNA level in normal liver samples was greater than in HCC. Data are mean ± SD for ratios of NEP to β‐actin. The experiment for mRNA expression was performed in triplicate. *P < 0.05.
Figure 4.
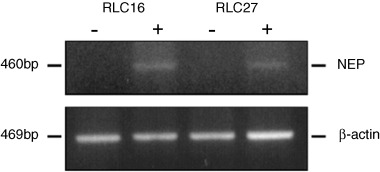
Increased expression of NEP transcripts in rat liver‐derived cells after exposure to 5‐aza‐2′‐deoxycytidine. The mRNA level of NEP expression in the cells was determined using semiquantitative reverse transcription–polymerase chain reaction before (–) and after (+) treatment with 10 µM 5‐aza‐2′‐deoxycytidine for 4 days. Top panel, NEP primer products; bottom panel, β‐actin primer products.
Re‐expression of NEP by 5‐aza‐2′‐deoxycytidine treatment
To investigate whether suppression of NEP gene expression was mediated by promoter hypermethylation, the two rat liver cell lines RLC‐16 and RLC‐27 were treated with 5‐aza‐2′‐deoxycytidine at 10 µM for 4 days. The promoter of these cell lines was heavily methylated (Fig. 2a,b) and the mRNA expression of NEP was negative under basal conditions, but gene expression at the mRNA level was restored in both cell lines after treatment (Fig. 4).
Discussion
The present study provides clear evidence that methylation of CpG dinucleotides in the promoter region of NEP is a common event in a rat HCC model. Furthermore, on quantitation of the extent of methylation in HCC samples, an inverse correlation with mRNA expression of NEP was observed and in vitro studies demonstrated that exposure of cultured rat liver cell lines with complete promoter methylation to 5‐aza‐2′‐deoxycytidine restored mRNA expression. The results also indicated that methylation of the NEP gene is not an effect of aging, because age‐matched normal livers were almost completely unmethylated. In HCC samples that showed positive methylation patterns, their surrounding, non‐cancerous, cirrhotic areas also gave weak methylation patterns, indicating that methylation of the NEP gene is a progressive change, caused by administration of the CDAA diet. In the current study, nine out of 12 HCC samples were methylated (75%) and therefore, the frequency of methylation seems higher than in patients with hormone‐naïve prostate cancer (three out of 21 patients, 14%)( 15 ) or adult acute lymphocytic leukemia (eight out of 80 patients, 10%).( 17 ) Promoter methylation has also been reported for several tumor‐suppressor genes in human HCC. The most frequently methylated genes include SOCS‐1, GSTP, APC, E‐cadherin and p15.( 30 , 31 , 32 ) One study indicated that the SOCS‐1, GSTP and APC genes were methylated in 65%, 54% and 53% of patients with HCC, respectively,( 33 ) so it appears that in rat HCC the NEP gene may be methylated with similarly high frequency. In HCC, expression of two crucial methyltransferases, DNMT1 and DNMT3a, was shown to be elevated compared to non‐cancerous liver,( 34 ) suggesting that epigenetic mechanisms may be important. It is thus important to detect genes regulated by methylation of the promoter region during HCC development.
Hypermethylation of the NEP promoter and reduced expression has been observed in androgen‐independent prostate cells, but not in androgen‐sensitive cells,( 15 ) and it has been suggested that androgen withdrawal triggers silencing of the NEP gene in prostate cancers.( 5 ) Androgen withdrawal is the primary therapy for patients who develop locally advanced or metastatic prostate cancer. Although responses to androgen withdrawal occur in the majority of people with the disease, treatment failure and tumor growth develop within 2 years. Long‐term use of oral contraceptives and androgenic steroids can induce benign and malignant hepatocellular tumors.( 35 ) In addition, HCC is more prevalent in men than in women, and the CDAA diet used in the current study induces HCC in male rats. These findings have suggested the possibility that androgen is involved in its pathogenesis,( 36 ) and at least portions of human HCC are considered to be androgen‐dependent.( 37 , 38 , 39 ) As in prostate cancer, anti‐androgen therapy for unresectable human HCC has been tried, but did not prove effective at prolonging survival when tested in a randomized controlled trial.( 40 ) The mechanisms of these observations are still obscure, but they suggest that HCC may use alternative sources to stimulate cell proliferation. Involvement of sex hormones and their receptors as a mechanism that may trigger inactivation of NEP need to be investigated in the future.
Several lines of evidence support the involvement of biologically active small peptides and their receptors in hepatocarcinogenesis. In human HCC, altered expression of endothelin‐1, a substrate of NEP, is observed.( 23 , 24 , 25 ) Neurotensin is another substrate of NEP, which stimulates the growth of hepatocytes.( 41 ) Aberrant expression of regulatory peptide receptors, such as somatostatin, vasoactive intestinal peptide, endothelin B, and substance P receptors, all of which are members of the GPCR superfamily, has also been reported.( 42 ) Although the contribution of cell‐surface peptidases, including NEP, to the regulation of bioactive small peptides in the liver remains to be elucidated, reduced expression of NEP has been observed in HCC.( 22 ) Moreover, elevated expression of NEP in HCC explored by immunohistochemistry has been correlated with longer survival.( 26 ) These may suggest its possible roles in HCC progression. As cell‐surface peptidases modulate the activity of peptide factors and regulate access to adjacent cells, their loss of function may result in an inability to inactivate stimulatory peptides. Whereas loss or decreased expression of NEP has been observed in various malignancies, the lack of tools to simply evaluate concordant changes of regulatory molecules in these processes has hitherto made investigation of the underlying mechanisms difficult. Recently developed technologies such as microarray systems may facilitate studies in this direction.
In the above‐mentioned oligonucleotide microarray analysis using rat liver HCC induced by the CDAA diet, we detected 146 differentially expressed genes, which were classified into four clusters solely based on their expression patterns.( 21 ) Genes belonging to the categories cytokines and cell receptors were both enriched in a cluster characterized by reduced mRNA in HCC compared with age‐matched normal liver (P < 0.05, Fisher's exact test).( 21 ) The GPCR in this cluster included opioid receptor κ1, somatostatin receptor 1, endothelin receptor A and endothelin receptor B. The study thus elucidated concordant changes in expression of certain small peptides and cell receptors during hepatocarcinogenesis. The causes of the altered expression are largely unknown, but based on expression of bioactive peptides and receptors detected with the microarrays, we hypothesize that a negative feedback mechanism triggered by loss or reduced expression of cell‐surface peptidases could be involved. In the HCC, peptidase expression is attenuated, and cells may not be able to inactivate bioactive peptides. The signals initiated by binding of the ligands to receptors stimulate cell growth. Excessive signaling may in turn trigger a negative feedback system, resulting in a suppression of transcription of peptides and receptors in HCC.
In summary, we here detected a high incidence of aberrant methylation of the promoter of the NEP gene in rat HCC, resulting in its reduced expression in cancer cells. We propose that altered expression of NEP may be a factor that can cause concordant changes in signal‐transducing molecules during the development of HCC in the CDAA rat model. Because of the high incidence of promoter methylation and reduced expression of the NEP gene, this model should find application for further exploration of the involvement of cell‐surface peptidases in carcinogenesis.
Acknowledgments
We thank Dr. Yoshiyuki Hashimoto (Kyoritsu University of Pharmacy) for constant advice, and Ms Hiromi Asako and Ms Chinami Kajiwara (Department of Pathology, Sasaki Institute) for expert technical assistance. This work was supported by a Research Grant of the Princess Takamatsu Cancer Research Fund of Japan (01‐23308).
References
- 1. Nanus DM. Of peptides and peptidases: the role of cell surface peptidases in cancer. Clin Cancer Res 2003; 9: 6307–9. [PubMed] [Google Scholar]
- 2. Shipp MA, Tarr GE, Chen CY et al. CD10/neutral endopeptidase 24.11 hydrolyzes bombesin‐like peptides and regulates the growth of small cell carcinomas of the lung. Proc Natl Acad Sci USA 1991; 88: 10 662–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen AJ, Franklin WA, Magill C, Sorenson J, Miller YE. Low neutral endopeptidase levels in bronchoalveolar lavage fluid of lung cancer patients. Am J Respir Crit Care Med 1999; 159: 907–10. [DOI] [PubMed] [Google Scholar]
- 4. Pekonen F, Nyman T, Ammala M, Rutanen EM. Decreased expression of messenger RNAs encoding endothelin receptors and neutral endopeptidase 24.11 in endometrial cancer. Br J Cancer 1995; 71: 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Papandreou CN, Usmani B, Geng Y et al. Neutral endopeptidase 24.11 loss in metastatic human prostate cancer contributes to androgen‐independent progression. Nat Med 1998; 4: 50–7. [DOI] [PubMed] [Google Scholar]
- 6. Gohring B, Holzhausen HJ, Meye A et al. Endopeptidase 24.11/CD10 is down‐regulated in renal cell cancer. Int J Mol Med 1998; 2: 409–14. [DOI] [PubMed] [Google Scholar]
- 7. Mari B, Guerin S, Maulon L et al. Endopeptidase 24.11 (CD10/NEP) is required for phorbol ester‐induced growth arrest in Jurkat T cells. FASEB J 1997; 11: 869–79. [DOI] [PubMed] [Google Scholar]
- 8. Terauchi M, Kajiyama H, Shibata K, Ino K, Mizutani S, Kikkawa F. Anti‐progressive effect of neutral endopeptidase 24.11 (NEP/CD10) on cervical carcinoma in vitro and in vivo . Oncology 2005; 69: 52–62. [DOI] [PubMed] [Google Scholar]
- 9. Deddish PA, Marcic BM, Tan F, Jackman HL, Chen Z, Erdos EG. Neprilysin inhibitors potentiate effects of bradykinin on b2 receptor. Hypertension 2002; 39: 619–23. [DOI] [PubMed] [Google Scholar]
- 10. Dai J, Shen R, Sumitomo M et al. Tumor‐suppressive effects of neutral endopeptidase in androgen‐independent prostate cancer cells. Clin Cancer Res 2001; 7: 1370–7. [PubMed] [Google Scholar]
- 11. Okamoto A, Lovett M, Payan DG, Bunnett NW. Interactions between neutral endopeptidase (EC 3.4.24.11) and the substance P (NK1) receptor expressed in mammalian cells. Biochem J 1994; 299: 683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sumitomo M, Shen R, Nanus DM. Involvement of neutral endopeptidase in neoplastic progression. Biochim Biophys Acta 2005; 1751: 52–9. [DOI] [PubMed] [Google Scholar]
- 13. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999; 21: 163–7. [DOI] [PubMed] [Google Scholar]
- 14. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 2002; 21: 5427–40. [DOI] [PubMed] [Google Scholar]
- 15. Usmani BA, Shen R, Janeczko M et al. Methylation of the neutral endopeptidase gene promoter in human prostate cancers. Clin Cancer Res 2000; 6: 1664–70. [PubMed] [Google Scholar]
- 16. Osman I, Yee H, Taneja SS et al. Neutral endopeptidase protein expression and prognosis in localized prostate cancer. Clin Cancer Res 2004; 10: 4096–100. [DOI] [PubMed] [Google Scholar]
- 17. Garcia‐Manero G, Daniel J, Smith TL et al. DNA methylation of multiple promoter‐associated CpG islands in adult acute lymphocytic leukemia. Clin Cancer Res 2002; 8: 2217–24. [PubMed] [Google Scholar]
- 18. Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002; 31: 339–46. [DOI] [PubMed] [Google Scholar]
- 19. Nakae D. Endogenous liver carcinogenesis in the rat. Pathol Int 1999; 49: 1028–42. [DOI] [PubMed] [Google Scholar]
- 20. Tsujiuchi T, Tsutsumi M, Sasaki Y, Takahama M, Konishi Y. Hypomethylation of CpG sites and c‐myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline‐deficient 1‐amino acid‐defined diet in rats. Jpn J Cancer Res 1999; 90: 909–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uematsu F, Takahashi M, Yoshida M et al. Distinct patterns of gene expression in hepatocellular carcinomas and adjacent non‐cancerous, cirrhotic liver tissues in rats fed a choline‐deficient, l‐amino acid‐defined diet. Cancer Sci 2005; 96: 414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rocken C, Carl‐McGrath S, Grantzdorffer I, Mantke R, Roessner A, Lendeckel U. Ectopeptidases are differentially expressed in hepatocellular carcinomas. Int J Oncol 2004; 24: 487–95. [PubMed] [Google Scholar]
- 23. Kar S, Yousem SA, Carr BI. Endothelin‐1 expression by human hepatocellular carcinoma. Biochem Biophys Res Commun 1995; 216: 514–19. [DOI] [PubMed] [Google Scholar]
- 24. Ishibashi M, Fujita M, Nagai K et al. Production and secretion of endothelin by hepatocellular carcinoma. J Clin Endocrinol Metab 1993; 76: 378–83. [DOI] [PubMed] [Google Scholar]
- 25. Nakamuta M, Ohashi M, Tabata S et al. High plasma concentrations of endothelin‐like immunoreactivities in patients with hepatocellular carcinoma. Am J Gastroenterol 1993; 88: 248–52. [PubMed] [Google Scholar]
- 26. Mondada D, Bosman FT, Fontolliet C, Seelentag WK. Elevated hepatocyte paraffin 1 and neprilysin expression in hepatocellular carcinoma are correlated with longer survival. Virchows Arch 2006; 448: 35–45. [DOI] [PubMed] [Google Scholar]
- 27. Takaoka T, Yasumoto S, Katsuta H. A simple method for the cultivation of rat liver cells. Jpn J Exp Med 1975; 45: 317–26. [PubMed] [Google Scholar]
- 28. Li C, Hersh LB. Characterization of the promoter region of the rat neprilysin gene. Arch Biochem Biophys 1998; 358: 189–95. [DOI] [PubMed] [Google Scholar]
- 29. Gardiner‐Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196: 261–82. [DOI] [PubMed] [Google Scholar]
- 30. Wong IH, Lo YM, Yeo W, Lau WY, Johnson PJ. Frequent p15 promoter methylation in tumor and peripheral blood from hepatocellular carcinoma patients. Clin Cancer Res 2000; 6: 3516–21. [PubMed] [Google Scholar]
- 31. Yoshikawa H, Matsubara K, Qian GS et al. SOCS‐1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth‐suppression activity. Nat Genet 2001; 28: 29–35. [DOI] [PubMed] [Google Scholar]
- 32. Tchou JC, Lin X, Freije D et al. GSTP1 CpG island DNA hypermethylation in hepatocellular carcinomas. Int J Oncol 2000; 16: 663–76. [DOI] [PubMed] [Google Scholar]
- 33. Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol 2003; 163: 1101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Expression of mRNA for DNA methyltransferases and methyl‐CpG‐binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology 2001; 33: 561–8. [DOI] [PubMed] [Google Scholar]
- 35. D’Arville CN, Johnson PJ. Growth factors, endocrine aspects and hormonal treatment in hepatocellular carcinoma − an overview. J Steroid Biochem Mol Biol 1990; 37: 1007–12. [DOI] [PubMed] [Google Scholar]
- 36. De Maria N, Manno M, Villa E. Sex hormones and liver cancer. Mol Cell Endocrinol 2002; 193: 59–63. [DOI] [PubMed] [Google Scholar]
- 37. Iqbal MJ, Wilkinson ML, Johnson PJ, Williams R. Sex steroid receptor proteins in foetal, adult and malignant human liver tissue. Br J Cancer 1983; 48: 791–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagasue N, Yukaya H, Chang YC, Ogawa Y, Kohno H, Ito A. Active uptake of testosterone by androgen receptors of hepatocellular carcinoma in humans. Cancer 1986; 57: 2162–7. [DOI] [PubMed] [Google Scholar]
- 39. Tavian D, De Petro G, Pitozzi A, Portolani N, Giulini SM, Barlati S. Androgen receptor mRNA under‐expression in poorly differentiated human hepatocellular carcinoma. Histol Histopathol 2002; 17: 1113–19. [DOI] [PubMed] [Google Scholar]
- 40. Grimaldi C, Bleiberg H, Gay F et al. Evaluation of antiandrogen therapy in unresectable hepatocellular carcinoma: results of a European Organization for Research and Treatment of Cancer multicentric double‐blind trial. J Clin Oncol 1998; 16: 411–17. [DOI] [PubMed] [Google Scholar]
- 41. Hasegawa K, Kar S, Carr BI. Stimulation of hepatocyte DNA synthesis by neurotensin. J Cell Physiol 1994; 158: 215–22. [DOI] [PubMed] [Google Scholar]
- 42. Reubi JC, Zimmermann A, Jonas S et al. Regulatory peptide receptors in human hepatocellular carcinomas. Gut 1999; 45: 766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]