

Abstract
The cellular response to genotoxic stress that damages DNA includes cell cycle arrest, activation of DNA repair, and in the event of irreparable damage, induction of apoptosis. However, the signals that determine cell fate, that is, survival or apoptosis, are largely unclear. The tumor suppressor p53 has been implicated in many important cellular processes, including regulation of apoptotic cell death. When cells encounter genotoxic stress, certain sensors for DNA lesions eventually stabilize and activate p53. Subsequently, p53 exerts its tumor suppressor function by transactivating numerous target genes. Active p53 is subjected to a complex and diverse array of covalent post‐translational modifications, which selectively influence the expression of p53 target genes. In this regard, the molecular basis for how p53 induces apoptosis has been extensively studied; however, the relative contribution of each downstream effecter is still to be explored. Moreover, little is known about precise mechanisms by which modified p53 is capable of apoptosis induction. A thorough understanding for the whole picture of p53 modification in apoptosis will be extremely valuable in the development of highly effective and specific therapies for caner patients. This review is focused on the current views regarding the regulation of cell fate by p53 in the apoptotic response to DNA damage.
(Cancer Sci 2010; 101: 831–835)
Genotoxic stress that damages DNA induces cell cycle arrest, activation of DNA repair, and in the event of irreparable damage, induction of apoptosis. The decision by cells either to repair DNA lesions and continue through the cell cycle or to undergo apoptosis is relevant to the incidence of mutagenesis and, subsequently, carcinogenesis.( 1 ) In this context, incomplete repair of DNA damage prior to replication or mitosis can result in the accumulation of heritable genetic changes. Therapeutic anti‐cancer treatments that use DNA‐damaging agents must strike a balance between induction of repair and apoptosis in order to maximize the therapeutic effect. However, the nature of the cellular signaling response that determines cell survival or death is far from being understood. Certain insights have been derived from the findings that the tumor suppressor p53 plays a central role in response to DNA damage. The p53 gene is one of the most common sites for genetic alterations in human solid cancers since it is mutated in more than 50% cancer cases worldwide.( 2 ) The level of p53 protein is basically undetectable in normal cells but rapidly increases in response to a variety of stress signals. The mechanism by which the p53 protein is stabilized is not completely understood, but post‐translational modification plays a pivotal role.( 3 ) The p53 pathway can be impaired by numerous oncogenic proteins.( 4 ) Mice engineered to have the p53 gene knocked‐out develop tumors at an increased rate.( 5 ) Subsequent research on wild‐type p53 clearly demonstrates that the transcription factor is a key controller in cell cycle and determines the cell fate in response to oncogenic and to other stresses to maintain genomic integrity. The p53 has thus provided the name of “guardian of the genome”.( 6 ) In the complexity of p53 network, there are various crucial post‐translational modifications that induce p53 activation as a transcription factor. In the past 10 years, understanding of the cellular mechanisms of apoptosis mediated by p53 has advanced considerably, and the primary focus of this review is to provide an overview of p53, its mode of action and its physiological role in genotoxic stress‐induced apoptosis.
Functions of p53
The biological role of p53 is to ensure genome integrity of cells. p53 can stimulate the repair processes and protective mechanisms, or the cessation of cell division and the induction of acquired cell death. To achieve its aims, p53 may use a wide spectrum of activities, such as its ability to function as transcription factor, by inducing or repressing different genes or as a regulatory protein, within numerous signaling pathways. The functional consequence for these different activities of p53 fits into the enforcement of genetic stability. The activity of p53 is stimulated in response to DNA damage and to various genotoxic insults that eventually compromise genome integrity.( 7 ) Following genotoxic stress, p53 primarily determines the cell fate to induce growth arrest, DNA repair, or apoptosis. In particular, upon exposure to severe DNA damage, p53 conducts to elicit cell death by apoptosis (Fig. 1). The loss of p53 functions and the deregulated activities of p53 are involved not only in the development of malignant diseases, but also in relation to cardiovascular, neurodegenerative, infectious and metabolic diseases, as well as participate in the process of organism aging. p53 has the ability to bind specific responsive DNA elements and the specificity of the transcriptional activation depends on the ability of p53 DNA‐binding domain to interact with the regulatory regions of certain genes. The transcriptional activation is determined by the N‐terminus of p53, which contains several regions interacting with the transcription machinery and recruiting factors that modify local chromatin structure.( 8 )
Figure 1.
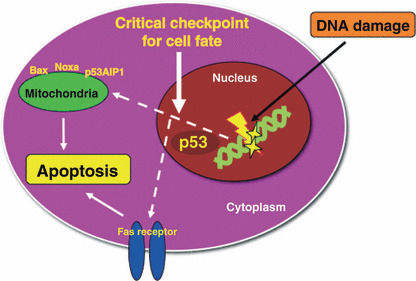
A hypothetical schema of a role for p53 on the critical checkpoint of cell fate in the apoptotic response to DNA damage. Following DNA damage, the cellular signaling is targeted to the nucleus, then the cell fate may be determined by p53 in the nucleus. The death signals are transmitted into the mitochondria or the receptors to induce apoptosis.
Stabilization of p53 in Response to DNA Damage
p53 is regulated primarily through post‐translational modifications, especially phosphorylation, and the accumulation of p53 is the first step in response to cellular stress (Fig. 2).( 9 ) The N‐terminus is heavily phosphorylated, while the C‐terminus contains phosphorylated, acetylated, methylated and sumoylated residues. Post‐translational modifications on the N‐terminus are important for stabilizing p53 while those on the C‐terminus inhibit the ability for regulation of sequence‐specific DNA binding, the oligomerization state, the nuclear import/export process, and the ubiquitination.( 10 ) The mdm2 gene is a transcriptional target of p53, and once synthesized, the MDM2 protein can bind to p53 at the N‐terminus leading to its rapid degradation through the ubiquitin‐proteasome machinery.( 9 , 11 , 12 ) In response to DNA damage, the ATM kinase rapidly phosphorylates p53 at Ser15. The serine/threonine kinase Chk2 acts downstream of ATM phosphorylating p53 at Ser20. These phopshorylated sites in the N‐terminus of p53 are close to the Mdm2‐binding region of the protein, thereby they block the interaction with Mdm2, leading to stabilization of p53, that escapes from proteasomal degradation.( 10 ) Recent findings suggested that constitutive phosphorylation of p53 by protein kinase C (PKC) at the C‐terminal domain contributes to its degradation via the ubiquitin‐proteasome pathway.( 13 )
Figure 2.
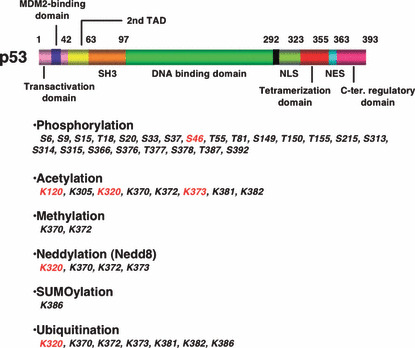
Functional domain and modification residues in p53. TAD indicates transactivation domain. NLS and NES mean nuclear localization and export signals, respectively. Modifications related to apoptosis are highlighted in red.
p53 and Apoptosis
Apoptosis is a well‐studied and well‐understood process that has been considered to play an important role in the tumor suppression. Apoptosis is triggered in response to a variety of signals, which can activate the extrinsic and/or intrinsic death pathways, or when cells are deprived of pro‐survival signals.( 14 ) p53 acts at multiple levels of the intrinsic and extrinsic pathways,( 15 ) via the induction of multiple apoptotic target genes, as well as via transcription‐independent mechanisms.( 16 , 17 ) For example, p53 activates several important genes that are crucial for the execution of the intrinsic pathway of apoptosis including pro‐apoptotic genes such as Bax, Noxa, Puma, and Apaf–1.( 18 , 19 , 20 , 21 ) When p53‐dependent apoptosis is employed by cells, these cells typically undergo the intrinsic cell death pathway.( 16 ) Further, p53 represses the apoptosis repressor with caspase recruitment domain protein, which counteracts the apoptotic functions of Puma and Bad.( 22 ) p53 can also promote cytochrome c release by inducing the expression of the OKL38 tumor suppressor gene, which localizes to the mitochondria and augments cytochrome c release. Silencing OKL38 correlates with tumorigenesis, and its overexpression induces apoptosis in several carcinoma cell lines.( 23 ) Among the extrinsic pathway, p53 induces the expression of the death receptor Fas and DR5.( 15 ) Moreover, p53 induces the expression of the TRAIL death ligand and the Fas ligand.( 24 , 25 ) In addition to its activity as a transcriptional regulator, p53 can also induce cell death in a transcription‐independent manner. It has been shown that p53 is able to physically interact with anti‐apoptotic proteins including bcl‐2, bcl‐XL and mcl‐1 at the mitochondrial membrane.( 26 ) When these anti‐apoptotic proteins bind p53, their ability to stabilize the mitochondrial membrane is compromised resulting in permeability changes and the release of cytochrome c. Additionally, p53 can interact with bak and thus directly induce the release of cytochrome c from the intermembrane space of the mitochondrion.( 26 ) Taken together, these studies suggest a role for mitochondrial p53 in the induction of apoptosis.( 27 ) In addition, it should be noted that a new p53 cofactor, human cellular apoptosis susceptibility protein (hCAS/CSE1L) suppresses p53‐mediated transcription by binding directly to the promoters of certain p53 target genes in a p53‐independent manner.( 28 ) The promoters bound by hCAS/CSE1L do not appear to correlate with a particular physiological outcome whereas silencing of hCAS/CSE1L inhibits ultraviolet (UV) irradiation‐induced death. Finally, It was also recently shown that p53 regulates the expression of microRNAs (miRNAs), where a primary role has been attributed to the miR‐34 family.( 29 , 30 ) Inactivation of miR‐34 attenuates p53‐mediated apoptosis in cells exposed to genotoxic stress, suggesting a role for this microRNA in regulating p53 responses.
Transcriptional Regulation of p53 in the Apoptotic Response to DNA Damage
Recent reports document that PKCδ transactivates p53 expression at the transcriptional level.( 1 , 31 , 32 ) The tumor‐promoting phorbol ester 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA) prevents DNA damage‐induced up‐regulation of p53 by down‐regulating PKCδ. TPA promotes tumor formation in various mice and cell culture models, and this has been correlated with the down‐regulation of PKC.( 33 ) TPA is known to activate but then down‐regulate the diacylglyceroldependent PKC isoforms.( 33 , 34 ) Previous studies demonstrated that the tumor‐promoting activities of TPA are mediated at least in part by down‐regulating PKCδ.( 35 ) Moreover, transgenic mice overexpressing PKCδ in their epidermis are resistant to tumor promotion by TPA.( 36 ) Previous studies have suggested that TPA can inhibit the DNA damage‐mediated induction of p53.( 37 ) Moreover, other studies with protein kinase inhibitors have suggested that PKCδ regulates the p53 pathway.( 38 ) Regulation of p53 in response to genotoxic stress commonly occurs by hampering ubiquitination and subsequent degradation of the p53 protein. By contrast, suppression of p53 expression by inhibition of PKCδ is caused by the inhibition of p53 synthesis, not increased degradation of p53 protein. Inhibiting PKCδ blocks both basal transcription of the human p53 gene and initiation of transcription from the human p53 promoter. The DNA damage‐induced increase in p53 accumulation is dramatically inhibited by pretreatment of cells with the tumor promoter TPA. In addition, the PKCδ inhibitor, rottlerin, is also able to block the DNA damage‐mediated induction of p53. More importantly, pretreatment of cells with TPA or with rottlerin results in the inhibition of basal p53 transcription. In this regard, accumulation of p53 can not be achieved any longer following TPA or rottlerin stimulation, because p53 transcription is blocked. It is thus conceivable that the tumor‐suppressing effects of PKCδ are mediated at least in part through activating p53 transcription. Indeed, repression of the p53 promoter has been suggested as a mechanism for tumor promotion.( 39 , 40 )
Recent study also demonstrated that PKCδ induces the promoter activity of p53 through the p53 core promoter element (CPE‐p53) and that such induction is enhanced in response to DNA damage. Upon exposure to genotoxic stress, PKCδ activates and interacts with the death‐promoting transcription factor Btf (Bcl‐2‐associated transcription factor) to co‐occupy CPE‐p53. Inhibition of PKCδ activity decreases the affinity of Btf for CPE‐p53, thereby reducing p53 expression at both the mRNA and protein levels. In concert with these results, disruption of Btf‐mediated p53 transcription by RNA interference leads to suppression of p53‐mediated apoptosis following genotoxic stress. These findings provide evidence that activation of p53 transcription by PKCδ triggers p53‐dependent apoptosis in response to DNA damage (Fig. 3).( 32 ) Subsequently, we also found that RAS‐responsive element‐binding protein‐1 (RREB‐1) efficiently binds to the p53 promoter via CPE‐p53 and transactivates p53 expression.( 41 ) Notably, disruption of RREB‐1‐mediated p53 transcription suppresses the expression of the p53 target genes. We also show that, upon exposure to genotoxic stress, RREB‐1 controls apoptosis in a p53‐dependent manner (Fig. 3). These findings provide evidence that RREB‐1 participates in modulating p53 transcription in response to DNA damage.
Figure 3.
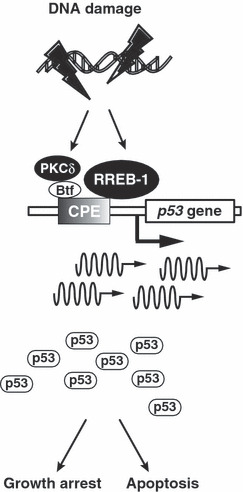
DNA damage‐induced transcriptional regulation of p53 gene by PKCδ and Btf or RREB‐1. Upon exposure to genotoxic stress, activated PKCδ induces Btf for co‐occupancy to CPE‐p53, thereby up‐regulating the expression of p53 at mRNA levels. p53 transcription is also induced by RREB‐1 via CPE‐p53 in response to DNA damage.
Post‐translational Modifications that Influence p53‐mediated Apoptosis
Accumulating studies have revealed that some of post‐translational modifications are specifically linked to the apoptotic function of p53 (Fig. 2). Exploiting pathways that specifically enhance p53‐mediated apoptosis have been a goal of cancer therapeutics. Some of the better‐understood examples from this subset will be discussed below.
Phosphorylation of serine at 46. Most important modification for the induction of apoptosis is the phosphorylation at serine 46 (Ser46). Enhanced apoptosis occurs as a result of this phosphorylated form of p53 selectively activating pro‐apoptotic target gene transcription.( 42 ) In response to UV irradiation, Ser46 phosphorylation is mediated by homeodomain‐interacting protein kinase 2.( 43 , 44 ) By contrast, little was known about Ser46 kinase(s) upon exposure to genotoxic stress. Recently, we found that dual‐specificity tyrosine‐phosphorylation‐regulated kinase 2 (DYRK2) has the characteristics of an in vitro direct Ser46 kinase.( 45 ) Moreover, our results demonstrated that DYRK2 phosphorylates p53 at Ser46 in cells exposed to genotoxic stress. Significantly, DYRK2 phosphorylation of Ser46 was associated with the induction of apoptosis following DNA damage (Fig. 4). These findings provided support a novel signaling mechanism in which phosphorylation of p53 at Ser46 by DYRK2 regulates apoptotic cell death in response to DNA damage. However, a previous study demonstrated that DYRK2 is predominantly expressed in the cytoplasm.( 46 , 47 ) We confirmed the cytoplasmic localization of DYRK2 in unstimulated cells. Furthermore, we found that DYRK2 translocates from the cytoplasm into the nucleus in response to DNA damage. The mechanism for nuclear targeting of DYRK2 is, at present, not known. Importantly, however, nuclear translocation may be required for efficient phosphorylation of p53 at Ser46 and, therefore it is conceivable that Ser46 phosphorylation occurs in relatively later periods following genotoxic stress.( 42 , 48 ) Indeed, our findings that nuclear re‐distribution of DYRK2 coincides with Ser46 phosphorylation of nuclear p53 further support the mechanism in which nuclear targeting of DYRK2 is, at least in part, required for sufficient phosphorylation of p53 at Ser46 in the nucleus.( 45 , 49 ) Importantly, upon exposure to genotoxic stress, p53DINP1 is expressed and then recruits a kinase(s) to p53 that specifically phosphorylates Ser46.( 48 ) Our recent data also showed that PKCδ is, presumably indirectly, involved in phosphorylation of p53 at Ser46.( 50 ) PKCδ‐mediated phosphorylation was required for the interaction of PKCδ with p53. The results also demonstrated that p53DINP1 associates with PKCδ upon exposure to genotoxic agents. In concert with these results, PKCδ potentiates p53‐dependent apoptosis by affecting Ser46 phosphorylation in response to genotoxic stress. These findings indicate that PKCδ regulates p53 to induce apoptotic cell death in the cellular response to DNA damage.( 50 ) Moreover, we recently found that PKCδ regulates MDM2 expression by controlling Akt‐mediated phosphorylation. As a result, PKCδ could control p53 expression indirectly by modulating MDM2 function in response to DNA damage (Hew HC, et al. unpublished observation). As mentioned above, Ser46 phosphorylation occurs in the later stages of p53 activation and influences the response by specifically promoting the induction of the apoptotic gene p53AIP1.( 42 ) This is accompanied by downregulation of p21 expression, ultimately resulting in p53‐dependent apoptosis. One possible mechanism for this enhancement is that phosphorylation of Ser46 enables prolyl isomerase Pin1‐mediated conformational change and dissociation of p53 from the inhibitor of apoptosis, iASPP, thereby promoting cell death.( 51 ) The isomerization of p53 by Pin1 facilitates the binding and acetylation of p53 by the acetyltransferase p300.( 51 ) However, the interaction with Pin1 is not sufficient to explain the effect of Ser46 phosphorylation on p53 function, since Pin1 loss also diminishes the ability of p53 to bind to the promoters of the non‐apoptotic target genes p21 and MDM2.( 52 ) Further studies are definitely required toward a better understanding of how Ser46 phosphorylation confers the ability of p53 to specifically transactivate pro‐apoptotic genes and to induce apoptosis. Nevertheless, a recent study demonstrated that the expression of a p53‐46F mutant, in which Ser46 is replaced with phenylalanine, induces specific p53‐target genes associated with apoptosis, including Noxa, p53AIP1, and p53RFP.( 53 ) Moreover, transduction of the p53‐46F enhanced apoptosis and suppressed tumor growth.( 53 ) These findings thus provide a model in which gene‐transfer of the p53‐46F mutant contributes to a more effective strategy for cancer therapy. Although it is unclear whether the p53‐46F is mimicked to the phosphorylation of Ser46, modification of Ser46 may be of importance for the possibility to develop a new therapeutic strategy based on the ‘Super‐p53′ against cancer.
Figure 4.
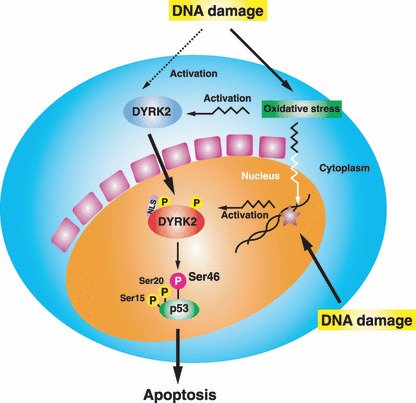
A proposed model for DYRK2‐mediated apoptosis in the cellular response to DNA damage. Upon exposure to genotoxic stress, p53 is stabilized and activated by phosphorylation at Ser15 and Ser20. Cytoplasmic DYRK2 is activated and targeted to the nucleus, and then phosphorylates p53 at Ser46. Ser46 phosphorylation of p53 triggers induction of apoptosis by up‐regulation of apoptosis‐related gene expression, such as p53AIP1.
Modifications of lysines at 320 and 373. Lys320 of p53 can be modified independently by both acetylation and ubiquitination to influence promoter selectivity. p53 transcriptional activity and growth arrest function is enhanced by acetylation at Lys320 by the acetyltransferase PCAF.( 54 , 55 ) On the other hand, the E3 ligase E4F1 ubiquitinates p53 at Lys320, and specifically increases the activation of cell‐cycle‐arrest genes, such as p21, Gadd45, and cyclin G1 while the expression of apoptotic targets remains unchanged.( 56 ) Chromatin immunoprecipitation experiments demonstrate that Lys320‐ubiquitylated p53 is bound to the p21 gene promoter, but not to that of the apoptotic target gene Noxa. Similarly, acetylation of Lys320 following drug‐induced DNA‐damage promotes cell survival, with Lys320‐acetylated p53 binding more efficiently to the p21 promoter than does non‐acetylated p53.( 57 ) Interestingly, for the Lys320 equivalent in mice, Lys317, mutation to an arginine leads to p53‐mediated transcription of pro‐apoptotic target genes, such as Puma, and Noxa.( 58 ) These observations are consistent with a loss of E4F1‐dependent ubiquitination of Lys320, which facilitates growth arrest.( 56 ) Furthermore, FBXO11‐mediated neddylation of human p53 at Lys320 represses p53 activity and therefore the observations made in p53K317R mice are also representative of a loss of neddylation.( 59 ) Thus, the increased apoptotic activity of mouse p53K317R may be explained by impaired acetylation, neddylation, or ubiquitination at this residue. In contrast, acetylation of Lys373, catalyzed by p300 and/or CBP, contributes to increased phosphorylation of N‐terminal residues of p53, and increased ability to transactivate lower affinity binding sites including the pro‐apoptotic target genes, such as PIG3, Bax, and p53AIP1.( 57 ) These results are consistent with a model whereby acetylation at Lys320 and Lys373 exerts as a sensor system that enables p53 to determine between growth arrest and cell death, and to coordinate gene expression patterns appropriately following DNA damage.
Acetylation of lysine at 120. p53‐dependent apoptosis is also specifically enhanced after DNA damage via acetylation at Lys120 by the MYST family acetyltransferases MOF and TIP60. Lys120 lies in the DNA‐binding domain of p53, and its acetylation leads to increased recruitment of p53 specifically to pro‐apoptotic target genes, such as Puma and Bax, suggesting that this modification alone can influence how p53 responds to the DNA‐damaging signals. This modification seems to be required for p53‐dependent apoptosis, as mutants that can no longer be modified in this way exhibit impaired apoptotic activity while maintaining the proper regulation of Mdm2 and growth‐arrest genes.( 60 , 61 ) Collectively, these data indicate that a defined p53 modification can be linked to a specific cellular outcome.
Concluding Remarks
The molecular basis for how p53 induces apoptosis has been extensively studied; however, the relative contribution of each downstream effecter is still to be explored. In this regard, recent studies have shed some light on post‐translational modifications and factors that can influence apoptosis whereas the answers to many questions still need to be completed. Moreover, through in vivo experimental approach, we need to verify the biological significance of each modification site both in p53 wild‐type and mutant. If we could understand the whole picture of p53 modification in apoptosis, it would be then possible to develop highly effective and specific strategies for the cancer treatment.
Acknowledgments
This work was supported by grants from the Ministry of Education, Science and Culture of Japan, Takeda Science Foundation, and Kowa Life Science Foundation.
References
- 1. Yoshida K. Nuclear trafficking of pro‐apoptotic kinases in response to DNA damage. Trends Mol Med 2008; 14: 305–13. [DOI] [PubMed] [Google Scholar]
- 2. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253: 49–53. [DOI] [PubMed] [Google Scholar]
- 3. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage‐induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91: 325–34. [DOI] [PubMed] [Google Scholar]
- 4. Oren M, Damalas A, Gottlieb T et al. Regulation of p53: intricate loops and delicate balances. Biochem Pharmacol 2002; 64: 865–71. [DOI] [PubMed] [Google Scholar]
- 5. Donehower LA, Harvey M, Slagle BL et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356: 215–21. [DOI] [PubMed] [Google Scholar]
- 6. Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358: 15–6. [DOI] [PubMed] [Google Scholar]
- 7. Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene 2007; 26: 1306–16. [DOI] [PubMed] [Google Scholar]
- 8. Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene 2005; 24: 2899–908. [DOI] [PubMed] [Google Scholar]
- 9. Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem 1999; 274: 36031–4. [DOI] [PubMed] [Google Scholar]
- 10. Appella E, Anderson CW. Post‐translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 2001; 268: 2764–72. [DOI] [PubMed] [Google Scholar]
- 11. Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol 2001; 13: 332–7. [DOI] [PubMed] [Google Scholar]
- 12. Kubbutat MH, Vousden KH. Keeping an old friend under control: regulation of p53 stability. Mol Med Today 1998; 4: 250–6. [DOI] [PubMed] [Google Scholar]
- 13. Chernov MV, Bean LJ, Lerner N, Stark GR. Regulation of ubiquitination and degradation of p53 in unstressed cells through C‐terminal phosphorylation. J Biol Chem 2001; 276: 31819–24. [DOI] [PubMed] [Google Scholar]
- 14. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432: 307–15. [DOI] [PubMed] [Google Scholar]
- 15. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis ‐ the p53 network. J Cell Sci 2003; 116: 4077–85. [DOI] [PubMed] [Google Scholar]
- 16. Chipuk JE, Green DR. Dissecting p53‐dependent apoptosis. Cell Death Differ 2006; 13: 994–1002. [DOI] [PubMed] [Google Scholar]
- 17. Meulmeester E, Jochemsen AG. p53: a guide to apoptosis. Curr Cancer Drug Targets 2008; 8: 87–97. [DOI] [PubMed] [Google Scholar]
- 18. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995; 80: 293–9. [DOI] [PubMed] [Google Scholar]
- 19. Oda E, Ohki R, Murasawa H et al. Noxa, a BH3‐only member of the Bcl‐2 family and candidate mediator of p53‐induced apoptosis. Science 2000; 288: 1053–8. [DOI] [PubMed] [Google Scholar]
- 20. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7: 683–94. [DOI] [PubMed] [Google Scholar]
- 21. Robles AI, Bemmels NA, Foraker AB, Harris CC. APAF‐1 is a transcriptional target of p53 in DNA damage‐induced apoptosis. Cancer Res 2001; 61: 6660–4. [PubMed] [Google Scholar]
- 22. Li YZ, Lu DY, Tan WQ, Wang JX, Li PF. p53 initiates apoptosis by transcriptionally targeting the antiapoptotic protein ARC. Mol Cell Biol 2008; 28: 564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yao H, Li P, Venters BJ et al. Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J Biol Chem 2008; 283: 20060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuribayashi K, Krigsfeld G, Wang W et al. TNFSF10 (TRAIL), a p53 target gene that mediates p53‐dependent cell death. Cancer Biol Ther 2008; 7: 2034–8. [DOI] [PubMed] [Google Scholar]
- 25. Maecker HL, Koumenis C, Giaccia AJ. p53 promotes selection for Fas‐mediated apoptotic resistance. Cancer Res 2000; 60: 4638–44. [PubMed] [Google Scholar]
- 26. Wolff S, Erster S, Palacios G, Moll UM. p53′s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res 2008; 18: 733–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta 2009; 1787: 414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell 2007; 130: 638–50. [DOI] [PubMed] [Google Scholar]
- 29. Hermeking H. p53 enters the microRNA world. Cancer Cell 2007; 12: 414–8. [DOI] [PubMed] [Google Scholar]
- 30. He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network – another piece in the tumour‐suppression puzzle. Nat Rev Cancer 2007; 7: 819–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abbas T, White D, Hui L, Yoshida K, Foster DA, Bargonetti J. Inhibition of human p53 basal transcription by down‐regulation of protein kinase Cdelta. J Biol Chem 2004; 279: 9970–7. [DOI] [PubMed] [Google Scholar]
- 32. Liu H, Lu ZG, Miki Y, Yoshida K. Protein kinase C delta induces transcription of the TP53 tumor suppressor gene by controlling death‐promoting factor Btf in the apoptotic response to DNA damage. Mol Cell Biol 2007; 27: 8480–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hansen LA, Monteiro‐Riviere NA, Smart RC. Differential down‐regulation of epidermal protein kinase C by 12‐O‐tetradecanoylphorbol‐13‐acetate and diacylglycerol: association with epidermal hyperplasia and tumor promotion. Cancer Res 1990; 50: 5740–5. [PubMed] [Google Scholar]
- 34. Fournier A, Murray AW. Application of phorbol ester to mouse skin causes a rapid and sustained loss of protein kinase C. Nature 1987; 330: 767–9. [DOI] [PubMed] [Google Scholar]
- 35. Lu Z, Hornia A, Jiang YW, Zang Q, Ohno S, Foster DA. Tumor promotion by depleting cells of protein kinase C delta. Mol Cell Biol 1997; 17: 3418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reddig PJ, Dreckschmidt NE, Ahrens H et al. Transgenic mice overexpressing protein kinase C delta in the epidermis are resistant to skin tumor promotion by 12‐O‐tetradecanoylphorbol‐13‐acetate. Cancer Res 1999; 59: 5710–8. [PubMed] [Google Scholar]
- 37. Magnelli L, Cinelli M, Chiarugi V. Phorbol esters attenuate the expression of p53 in cells treated with doxorubicin and protect TS‐P53/K562 from apoptosis. Biochem Biophys Res Commun 1995; 215: 641–5. [DOI] [PubMed] [Google Scholar]
- 38. Ghosh JC, Suzuki K, Kodama S, Watanabe M. Effects of protein kinase inhibitors on the accumulation kinetics of p53 protein in normal human embryo cells following X‐irradiation. J Radiat Res (Tokyo) 1999; 40: 23–37. [DOI] [PubMed] [Google Scholar]
- 39. Raman V, Martensen SA, Reisman D et al. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000; 405: 974–8. [DOI] [PubMed] [Google Scholar]
- 40. Stuart ET, Haffner R, Oren M, Gruss P. Loss of p53 function through PAX‐mediated transcriptional repression. EMBO J 1995; 14: 5638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu H, Hew HC, Lu ZG, Yamaguchi T, Miki Y, Yoshida K. DNA damage signalling recruits RREB‐1 to the p53 tumour suppressor promoter. Biochem J 2009; 422: 543–51. [DOI] [PubMed] [Google Scholar]
- 42. Oda K, Arakawa H, Tanaka T et al. p53AIP1, a potential mediator of p53‐dependent apoptosis, and its regulation by Ser‐46‐phosphorylated p53. Cell 2000; 102: 849–62. [DOI] [PubMed] [Google Scholar]
- 43. Hofmann TG, Moller A, Sirma H et al. Regulation of p53 activity by its interaction with homeodomain‐interacting protein kinase‐2. Nat Cell Biol 2002; 4: 1–10. [DOI] [PubMed] [Google Scholar]
- 44. D’Orazi G, Cecchinelli B, Bruno T et al. Homeodomain‐interacting protein kinase‐2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 2002; 4: 11–9. [DOI] [PubMed] [Google Scholar]
- 45. Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 2007; 25: 725–38. [DOI] [PubMed] [Google Scholar]
- 46. Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. Sequence characteristics, subcellular localization, and substrate specificity of DYRK‐related kinases, a novel family of dual specificity protein kinases. J Biol Chem 1998; 273: 25893–902. [DOI] [PubMed] [Google Scholar]
- 47. Yoshida K. Role for DYRK family kinases on regulation of apoptosis. Biochem Pharmacol 2008; 76: 1389–94. [DOI] [PubMed] [Google Scholar]
- 48. Okamura S, Arakawa H, Tanaka T et al. p53DINP1, a p53‐inducible gene, regulates p53‐dependent apoptosis. Mol Cell 2001; 8: 85–94. [DOI] [PubMed] [Google Scholar]
- 49. Taira N, Yamamoto H, Yamaguchi T, Miki Y, Yoshida K. ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. J Biol Chem 2010; 285: 4909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoshida K, Liu H, Miki Y. Protein Kinase C delta regulates Ser46 phos‐phorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J Biol Chem 2006; 281: 5734–40. [DOI] [PubMed] [Google Scholar]
- 51. Mantovani F, Tocco F, Girardini J et al. The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat Struct Mol Biol 2007; 14: 912–20. [DOI] [PubMed] [Google Scholar]
- 52. Zheng H, You H, Zhou XZ et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002; 419: 849–53. [DOI] [PubMed] [Google Scholar]
- 53. Nakamura Y, Futamura M, Kamino H, Yoshida K, Nakamura Y, Arakawa H. Identification of p53‐46F as a super p53 with an enhanced ability to induce p53‐dependent apoptosis. Cancer Sci 2006; 97: 633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu L, Scolnick DM, Trievel RC et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol 1999; 19: 1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barlev NA, Liu L, Chehab NH et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell 2001; 8: 1243–54. [DOI] [PubMed] [Google Scholar]
- 56. Le Cam L, Linares LK, Paul C et al. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 2006; 127: 775–88. [DOI] [PubMed] [Google Scholar]
- 57. Knights CD, Catania J, Di Giovanni S et al. Distinct p53 acetylation cassettes differentially influence gene‐expression patterns and cell fate. J Cell Biol 2006; 173: 533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chao C, Wu Z, Mazur SJ et al. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol 2006; 26: 6859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abida WM, Nikolaev A, Zhao W, Zhang W, Gu W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J Biol Chem 2007; 282: 1797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sykes SM, Mellert HS, Holbert MA et al. Acetylation of the p53 DNA‐binding domain regulates apoptosis induction. Mol Cell 2006; 24: 841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tang Y, Luo J, Zhang W, Gu W. Tip60‐dependent acetylation of p53 modulates the decision between cell‐cycle arrest and apoptosis. Mol Cell 2006; 24: 827–39. [DOI] [PubMed] [Google Scholar]