

Abstract
Centrosome amplification can be detected in the tissues of p53−/– mice. In contrast, loss of p53 does not induce centrosome amplification in cultured human cells. However, examination of human cancer tissues and cultured cells has revealed a significant correlation between loss or mutational inactivation of p53 and occurrence of centrosome amplification, supporting the notion that p53 mutation alone is insufficient to induce centrosome amplification in human cells, and that additional regulatory mechanisms are involved. It has recently been shown that gamma irradiation of tumor cells induces centrosome amplification. However, the precise mechanism of radiation‐induced centrosome amplification is not fully understood. In the present study, CCD32SK diploid normal human fibroblasts were transfected transiently with short interfering RNA (siRNA) specific for human p53 (CCD/p53i). There was a small increase in the frequency of centrosome amplification in CCD/p53i cells (4.0%) without irradiation. In contrast, CCD/p53i cells after 5‐Gy irradiation showed a marked increase in abnormal nuclear shapes and pronounced amplification of centrosomes (46.0%). At 12 h after irradiation, irradiated CCD/p53i cells were arrested in G2 phase. By laser scanning cytometry, abnormal mitosis with amplified centrosomes was observed frequently in the accumulating G2/M population at 48 h after irradiation. In the present study, we found that siRNA‐mediated silencing of p53 in normal human fibroblasts, together with DNA damage by irradiation, efficiently induced centrosome amplification and nuclear fragmentation, but these phenomena were not observed with either siRNA‐mediated silencing of p53 or irradiation alone. (Cancer Sci 2006; 97: 252–258)
The centrosome, which is composed of a pair of centrioles and surrounding amorphous pericentriolar material, is the major microtubule‐organizing center in animal cells. Its most prominent function is observed in mitosis, during which the duplicated centrosomes form the spindle poles that direct spindle microtubule assembly and establish bipolarity, both of which are required for accurate segregation of chromosomes into daughter cells.( 1 , 2 )
Chromosomal instability (CIN) is considered to be one of the most formidable forces driving tumor progression. The loss or gain of a single chromosome can introduce the multiple genetic alterations required for acquisition of malignant phenotypes.( 3 ) Centrosome amplification, which occurs frequently in human cancers, is believed to contribute to CIN by causing abnormal mitotic processes, thus increasing the frequency of chromosome transmission errors.( 4 ) We reported previously that centrosome amplification is observed frequently in histologically high‐grade bladder cancer.( 5 , 6 , 7 ) Similar findings have also been reported in other types of cancer, including breast,( 8 , 9 , 10 ) brain,( 11 , 12 ) lung,( 11 ) bile duct( 13 ) and colon.( 11 )
The tumor suppressor protein p53 is a critical regulator of the G1/S checkpoint. DNA damage causes stabilization of p53, leading to G1 arrest via induction of p21.( 14 ) Centrosome amplification is known to be associated with loss or mutational inactivation of p53.( 15 , 16 ) In mouse cells, centrosome amplification can be induced readily by loss or mutational inactivation of p53.( 16 ) In human cells, however, silencing of endogenous p53 alone does not induce centrosome amplification or CIN,( 17 , 18 ) but strong correlations between p53 mutation and CIN/centrosome amplification in human cancer have been detected,( 7 , 8 ) thus suggesting the presence of additional regulatory mechanisms in human cells that ensure the numerical integrity of centrosomes and genomic integrity.
It has been shown that irradiation has lethal effects in mammalian cells, and thus irradiation is used as a cancer treatment.( 19 ) However, the precise mechanism of radiation‐induced cell death is not yet fully understood. It has recently been shown that gamma irradiation of tumor cells induces centrosome amplification, which may lead to lethal nuclear fragmentation via establishment of multipolar mitotic spindles.( 20 , 21 , 22 )
In the present study, we found that short interfering RNA (siRNA)‐mediated silencing of p53 in normal human fibroblasts, together with DNA damage by irradiation, efficiently induces centrosome amplification and nuclear fragmentation. However, these phenomena are not induced by either siRNA‐mediated silencing of p53 or irradiation alone.
Materials and Methods
Cell line
The CCD32SK (human skin fibroblast) cell line was obtained from American Type Culture Collection (Manassas, VA, USA). Cells were maintained in medium (Delbucco's modified eagle medium [DMEM] supplemented with 10% fetal bovine serum containing penicillin [100 units/mL] and streptomycin [100 µg/mL]) and were grown in an atmosphere containing 10% CO2.
Irradiation
Cells were irradiated at room temperature using an X‐ray irradiation system (MBR‐1520A‐TW; Hitachi, Tokyo, Japan).
Indirect immunofluorescence
Cells were examined for centrosome amplification by immunostaining for γ‐tubulin.( 23 ) Cultured cells grown on slides were washed twice with phosphate‐buffered saline (PBS) and were fixed with 10% formalin for 20 min at room temperature. Samples were then washed with PBS and permeabilized with 1% NONIDET P‐40 (NP‐40) in PBS for 5 min at room temperature. After incubation with blocking solution (10% normal goat serum in PBS) for 1 h at room temperature, cells were probed with anti γ‐tubulin polyclonal antibody (obtained from Professor Kenji Fukasawa, University of Cincinnati, OH, USA( 15 , 16 )) for 1 h at 37°C. Negative controls were produced by omitting primary antibody. Antibody–antigen complexes were detected by incubating cells with Alexa 488 goat antirabbit IgG antibody (Molecular Probes, Eugene, OR, USA) for 1 h at room temperature. After incubation, samples were washed three times with Tris‐buffered saline (TBS) and were then counterstained with 4′‐diamidino 2‐phenylindole (DAPI) or propidium iodide (PI). For morphological determination of abnormal nuclear shape, irradiated cells stained with PI or DAPI were observed under a microscope. Giant cells containing multiple nuclei, small nuclear fragments (micronuclei) and lobulated nuclei were considered to represent abnormal nuclear shape.
Immunoblot analysis
Whole‐cell extracts prepared by lysing cells in sodium dodecylsulfate (SDS) sample buffer were resolved by SDS‐polyacrylamide gel electrophoresis, and transferred to Immobilon sheets (Milipore, Billerica MA, USA). Blots were incubated in blocking buffer (5%[w/v] non‐fat dry milk in Tris‐buffered saline + Tween 20 [TBS‐T]) for 1 h, and were then probed with primary antibody (anti‐p53 monoclonal antibody, Pab‐1801; Santa Cruz, CA, USA). To control for protein loading, blots were also probed with an actin‐specific antibody (SC‐1616; Santa Cruz). Blots were rinsed in TBS‐T and incubated with the appropriate horseradish peroxidase‐conjugated goat secondary antibody for 1 h. Blots were then rinsed in TBS‐T, and antibody–antigen complexes were visualized by ECL chemiluminescence (Amersham, Piscataway, NJ, USA).
Transfection procedure: Silencing of p53 protein by siRNA
One day before transfection, cells were plated in an appropriate amount of growth medium without antibiotics in order to reach 30% confluence at the time of transfection. siRNA oligomers (p53 siRNA, catalog no. 1024849; Qiagen, Hilden, Germany; negative control siRNA: catalog no. 1022563; Qiagen) were diluted in appropriate amounts of Opti‐MEM I (Gibco, Carlsbad, CA, USA) without serum. LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, USA) was diluted with an appropriate amount of Opti‐MEM I. After a 5‐min incubation, the diluted oligomers were combined with diluted LipofectamineTM 2000, and were incubated for 20 min at room temperature in order to allow complex formation to occur. Oligomer–LipofectamineTM 2000 complexes were added to each well containing cells and medium. Cells were transfected with siRNA at a concentration of 100 nM, and were incubated at 37°C in a CO2 incubator for 24 h.
Laser scanning cytometry
We analyzed the centrosome duplication cycle of cultured cells using laser scanning cytometry (LSC) to measure DNA content and examine the centrosome. The cytometry procedure is described in detail elsewhere.( 24 , 25 ) Cells were first fixed with 100% ethanol for 20 min at room temperature. Centrosome staining was carried out as described above using anti‐γ‐tubulin polyclonal antibody.( 15 , 16 ) Cell nuclei were stained with PI solution (25 µg/mL; Sigma Chemical Co., St Louis, MO, USA) containing 1 mg/mL RNase (Sigma), and DNA ploidy was analyzed using a laser scanning cytometer (LSC‐01; Olympus; Tokyo, Japan). An excitation wavelength of 488 nm was used, and emission by PI or Alexa was measured using standard long‐pass (570 nm) or band‐pass (530 nm) filters, respectively. At least 5000 cells were measured on each slide. For each cell, we measured DNA content and examined the Alexa 488 fluorescence profile. It is possible to discriminate between mitotic cells and G2 cells, between post‐mitotic cells and G1 cells, and between quiescent cells and cycling cells based on PI fluorescence peak (chromatin condensation) versus fluorescence value (DNA content) cytograms for cells stained with PI.( 26 , 27 ) Using a quadrant cell cycle analysis program for the LSC, we were able to identify four cell subpopulations among the PI‐stained cells: interphase (G1 + S), G2, M and post‐mitotic. For each stage of the cell cycle, the presence or absence of centrosome amplification was confirmed by microscopy. Samples were photographed using a digital camera (COOLPIX‐5000; Nikon, Tokyo, Japan) mounted on the LSC microscope.
Results
Silencing of p53 protein by siRNA
CCD32SK diploid normal human fibroblasts were transfected transiently with siRNA specific for human p53 (CCD/p53i) and negative siRNA (CCD/NEGi) for 24 h. After p53 siRNA transfection (final concentration 100 nM), CCD32SK cells were irradiated with a single dose of 5 Gy. At 24 h after irradiation, lysates prepared from these cell lines were analyzed for p53 expression by immunoblotting with anti‐p53 antibody (Pb‐1801). As a loading control, lysates also were probed with anti‐actin antibody. Expression of p53 in cells transfected with p53 siRNA was silenced successfully (Fig. 1).
Figure 1.
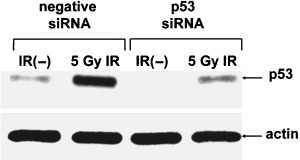
Western blotting analysis of CCD32SK cells following irradiation (IR). Whole‐cell lysates (10 µg) were loaded into each lane. Actin was used as the loading control. siRNA, small interfering RNA.
Radiation‐induced abnormal nuclear shapes and centrosome amplification in p53 siRNA‐treated CCD32SK cells
We investigated the induction of abnormal nuclear shapes by p53 siRNA transfection after irradiation (Fig. 2a). CCD/NEGi and CCD/p53i cells exhibited normal nuclear shapes after siRNA transfection. After 5‐Gy irradiation, CCD/NEGi cells exhibited normal nuclear shapes, and almost all cells were dead at 120 h after irradiation. In contrast, CCD/p53i cells showed a marked increase in abnormal nuclear shapes after irradiation. DNA histograms showed that the CCD/p53i cells were diploid, but at 24–48 h after irradiation, there was a broad peak in G2/M cells, and the proportion of G2/M cells had increased (Fig. 2b).
Figure 2.
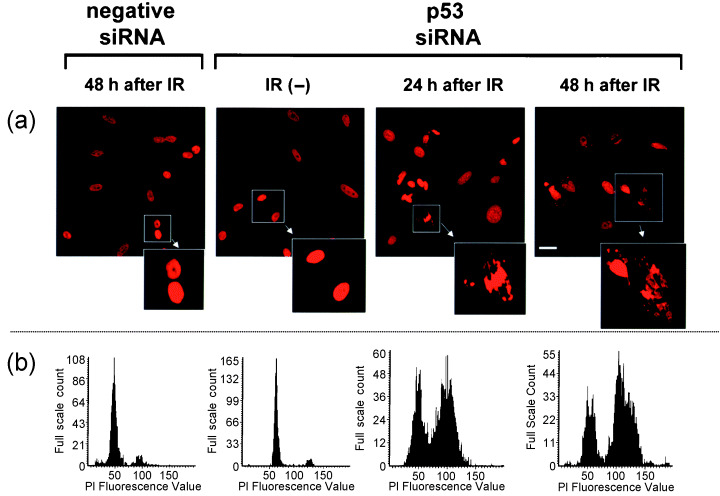
Radiation‐induced abnormal nuclear shapes in p53 short interfering RNA (siRNA)‐treated CCD32SK cells. (a) Propidium iodide (PI) staining (×400, scale bar = 10 µm). (b) DNA histogram produced using laser scanning cytometry. IR, irradiation; siRNA, small interfering RNA.
We then investigated the induction of centrosome amplification by p53 siRNA transfection after irradiation (Fig. 3). The frequency of centrosome amplification (n ≥ 3) in untreated CCD32SK cells (0.5%) and CCD/NEGi cells (2.0%) was low. At 72 h after 5‐Gy irradiation, a small proportion (5.0%) of CCD/NEGi cells exhibited centrosome amplification. There was a small increase in the frequency of centrosome amplification in CCD/p53i cells (4.0%) without irradiation. In contrast, 72 h after 5‐Gy irradiation, CCD/p53i cells exhibited pronounced centrosome amplification (46%). After 5‐Gy irradiation, the fraction of CCD/p53i cells with centrosome amplification increased in 48 h from 4.0 to 45.0% (Table 1). Figure 4 shows centrosome staining results for CCD/p53i cells after irradiation. At 24 h after irradiation, most of the centrosome amplification was observed in interphase cells. At 36–48 h after irradiation, numerous multipolar mitotic spindle cells were observed.
Figure 3.
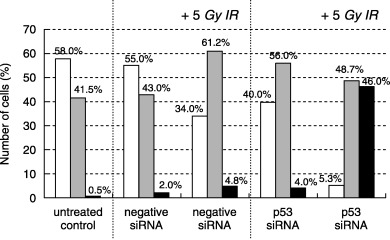
Radiation‐induced abnormal amplification of centrosomes in p53 short interfering RNA (siRNA)‐treated CCD32SK cells. (□), n = 1; (▒), n = 2; (▪), n ≥ 3. DAPI, 4′‐diamidino 2‐phenylindole; IR, irradiation; PI, propidium iodide.
Table 1.
Percentage of CCD/p53i cells with centrosome amplification following 5‐Gy irradiation
| Treatment | 1 centrosome (%) | 2 centrosomes (%) | 3 centrosomes (%) |
|---|---|---|---|
| Untreated control | 40.0 | 56.0 | 4.0 |
| 3 h after 5‐Gy irradiation | 30.4 | 65.6 | 4.0 |
| 12 h after 5‐Gy irradiation | 27.0 | 60.0 | 13.0 |
| 24 h after 5‐Gy irradiation | 5.6 | 60.7 | 33.6 |
| 36 h after 5‐Gy irradiation | 9.0 | 53.0 | 38.0 |
| 48 h after 5‐Gy irradiation | 4.0 | 51.0 | 45.0 |
Figure 4.
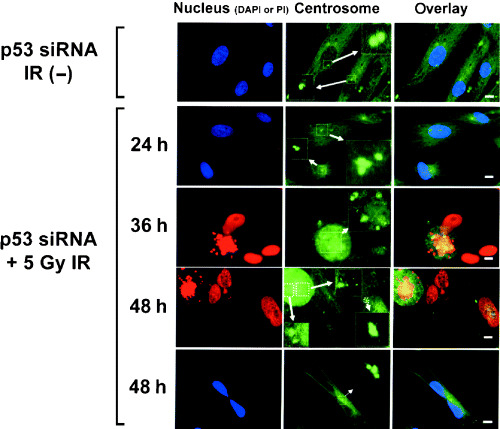
Representative γ‐tubulin immunostaining images of radiation‐induced abnormal amplification of centrosomes in p53 short interfering RNA (siRNA)‐treated CCD32SK cells (scale bar = 10 µm).
We found that siRNA‐mediated silencing of p53 in normal human fibroblasts together with DNA damage by irradiation efficiently induced abnormal nuclear shapes and centrosome amplification. However, these phenomena were not observed with either siRNA‐mediated silencing of p53 or irradiation alone.
Cell cycle progression of CCD32SK cells after irradiation
We investigated the changes in cell cycle progression after irradiation in CCD32SK cells (Fig. 5). With CCD/NEGi cells, the proportion of M‐phase cells was 0% 24 h after irradiation, and most of the cells at 24 h after irradiation were arrested in G1 or G2 phase. At 24 h after 5‐Gy irradiation, approximately 15% of the CCD/NEG cells were arrested in G2 phase. In contrast, at 12 h after 5‐Gy irradiation, approximately 40% of the CDD/p53i cells were arrested in G2 phase. Beginning at 24 h after irradiation, the proportion of M‐phase cells began to increase, and at 36 h after irradiation, the proportion of M‐phase cells was 36%.
Figure 5.
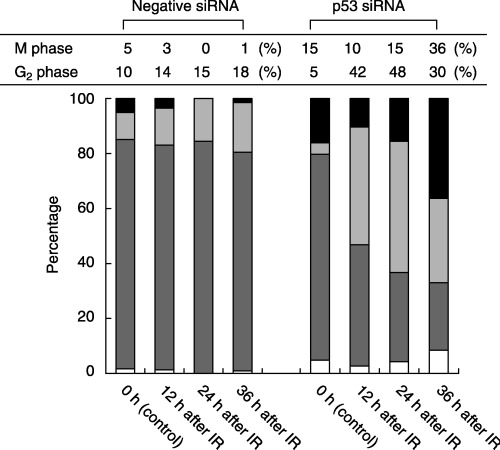
Cell cycle analysis of short interfering RNA (siRNA)‐treated CCD32SK cells. (▪), M phase; (▒), G2 phase; (▓), interphase (G1 + S); (□), post‐mitotic cells. IR, irradiation.
We used LSC to analyze the centrosome replication cycle. Figure 6 shows the centrosome replication state of CCD/p53i cells without irradiation during the cell cycle. Cells at G1 phase contained one centrosome juxtaposed with the nucleus (Fig. 6a). S‐phase cells contained two centrosomes juxtaposed with the nucleus (Fig. 6b). Duplication begins near the G1/S boundary and is completed in G2. Cells in mitosis displayed typical bipolar spindle poles organized by two centrosomes (Fig. 6d), and thus the centrosome duplication cycle was well regulated in CCD/p53i cells without irradiation.
Figure 6.
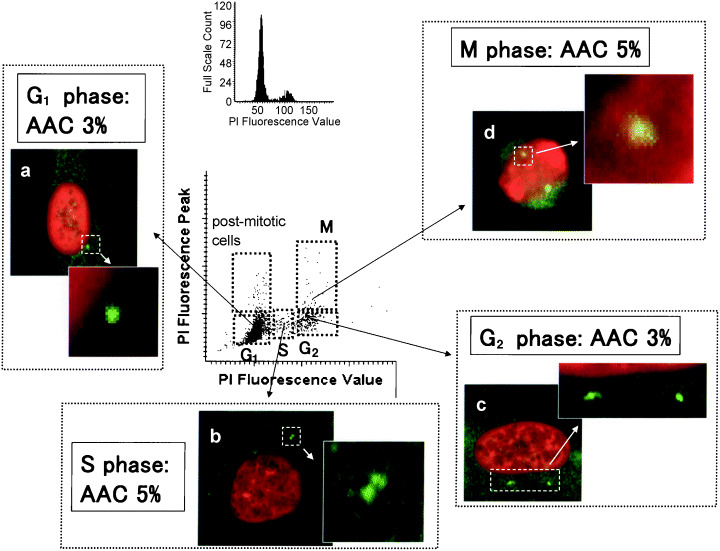
Centrosome duplication throughout the cell cycle determined by laser scanning cytometry in p53 short interfering RNA‐treated CCD32SK cells without irradiation. (a) G1 phase; (b) S phase; (c) G2 phase; (d) M phase. Numbers (%) in the figure represent the incidence of abnormal amplification of centrosomes (AAC) for each cell‐cycle stage (original magnification ×600). PI, propidium iodide.
In contrast, abnormal mitosis with amplified centrosomes was observed frequently in the accumulating G2/M population after irradiation (Fig. 7e–g). Among the CCD/p53i cells at 48 h after irradiation, 78% of G2 cells (Fig. 7e) and 72% of M‐phase cells (Fig. 7f,g) contained more than two centrosomes. The incidence of abnormal amplification of the centrosome (AAC) in G1 phase (Fig. 7c) and S phase (Fig. 7d) was 5% and 10%, respectively, which was lower than the incidence in G2/M phase. Among the post‐mitotic cells (Fig. 7a) and the sub‐G1 population (Fig. 7b), abnormal nuclear shapes were seen in many cells.
Figure 7.
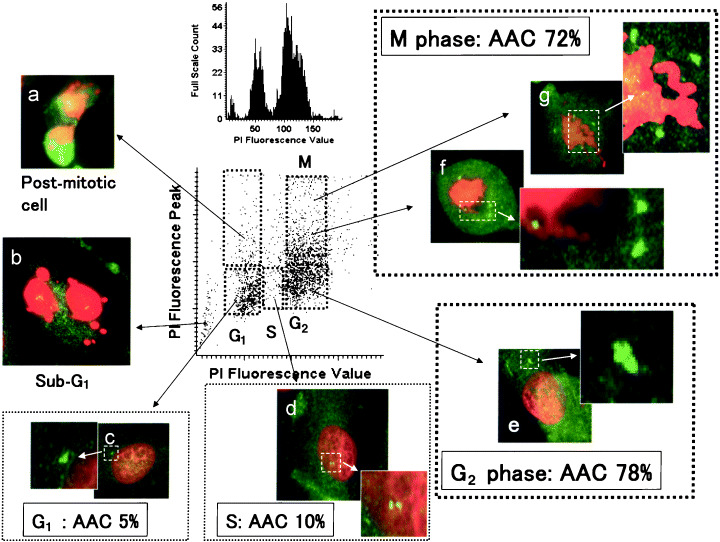
Centrosome duplication throughout the cell cycle determined by laser scanning cytometry in p53 short interfering RNA‐treated CCD32SK cells at 48 h after 5‐Gy irradiation. (a) Post‐mitotic cell; (b) sub‐G1 phase; (c) G1 phase; (d) S phase; (e) G2 phase; (f) prometa phase; (g) meta phase. Numbers (%) in the figure represent the incidence of abnormal amplification of centrosomes (AAC) for each cell cycle stage (original magnification ×600). PI, propidium iodide.
Discussion
Centrosome amplification can occur via several mechanisms, including deregulated centrosome duplication, uncontrolled splitting of centriole pairs and cytokinesis failure,( 28 , 29 ) and is induced by mutations in various cell cycle‐related proteins and tumor suppressor proteins, most notably p53 and BRCA1.( 30 , 31 ) Centrosome amplification caused by loss or mutational inactivation of p53 has been studied extensively in cultured mouse cells. For example, cells derived from p53‐deficient (p53−/–) mice( 15 ) and cells transfected with dominant‐negative p53 mutants( 17 , 18 ) show high frequencies of centrosome amplification. Centrosome amplification also can be detected in the tissues of p53−/– mice( 16 ) and human papillomavirus E6 (a potent destabilizer of p53) transgenic mice.( 32 ) In contrast, loss of p53 does not result in centrosome amplification in cultured human cells and appears to require additional mutations for efficient induction of centrosome amplification and CIN. For example, siRNA‐mediated silencing of p53 in normal human fibroblasts or expression of human papillomavirus E6 in normal human keratinocytes does not result in significant levels of centrosome amplification( 33 ) or CIN.( 34 ) However, examination of human cancer tissues and cultured cells has revealed a significant correlation between loss or mutational inactivation of p53 and occurrence of centrosome amplification, thus supporting the notion that p53 mutation alone is not sufficient to induce centrosome amplification in human cells, but rather that this phenomenon requires additional mutations.
CCD/p53i cells exhibit a well‐regulated centrosome duplication cycle. However, an excessive number of centrosomes, produced by irradiation and p53 siRNA transfection, resulted in multiple mitotic spindles and missegregation of chromosomes. After DNA damage, many cancer cells appear to enter a sustained arrest in the G2 phase of the cell cycle.( 22 ) Bunz et al.( 35 ) reported that this arrest could be sustained only when p53 was present in the cell and was capable of transcriptionally activating the cyclin‐dependent kinase inhibitor p21. After disruption of either the p53 or the p21 gene, γ‐irradiated cells progressed into mitosis and exhibited a G2 DNA content only because of a failure of cytokinesis. They concluded that p53 and p21 appear to be essential for maintaining the G2 checkpoint in human tumor cells. In the present study, we found that continual duplication of centrosomes occurred in the G2 arrested cells after irradiation, leading to centrosome amplification. At 12 h after irradiation, irradiated CCD/p53i cells were arrested in G2 phase. At this stage, although DNA replication is complete, centrosome duplication may still occur. Several studies have shown that a centrosome can be duplicated repeatedly, even if DNA replication or mitosis is blocked.( 36 ) It has been reported that reported that the centrosome duplication cycle is regulated in two ways.( 28 , 30 ) The first ensures that initiation of centrosome duplication and initiation of DNA replication occur in a coordinated fashion. The other suppresses reduplication of centrosomes after they are duplicated. Involvement of p53 in these regulatory pathways has been shown in cells derived from p53‐null mice.( 15 , 16 ) In CCD/p53i cells arrested in G2 phase, centrosome amplification may be caused by failure to suppress centrosome reduplication.
Thus, in the presence of p53, irradiation halts DNA replication as well as centrosome duplication. However, in the absence of p53, irradiation blocks DNA synthesis but not centrosome duplication in G2 phase.
In the present study, we examined the fate of irradiated human fibroblasts in which the p53 tumor suppressor protein was silenced, and focused on changes in the number of centrosomes present. We showed that loss of p53 allows centrosome amplification upon irradiation of human normal fibroblast cells. The implication of this finding is important in view of radiation treatment of cancer: (1) loss (or inactivation) of p53 has two opposing consequences in terms of the effectiveness of radiation treatment; (2) loss of p53 increases the frequency of centrosome amplification upon irradiation, leading to induction of cell death. On the other hand, if cells escape cell death after radiation treatment and continue cell cycling, abnormally amplified centrosomes in those cells will suffer extensive chromosomal instability, promoting acquisition of further malignant phenotypes. Thus, radiation treatment, if not successful, could facilitate the malignant progression of the original tumors.
In conclusion, the present findings indicate a relationship between centrosome amplification and chromosomal damage following irradiation in p53 siRNA‐treated CCD32SK cells. Research into the molecular mechanisms involved in centrosome dysregulation may help to clarify the mechanisms involved in radiation‐induced cell death.
References
- 1. Brinkley BR. Microtubule organizing centers. Annu Rev Cell Biol 1985; 1: 145–72. [DOI] [PubMed] [Google Scholar]
- 2. Lange BM, Faragher AJ, March P et al. Centriole duplication and maturation in animal cells. Curr Top Dev Biol 2000; 49: 235–49. [DOI] [PubMed] [Google Scholar]
- 3. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386: 623–7. [DOI] [PubMed] [Google Scholar]
- 4. Brinkley BR, Goepfert TM. Supernumerary centrosomes and cancer: Boveri's hypothesis resurrected. Cell Motil Cytoskeleton 1998; 41: 281–8. [DOI] [PubMed] [Google Scholar]
- 5. Kawamura K, Moriyama M, Shiba N et al. Centrosome hyperamplification and chromosomal instability in bladder cancer. Eur Urol 2003; 43: 505–15. [DOI] [PubMed] [Google Scholar]
- 6. Kawamura K, Moriyama M, Suga K et al. Centrosome isolation from cultured bladder cancer cells: p53 mutation and centrosome hyperamplification. Acta Urol Japonica 2003; 49: 69–74. [PubMed] [Google Scholar]
- 7. Kawamura K, Izumi H, Ma Z et al. Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res 2004; 64: 4800–9. [DOI] [PubMed] [Google Scholar]
- 8. Carroll PE, Okuda M, Horn FH et al. Centrosome hyperamplification in human cancer: chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene 1999; 18: 1935–44. [DOI] [PubMed] [Google Scholar]
- 9. Lingle WL, Lutz WH, Ingle JN et al. Centrosome hypertrophy in human breast tumors: Implications for genomic stability and cell polarity. Cell Biol 1998; 95: 2950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lingle WL, Salisbury JL. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol 1999; 155: 1941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Phihan GA, Purohit A, Wallace J et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res 1998; 58: 3974–85. [PubMed] [Google Scholar]
- 12. Weber RG, Bridger JM, Benner A et al. Centrosome amplification as a possible mechanism for numerical chromosome aberrations in cerebral primitive neuroectodermal tumors with TP53 mutations. Cytogenet Cell Genet 1998; 83: 266–8. [DOI] [PubMed] [Google Scholar]
- 13. Kuo KK, Sato N, Mizumoto K et al. Centrosome abnormalities in human carcinomas of the gallbladder and intrahepatic and extrahepatic bile ducts. Hepatology 2000; 31: 59–64. [DOI] [PubMed] [Google Scholar]
- 14. Bae I, Fan S, Bhatia K et al. Relationships between G1 arrest and stability of the p53 and p21Cip1/Waf1 proteins following gamma‐irradiation of human lymphoma cells. Cancer Res 1995; 55: 2387–93. [PubMed] [Google Scholar]
- 15. Fukasawa K, Choi T, Kuriyama R et al. Abnormal centrosome amplification in the absence of p53. Science 1996; 271: 1744–7. [DOI] [PubMed] [Google Scholar]
- 16. Fukasawa K, Wiener F, Vande Woude GF et al. Genomic instability and apoptosis are frequent in p53 deficient mice. Oncogene 1997; 15: 1295–302. [DOI] [PubMed] [Google Scholar]
- 17. Wang XJ, Greenhalgh DA, Jiang A et al. Analysis of centrosome abnormalities and angiogenesis in epidermal‐targeted p53 172H mutant and p53‐knockout mice after chemical carcinogenesis: evidence for a gain of function. Mol Carcinog 1998; 23: 185–92. [DOI] [PubMed] [Google Scholar]
- 18. Murphy KL, Dennis AP, Rosen JM. A gain of function p53 mutant promotes both genomic instability and cell survival in a novel p53‐null mammary epithelial cell model. FASEB J 2000; 14: 2291–302. [DOI] [PubMed] [Google Scholar]
- 19. Jonathan EC, Bernhard EJ, McKenna WG. How does radiation kill cells? Curr Opin Chem Biol 1999; 3: 77–83. [DOI] [PubMed] [Google Scholar]
- 20. Sato N, Mizumoto K, Nakamura M et al. Radiation‐induced centrosome overduplication and multiple mitotic spindles in human tumor cells. Exp Cell Res 2000; 255: 321–6. [DOI] [PubMed] [Google Scholar]
- 21. Sato N, Mizumoto K, Nakamura M et al. A possible role for centrosome overduplication in radiation‐induced cell death. Oncogene 2000; 19: 5281–90. [DOI] [PubMed] [Google Scholar]
- 22. Kawamura K, Fujikawa‐Yamamoto K, Ozaki M et al. Centrosome hyperamplification and chromosomal damage after exposure to radiation. Oncology 2004; 67: 460–70. [DOI] [PubMed] [Google Scholar]
- 23. Joshi HC. Microtubule organizing centers and gamma‐tubulin. Curr Opin Cell Biol 1994; 6: 54–62. [DOI] [PubMed] [Google Scholar]
- 24. Kawamura K, Tanaka T, Ikeda R et al. DNA ploidy analysis of urinary tract epithelial tumors by laser scanning cytometry. Anal Quant Cytol Histol 2000; 22: 26–30. [PubMed] [Google Scholar]
- 25. Kawamura K, Kobayashi Y, Tanaka T et al. Intranuclear localization of proliferative cell nuclear antigen during the cell cycle in renal cell carcinoma. Anal Quant Cytol Histol 2000; 22: 107–13. [PubMed] [Google Scholar]
- 26. Kakino S, Sasaki K, Kurose A et al. Intracellular localization of cyclin B1 during the cell cycle in glioma cells. Cytometry 1996; 24: 49–54. [DOI] [PubMed] [Google Scholar]
- 27. Kawasaki M, Sasaki K, Satoh T et al. Laser scanning cytometry (LCS) allows detailed analysis of the cell cycle in PI stained human fibroblasts (TIG‐7). Cell Prolif 1997; 30: 139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fukasawa K. Centrosome. Oncogene 2002; 21: 6140–5. [DOI] [PubMed] [Google Scholar]
- 29. Mazia D. The chromosome cycle and the centrosome cycle in the mitotic cycle. Int Rev Cytol 1987; 100: 49–92. [DOI] [PubMed] [Google Scholar]
- 30. Tarapore P, Fukasawa K. Loss of p53 and centrosome hyperamplification. Oncogene 2002; 21: 6234–40. [DOI] [PubMed] [Google Scholar]
- 31. Deng CX. Roles of BRCA1 in centrosome duplication. Oncogene 2002; 21: 6222–7. [DOI] [PubMed] [Google Scholar]
- 32. Riley RR, Duensing S, Brake T et al. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res 2003; 63: 4862–71. [PubMed] [Google Scholar]
- 33. Duensing S, Lee LY, Duensing A et al. The human papillomavirus type 16, E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci USA 2000, 97: 10 002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bunz F, Fauth C, Speicher MR et al. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res 2002; 62: 1129–33. [PubMed] [Google Scholar]
- 35. Bunz F, Dutriaux A, Lengauer C et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497–501. [DOI] [PubMed] [Google Scholar]
- 36. Chang WP, Little JB. Delayed reproductive death in X‐irradiation Chinese hamster ovary cells. Int J Radiat Biol 1991; 60: 483–96. [DOI] [PubMed] [Google Scholar]