

Abstract
Class I phosphatidylinositol 3 kinases (PI3K) phosphorylate phosphatidylinositol 4,5‐bisphosphate to generate phosphatidylinositol 3,4,5‐trisphosphate. These molecules play an important role in fundamental cellular responses. Four isoforms of class I PI3K are known to have different functions, and abnormalities in their activities have been related to various diseases such as cancer and inflammation. We previously identified a novel PI3K inhibitor, ZSTK474, which showed potent antitumor activity in vivo against a human cancer xenograft without observable toxicity. However, the mode of its molecular action was not investigated in detail. Our previous study only suggested that ZSTK474 possibly competes with ATP for the ATP‐binding pocket of PI3Kγ. In the present study, we have used an in vitro homogenous time‐resolved fluorescence kinase assay to examine whether ZSTK474 is indeed an ATP‐competing inhibitor of PI3K, and also to determine whether the inhibitory activity of ZSTK474 was isoform‐specific. Lineweaver–Burk plot analysis revealed that ZSTK474 inhibits all four PI3K isoforms in an ATP‐competitive manner. Among all of the PI3K isoforms, PI3Kδ was inhibited most potently by ZSTK474 with a Ki of 1.8 nM, and the other isoforms were inhibited at higher doses. We have also used a kinase activity ELISA to determine whether ZSTK474 inhibits mammalian target of rapamycin, a key kinase acting downstream of PI3K to promote protein synthesis and cell proliferation. Even at a concentration of 100 µM, ZSTK474 inhibited mammalian target of rapamycin activity rather weakly. These results indicate that ZSTK474 is an ATP‐competitive pan‐class I PI3K inhibitor. (Cancer Sci 2007; 98: 1638–1642)
Phosphatidylinositol 3‐kinases (PI3K) are ubiquitously expressed lipid kinases that phosphorylate phosphoinositides at the 3‐hydroxyl of the inositol ring.( 1 ) The products of these enzymes serve as second messengers with key roles in fundamental cellular responses such as proliferation, survival, motility and metabolism.( 2 , 3 ) The PI3K were classified into three types based on their primary structure and substrate specificity. The class I PI3K phosphorylate phosphatidylinositol 4,5‐bisphosphate (PIP2) to generate phosphatidylinositol 3,4, 5‐trisphosphate (PIP3). This class was further divided into subclasses IA and IB based on the regulatory subunit. The class IA kinases are heterodimers composed of a regulatory subunit p85 and a catalytic subunit p110. The p85 binds to various tyrosine kinases to activate p110 and downstream molecules such as Akt. The catalytic subunit of class IA consists of the three isoforms p110α, ‐β and ‐δ. Class IB PI3K contains the catalytic subunit p110γ and the regulatory subunit p101, which is mainly activated by G‐protein‐coupled receptors.( 4 , 5 )
Previous studies showed that the four class I PI3K isoforms possess specialized functions. PI3Kα, which is known to play an important role in tumorigenesis because a high frequency of mutations was detected in the PIK3CA gene encoding the catalytic subunit p110α in human cancers,( 6 , 7 , 8 ) is also thought to be involved in insulin signaling and glucose metabolism.( 9 ) PI3Kβ was demonstrated to activate platelets and therefore might have a role in the development of thrombosis‐related diseases.( 10 ) An increasing body of evidence suggests that PI3Kγ and PI3Kδ play key roles in inflammation and the immune system.( 11 , 12 , 13 , 14 , 15 , 16 ) Therefore, in recent years, development of isoform‐specific inhibitors has attracted much attention among researchers and pharmaceutical companies, as they are expected to become novel drug candidates specifically for the treatment of cancer (PI3Kα), thrombosis (PI3Kβ) and inflammatory diseases (PI3Kδ and PI3Kγ) with minimal side‐effects.
Kinases, including PI3K, regulate signal transduction by phosphorylation.( 17 , 18 ) Despite having diverse primary sequences, a homologous and well‐defined ATP‐binding site is present in all kinases.( 19 ) Most of the kinase inhibitors under development are competitive inhibitors of ATP and target the ATP‐binding pocket. Understanding the mode of action of a competitive inhibitor helps in identifying the key functional groups in its chemical structure, which in turn can lead to synthesis of a more effective drug candidate by further modification of the inhibitor structure. However, by using the competitive inhibitor as a tool to investigate the structure and functions of the corresponding kinase, one could obtain some new information about the ATP‐binding site.
Several PI3K inhibitors were reported to show anticancer effects in vivo. As first‐generation PI3K inhibitors, LY294002( 20 ) and wortmannin( 21 ) failed to enter clinical trials because they caused dermal( 22 ) and liver toxicity,( 23 ) respectively. PX‐866, an analog of wortmannin, showed decreased liver toxicity,( 23 ) but caused hyperglycemia and decreased glucose tolerance.( 24 ) Another PI3K inhibitor, PI‐103, when administered intraperitoneally showed significant anticancer activity but no observable toxicity.( 25 )
Recently, we developed a novel PI3K inhibitor, ZSTK474, which showed potent antitumor activity in vivo against a human cancer xenograft without observable toxicity when administered orally.( 26 ) In our previous study, we also analyzed the inhibition activities of ZSTK474 against 139 protein kinases and showed that ZSTK474 specifically inhibited PI3K.( 26 ) However, we did not investigate its selectivity among class I PI3K isoforms in detail. Because different PI3K isoforms play various functional roles, it thus remained unclear whether the excellent in vivo efficacy and low toxicity of ZSTK474 was due to its possible isoform specificity. Our previous results suggested that ZSTK474 possibly competes with ATP to bind to the ATP‐binding pocket of PI3Kγ.( 26 ) The purpose of the present study is to verify this possibility and to examine whether ZSTK474 is an isoform‐specific inhibitor. To measure the PI3K activity, we used a homogenous time‐resolved fluorescence (HTRF) assay,( 27 , 28 ) instead of the traditional radioactive kinase assay.( 20 , 21 , 23 , 26 ) The HTRF assay was originally developed for high‐throughput screening of kinase inhibitors and has been applied to enzyme kinetic analysis in recent years.( 27 , 28 ) Compared with the radioactive assay, HTRF assay is homogeneous, more reproducible and easier to handle.
In addition, we examined whether ZSTK474 inhibited the mammalian target of rapamycin (mTOR), a serine–threonine kinase that acts downstream of PI3K to promote cell growth and proliferation by activating effectors such as p70S6K and 4EBP1.( 29 ) This was primarily to further verify the specificity of ZSTK474 as mTOR contains a conserved PI3K domain( 30 ) and is known to be inhibited by several PI3K inhibitors such as wortmannin.( 31 )
In the present study, we demonstrate that ZSTK474 is an ATP‐competitive inhibitor of all four PI3K isoforms. Our results indicate that ZSTK474 is a pan‐PI3K inhibitor, which inhibits the PI3Kδ isoform most potently and inhibits the other PI3K isoforms at higher doses. Furthermore, we demonstrate that ZSTK474 is a much weaker inhibitor of mTOR than the PI3K isoforms.
Materials and Methods
Materials. ZSTK474 was provided by Zenyaku Kogyo Co. (Tokyo, Japan). LY294002, ATP disodium salt and dl‐dithiothreitol were purchased from Sigma (St Louis, MO, USA). The PI3‐Kinase (human) HTRF Assay Kit and PI3‐kinase p110α, ‐β, ‐δ and ‐γ were purchased from Upstate (now Millipore, Billerica, MA, USA). The K‐LISA mTOR Activity Kit was purchased from EMD Biosciences (San Diego, CA, USA). An EnVision 2103 Multilabel Reader (PerkinElmer, Wellesley, MA, USA) was used to measure the HTRF signal. The Benchmark Plus microplate spectrophotometer was from BioRad (Hercules, CA, USA).
Phosphatidylinositol 3‐kinase HTRF assay. The principle of the PI3K HTRF assay has been described previously.( 27 ) Briefly, PI3K catalyzes the phosphorylation of PIP2 to PIP3 in the presence of ATP. The PIP3 product is detected by displacement of biotinylated PIP3 (biotin‐PIP3) from an energy transfer complex consisting of Europium‐labeled anti‐glutathione S‐transferase (GST) antibody, a GST‐tagged receptor for phosphoinositide‐1 (GRP1) pleckstrin homology (PH) domain, biotin‐PIP3 and streptavidin–allophycocyanin (APC). Excitation of Europium in the complex results in an energy transfer to the APC. Displacement of biotin‐PIP3 from the complex leads to a loss of energy transfer and a corresponding decrease in HTRF signal. The decreased signal is proportional to the amount of PIP3 produced in the reaction and therefore can be used to monitor the PI3K activity.
The kinase reaction was carried out in a reaction mixture of 20 µL. Each class I PI3K isoform protein was incubated in the assay buffer containing 10 µM PIP2 and ATP (concentration as required) in a 384‐well plate at room temperature. The reaction was initiated by the addition of ATP, and stopped by adding 5 µL stop solution containing ethylenediaminetetraacetic acid and biotin‐PIP3 after 20 min. Then, 5 µL detection buffer was added, which contained the Europium‐labeled anti‐GST antibody, GST‐tagged GRP1 PH domain and streptavidin–APC. After incubation at room temperature for 14 h, the plate was read using the EnVision 2103 Multilabel Reader in time‐resolved fluorescence mode and the HTRF signal was determined according to the formula:
| HTRF signal = 10 000 × (emission at 665 nm/emission at 620 nm). |
Enzyme kinetic studies. The linear phase of each kinetic reaction was defined at the respective enzyme amount (0.05, 0.1, 0.12 and 1 µg/mL for PI3Kα, ‐β, ‐δ and ‐γ, respectively) and reaction time (20 min). PI3K activity was assayed at various concentrations of ATP (5, 10, 25, 50, 100 µM) in the presence of increasing concentrations of ZSTK474. A Lineweaver–Burk plot was developed by plotting 1/v (the inverse of v, where v was obtained by subtracting the HTRF signal of the kinase test sample from the HTRF signal of the minus‐enzyme control) versus 1/[ATP] (the inverse of the ATP concentration). For the minus‐enzyme control, PIP2 was incubated with ATP in the absence of kinase. To determine the Ki value (inhibition constant) of ZSTK474 for each PI3K isoform, the slope of the respective Lineweaver–Burk plot was replotted against the ZSTK474 concentration. The Ki values were calculated by analysis using GraphPad Prism 4 (GraphPad Software, San Diego, CA, USA) and by fitting the curve to:
| v = Vmax[ATP]/((Km(1 + [ZSTK474]/Ki) + [ATP]), |
where v is the reaction velocity, Vmax is the maximal velocity, Km is the Michaelis constant, and [ZSTK474] and [ATP] are the concentrations of ZSTK474 and ATP, respectively.
Determination of the IC50 of ZSTK474 and LY294002 for each class I PI3K isoform. Each PI3K isoform protein was incubated with a series of concentrations of ZSTK474 and LY294002 in the presence of 10 µM ATP. Other reaction conditions were as described above in the section ‘Enzyme kinetic studies’. The PI3K activity (% control) of a certain sample was calculated using the following formula:
| PI3K activity (% control) = (sample – minus‐enzyme control)/(plus‐enzyme control – minus‐enzyme control) × 100. |
For the plus‐enzyme control, the kinase was incubated with PIP2 and ATP in the absence of inhibitor. In the case of the minus‐enzyme control, PIP2 was incubated with ATP without kinase and inhibitor. For each PI3K isoform, the kinase activity was plotted as a function of the inhibitor concentration (ZSTK474 and LY294002). The IC50 values were calculated by fitting these data to a logistic curve using GraphPad Prism 4 software.
K‐LISA mTOR assay. The K‐LISA mTOR assay is an enzyme‐linked immunosorbent assay (ELISA)‐based method that uses the p70S6K–GST fusion protein as the mTOR substrate. This substrate is first bound to a glutathione‐coated 96‐well plate, and then mTOR‐containing samples are incubated with ATP in the wells where active mTOR phosphorylates p70S6K at Thr389 (T389). To detect the phosphorylated substrate, the wells are first treated with anti‐p70S6K‐T389 antibody, followed by horseradish peroxidase (HRP)‐conjugated antibody and 3,3′,5,5′‐tetramethyl benzidine (TMB) substrate. Because the product of the HRP‐catalyzed reaction shows maximum absorbance at 450 nm, the activity of mTOR can be evaluated from the absorbance difference at 450 nm and 595 nm (background absorbance).
The assay was carried out according to the manufacturer's protocol. Briefly, 100 µL of recombinant p70S6K–GST fusion protein was preincubated at room temperature in the glutathione‐coated 96‐well plate and then removed 1 h later. Fifty microliters of ice‐chilled rat brain‐derived mTOR kinase in the presence of dimethylsulfoxide (DMSO; control), 5 µM wortmannin (positive control) or various concentrations of ZSTK474 was added to each well. The reaction was initiated by the addition of 50 µL kinase assay buffer containing 100 µM ATP, and incubated for 30 min at 30°C. After being washed, the plate was treated first with 100 µL of anti‐p70S6K‐T389 for 1 h and then with 100 µL of HRP‐conjugated antibody for 1 h to detect the T389‐phosphorylated p70S6K. Finally, 100 µL of TMB was added as HRP substrate and incubated for 20 min. The reaction was then stopped by the addition of 100 µL ELISA stop solution containing 2.5 N H2SO4. Absorbance was measured at 450 nm and 595 nm using a Benchmark Plus microplate spectrophotometer.
Results
Competitive inhibition of class I PI3K isoforms by ZSTK474. ZSTK474 was previously reported to inhibit PI3K at nanomolar concentrations, and the molecular modeling analysis suggested that ZSTK474 might be an ATP‐competitive inhibitor that binds to the ATP‐binding pocket of PI3Kγ.( 26 ) To verify this notion, we used an in vitro assay to measure the PI3K activity at various ATP concentrations in the presence of increasing concentrations of ZSTK474. As shown in Fig. 1a, Lineweaver–Burk plot analysis revealed that ZSTK474 behaved as an ATP‐competitive inhibitor for all PI3K isoforms, as for each isoform the plots (straight lines) intersected on the 1/v axis. The Ki values of ZSTK474 for each PI3K isoform were shown by replotting the slope of each Lineweaver–Burk plot versus the respective ZSTK474 concentration (Fig. 1b). To determine the Ki values accurately, the data were best‐fitted to v = Vmax[ATP]/(Km(1 +[ZSTK474]/Ki) + [ATP]) using the GraphPad Prism 4 software program. As a result, the Ki values for PI3Kα, ‐β, ‐δ and ‐γ were determined as 6.7, 10.4, 1.8 and 11.7 nM, respectively.
Figure 1.
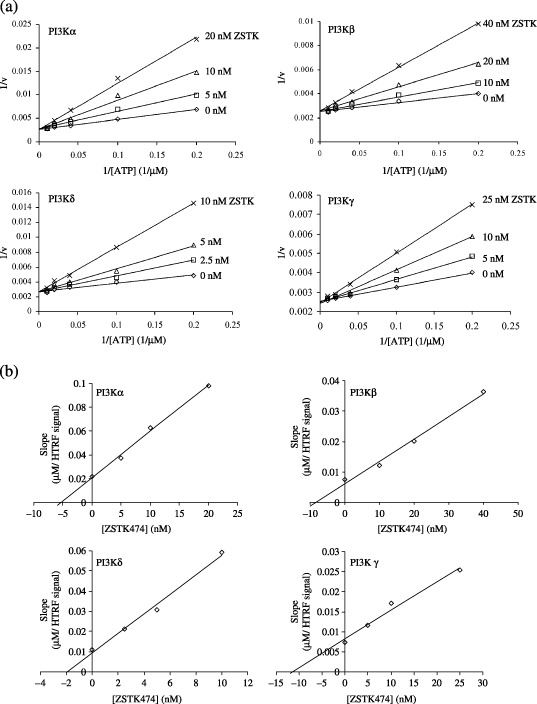
Inhibition mode of ZSTK474 against class I phosphatidylinositol 3‐kinase (PI3K) isoforms. (a) Lineweaver–Burk plot, 1/v versus 1/[ATP]. An inhibition assay was carried out using varying concentrations of ATP, fixed concentrations of a PI3K isoform protein (0.05, 0.1, 0.12 and 1 µg/mL for PI3Kα, ‐β, ‐δ and ‐γ, respectively) and 10 µM phosphorylate phosphatidylinositol 4,5‐bisphosphate (PIP2) in the absence or presence of increasing concentrations of ZSTK474. The homogenous time‐resolved fluorescence (HTRF) signal was measured using an EnVision 2103 Multilabel Reader. For the Lineweaver–Burk plot, 1/v (the inverse of v, where v is obtained by subtracting the HTRF signal of the sample from that of the minus‐enzyme control) was plotted versus 1/[ATP] (the inverse of ATP concentration). For the minus‐enzyme control, PIP2 was incubated with ATP in the absence of kinase. Each experiment was carried out in triplicate and the results shown are representative of two or three independent experiments. ZSTK474 behaved as an ATP‐competitive inhibitor for all PI3K isoforms because for each isoform, the 1/v versus 1/[ATP] plots at different ZSTK474 concentrations intersected on the 1/v axis. (b) Replot of the Lineweaver–Burk plot versus ZSTK474 concentration. The slopes, obtained from the Lineweaver–Burk plot (a), were plotted against ZSTK474 concentration. The Ki value of ZSTK474 for each PI3K isoform was determined from the intersection of the plot on the ZSTK474 concentration axis.
Comparison of inhibition of class I PI3K isoforms by ZSTK474 and LY294002. LY294002, a typical PI3K inhibitor, also competitively binds to the ATP‐binding pocket.( 20 ) Therefore, we compared the inhibition activities of ZSTK474 and LY294002 for each PI3K isoform. The dose–response inhibition profiles for both inhibitor are shown in Fig. 2. The IC50 values were calculated using GraphPad Prism 4 by fitting the data to a logistic curve. ZSTK474 was 30‐fold more potent than LY294002 in inhibiting the PI3K isoforms. Both inhibitors inhibited the PI3Kα and ‐δ isoforms more effectively than the PI3Kβ and ‐γ isoforms (Table 1).
Figure 2.
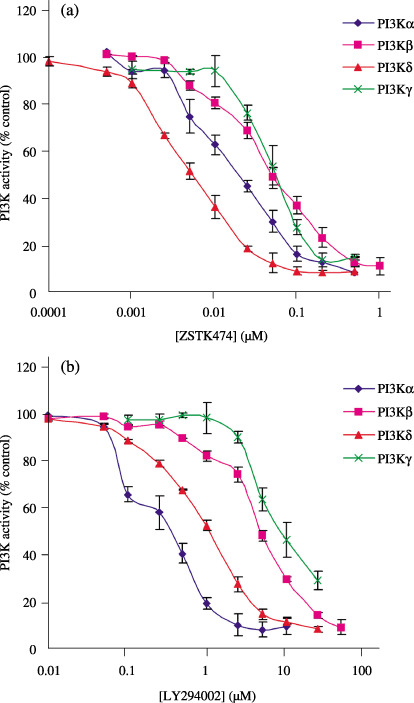
Inhibition profiles of (a) ZSTK474 and (b) LY294002. Each phosphatidylinositol 3‐kinase (PI3K) isoform (PI3Kα, 0.05 µg/mL; PI3Kβ, 0.1 µg/mL; PI3Kδ, 0.12 µg/mL; PI3Kγ, 1 µg/mL) was incubated with various concentrations of ZSTK474 or LY294002 in the presence of 10 µM ATP. The homogenous time‐resolved fluorescence (HTRF) signal was measured using the EnVision 2103 Multilabel Reader. The PI3K activity (% control) of a given sample was calculated by using the following formula: PI3K activity (% control) = ([sample – minus‐enzyme control)/(plus‐enzyme control – minus‐enzyme control]) × 100. The inhibition profile for each inhibitor was obtained by plotting the PI3K activity versus the inhibitor concentration. Data are mean ± SD of three independent experiments each carried out in triplicate.
Table 1.
IC50 values (M) of ZSTK474 and LY294002
| Inhibitor | PΙ3Κα | PI3Kβ | PI3Kδ | PI3Kγ |
|---|---|---|---|---|
| ZSTK474 | 1.6 × 10−8 | 4.4 × 10−8 | 4.6 × 10−9 | 4.9 × 10−8 |
| LY294002 | 5.5 × 10−7 | 1.1 × 10−5 | 1.6 × 10−6 | 1.2 × 10−5 |
PI3K, phosphatidylinositol 3‐kinase.
Inhibition of mTOR by ZSTK474. Inhibition of mTOR by ZSTK474 was investigated using the K‐LISA assay. Wortmannin, which was previously reported to inhibit mTOR,( 31 ) was used as a positive control. As shown in Fig. 3, ZSTK474 did not inhibit mTOR at 0.1 µM, a concentration that is higher than the IC50 for PI3K inhibition; even at a concentration of 100 µM, ZSTK474 inhibited mTOR activity less than 40%, suggesting that ZSTK474 is a much weaker inhibitor for mTOR than for PI3K.
Figure 3.
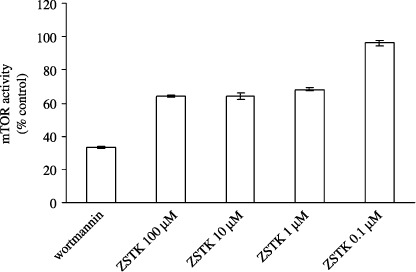
Inhibition of mammalian target of rapamycin (mTOR) by ZSTK474. An enzyme‐linked immunosorbent assay‐based K‐LISA kit was used to measure mTOR activity. Rat brain‐derived mTOR was incubated with 100 µM ATP at 30°C for 30 min in the presence of dimethylsulfoxide (DMSO; control), 5 µM wortmannin or various concentrations of ZSTK474 (0.1, 1, 10 and 100 µM). mTOR activity was measured as the absorbance at 450 nm minus the background absorbance at 595 nm. The activity in the presence of wortmannin or ZSTK474 was expressed as the percentage of the control (DMSO) activity. Data are mean ± SD of three independent experiments carried out in triplicate.
Discussion
In the present study, we demonstrated that ZSTK474 inhibits all four of the class I PI3K isoforms by competing with ATP, which is consistent with our previous prediction that ZSTK474 binds to the ATP‐binding site.( 26 ) This finding suggests that ZSTK474 can be used as a tool to analyze the structure and function of the ATP‐binding site of the class I PI3K isoforms. Additionally, by keeping the structure skeleton intact, the structure of ZSTK474 can be further modified to produce a more effective inhibitor analog. The Ki values determined for the four PI3K isoforms showed that ZSTK474 inhibited the PI3Kδ isoform most effectively with a Ki of 1.8 nM, whereas the other isoforms were inhibited with 4–10‐fold higher Ki values. Therefore, ZSTK474 should be regarded as a pan‐PI3K inhibitor. We also determined the IC50 values for inhibiting the four PI3K isoforms with ZSTK474 and LY294002. The IC50 values of ZSTK474 (16, 44, 4.6 and 49 nM for PI3Kα, ‐β, ‐δ and ‐γ, respectively) were shown to be consistent with the Ki values (6.7, 10.4, 1.8 and 11.7 nM for PI3Kα, ‐β, ‐δ and ‐γ, respectively), which further supported the idea that ZSTK474 inhibited PI3Kδ most potently.
ZSTK474 had excellent antitumor activity in the animal models and lower toxicity( 26 ) than other known PI3 kinase inhibitors such as LY294002 and wortmannin. In the present study, we compared ZSTK474 with LY294002 in an aspect of molecular pharmacology. ZSTK474 and LY294002 showed rather similar profiles in isoform specificity, but they were slightly different in that ZSTK474 most effectively inhibited PI3Kδ whereas LY294002 most effectively inhibited PI3Kα. More clearly, ZSTK474 was at least 30‐fold more effective than LY294002 at inhibiting the PI3K isoforms. In our previous study, ZSTK474 showed little inhibition to casein kinase,( 26 ) to which LY294002 is known to have cross reactivity.( 32 ) These differences may contribute to some extent to the superiority of ZSTK474 to LY294002 in efficacy and toxicity. However, what makes the differences in biological output between ZSTK474 and other PI3K inhibitors remains to be investigated.
Because PI3Kα is known to play an important role in tumorigenesis,( 6 , 7 , 8 ) we originally postulated that the antitumor efficacy of ZSTK474 might relate to the status of PIK3CA, which encodes PI3Kα. Recently, seven cell lines in the NCI‐60 cell line panel were found to express mutant PIK3CA.( 8 ) Our JFCR39 cancer cell line panel( 26 ) shares 26 common cell lines with the NCI‐60 panel; five out of the seven mutant PIK3CA‐expressing cell lines from the NCI‐60 panel are included in the JFCR39 panel. We thus compared the Log GI50 values (GI50 is the concentration to attain 50% growth inhibition of the cancer cells) of ZSTK474 for these five mutant PIK3CA‐expressing cell lines with those for the 21 cell lines that express normal PIK3CA in the JFCR39 panel.( 26 ) Our results showed no significant difference between the two groups (–6.452 versus –6.487), implying that there is no obvious correlation between the efficacy of ZSTK474 and the presence of a PIK3CA mutation in the cancer cells.
Among the four PI3K isoforms, ZSTK474 inhibited the PI3Kδ isoform most strongly. An increasing body of evidence suggests that PI3Kδ may play an essential role in the proliferation of some acute myeloid leukemia cells.( 33 , 34 ) Because ZSTK474 is a comparatively selective inhibitor of PI3Kδ, these tumor types may be therapeutic targets of ZSTK474.
ZSTK474 also inhibited PI3Kα, ‐β and ‐γ at IC50 lower than 50 nM (Table 1). Considering these activities, one may expect other therapeutic efficacies and adverse effects of ZSTK474. PI3Kγ together with ‐δ is well known to play an important role in inflammation and the immune system,( 11 , 12 , 13 , 14 , 15 , 16 ) and an inhibitor of PI3Kγ, AS605240, was indeed developed as an anti‐inflammatory reagent.( 14 , 15 ) Therefore, ZSTK474 may have anti‐inflammatory effects. An inhibitor of PI3Kβ, TGX‐221, was recently developed for the therapy of thrombosis, as PI3Kβ was demonstrated to activate platelets.( 10 ) The effect of ZSTK474 on platelet function is worth investigating. However, the inhibitors of PI3Kα may cause hyperglycemia because PI3Kα reportedly mediates insulin signals and plays a role in preventing hyperglycemia.( 9 ) The effect of ZSTK474 on glucose metabolism is now under investigation.
We have also shown that ZSTK474 inhibited mTOR much less effectively than the PI3K isoforms. As mTOR is a serine–threonine kinase containing a conserved PI3K domain, PI3K inhibitors are expected to be cross reactive to mTOR. In fact, other PI3K inhibitor such as LY294002 and PI‐103 are known to inhibit mTOR significantly.( 25 , 31 ) However, ZSTK474 did not inhibit mTOR. Our previous study showed that ZSTK474 inhibits PI3K more effectively than 139 other protein kinases.( 26 ) Taken together, these results further demonstrate the superior specificity of ZSTK474 to PI3K. The contribution of this selectivity to its reduced toxicity and its higher potency in vivo in animal models remains to be clarified.
In conclusion, we have demonstrated ZSTK474 is an ATP‐competitive inhibitor of all class I PI3K isoforms. ZSTK474 was indicated to be a pan‐class I PI3K inhibitor.
Acknowledgments
We thank Shinichi Yaguchi and his colleagues at Zenyaku Kogyo Co. for providing ZSTK474 and also for their helpful suggestions. This work was partly supported by: a grant from the National Institute of Biomedical Innovation, Japan, to T. Yamori (05‐13); grants‐in‐aid of the Priority Area ‘Cancer’ from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to T. Yamori (18015049); and grants‐in‐aid for Scientific Research (B) from Japan Society for the Promotion of Science to T. Yamori (17390032).
References
- 1. Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol‐3‐phosphate. Nature 1988; 332: 644–6. [DOI] [PubMed] [Google Scholar]
- 2. Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell 1997; 88: 435–7. [DOI] [PubMed] [Google Scholar]
- 3. Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3‐kinase: the key switch mechanism in insulin signalling. Biochem J 1998; 333: 471–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4: 988–1004. [DOI] [PubMed] [Google Scholar]
- 5. Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene‐targeted mice. Trends Biochem Sci 2005; 30: 194–204. [DOI] [PubMed] [Google Scholar]
- 6. Samuels Y, Wang Z, Bardelli A et al . High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. [DOI] [PubMed] [Google Scholar]
- 7. Levine DA, Bogomolniy F, Yee CJ et al . Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res 2005; 11: 2875–8. [DOI] [PubMed] [Google Scholar]
- 8. Whyte DB, Holbeck SL. Correlation of PIK3Ca mutations with gene expression and drug sensitivity in NCI‐60 cell lines. Biochem Biophys Res Commun 2006; 340: 469–75. [DOI] [PubMed] [Google Scholar]
- 9. Knight ZA, Gonzalez B, Feldman ME et al . A pharmacological map of the PI3‐K family defines a role for p110α in insulin signaling. Cell 2006; 125: 733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jackson SP, Schoenwaelder SM, Goncalves I et al . PI 3‐kinase p110β: a new target for antithrombotic therapy. Nat Med 2005; 11: 507–14. [DOI] [PubMed] [Google Scholar]
- 11. Hirsch E, Katanaev VL, Garlanda C et al . Central role for G protein‐coupled phosphoinositide 3‐kinase γ in inflammation. Science 2000; 287: 1049–53. [DOI] [PubMed] [Google Scholar]
- 12. Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC‐β2 and ‐β3 and PI3Kγ in chemoattractant‐mediated signal transduction. Science 2000; 287: 1046–9. [DOI] [PubMed] [Google Scholar]
- 13. Sasaki T, Irie‐Sasaki J, Jones RG et al . Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 2000; 287: 1040–6. [DOI] [PubMed] [Google Scholar]
- 14. Barber DF, Bartolome A, Hernandez C et al . PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med 2005; 11: 933–5. [DOI] [PubMed] [Google Scholar]
- 15. Camps M, Ruckle T, Ji H et al . Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med 2005; 11: 936–43. [DOI] [PubMed] [Google Scholar]
- 16. Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE. Essential role of phosphoinositide 3‐kinase δ in neutrophil directional movement. J Immunol 2003; 170: 2647–54. [DOI] [PubMed] [Google Scholar]
- 17. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298: 1912–34. [DOI] [PubMed] [Google Scholar]
- 18. Hunter T. Signaling – 2000 and beyond. Cell 2000; 100: 113–27. [DOI] [PubMed] [Google Scholar]
- 19. Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell 2002; 109: 275–82. [DOI] [PubMed] [Google Scholar]
- 20. Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3‐kinase, 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294002). J Biol Chem 1994; 269: 5241–8. [PubMed] [Google Scholar]
- 21. Okada T, Sakuma L, Fukui Y, Hazeki O, Ui M. Blockage of chemotactic peptide‐induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidylinositol 3‐kinase. J Biol Chem 1994; 269: 3563–7. [PubMed] [Google Scholar]
- 22. Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3‐kinase inhibitor (LY294002). Clin Cancer Res 2000; 6: 880–6. [PubMed] [Google Scholar]
- 23. Ihle NT, Williams R, Chow S et al . Molecular pharmacology and antitumor activity of PX‐866, a novel inhibitor of phosphoinositide‐3‐kinase signaling. Mol Cancer Ther 2004; 3: 763–72. [PubMed] [Google Scholar]
- 24. Ihle NT, Paine‐Murrieta G, Berggren MI et al . The phosphatidylinositol‐3‐kinase inhibitor PX‐866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A‐549 human non‐small cell lung cancer xenografts. Mol Cancer Ther 2005; 4: 1349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fan QW, Knight ZA, Goldenberg DD et al . A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006; 9: 341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yaguchi S, Fukui Y, Koshimizu I et al . Antitumor activity of ZSTK474, a new phosphatidylinositol 3‐kinase inhibitor. J Natl Cancer Inst 2006; 98: 545–56. [DOI] [PubMed] [Google Scholar]
- 27. Gray A, Olsson H, Batty IH, Priganica L, Downes C. Nonradioactive methods for the assay of phosphoinositide 3‐kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal Biochem 2003; 313: 234–45. [DOI] [PubMed] [Google Scholar]
- 28. Jia Y, Quinn CM, Gagnon AI, Talanian R. Homogeneous time‐resolved fluorescence and its applications for kinase assays in drug discovery. Anal Biochem 2006; 356: 273–81. [DOI] [PubMed] [Google Scholar]
- 29. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17: 596–603. [DOI] [PubMed] [Google Scholar]
- 30. Abraham RT. Phosphatidylinositol 3‐kinase related kinases. Curr Opin Immunol 1996; 8: 412–18. [DOI] [PubMed] [Google Scholar]
- 31. Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC Jr, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3‐kinase inhibitors, wortmannin and LY294002. EMBO J 1996; 15: 5256–67. [PMC free article] [PubMed] [Google Scholar]
- 32. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 2000; 351: 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sujobert P, Bardet V, Cornillet‐Lefebvre P et al . Essential role for the p110 δ isoform in phosphoinositide 3‐kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005; 106: 1063–6. [DOI] [PubMed] [Google Scholar]
- 34. Billottet C, Grandage VL, Gale RE et al . A selective inhibitor of the p110δ isoform of PI 3‐kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 2006; 25: 6648–59. [DOI] [PubMed] [Google Scholar]