

Abstract
Germ cell tumors (GCTs) are thought to arise from primordial germ cells (PGCs) that undergo epigenetic reprogramming: erasure of the somatic imprint in the genital ridge, and re‐establishment of the sex‐specific imprint at gametogenesis in the developing gonad. Previous studies suggested that GCTs show epigenetic patterns reflecting the reprogramming process of PGCs; however, epigenetic alterations of imprinted genes and their relationship with the methylation status of tumor suppressor genes (TSGs) have not been comprehensively studied. We analyzed the methylation status of the H19 and SNRPN differential methylated regions (DMRs) and the promoter region of 17 TSGs, and the expression status of H19, IGF2 and SNRPN in 45 GCTs, and found that 25 and 20 were in the normal and abnormal reprogramming pathways, respectively, defined on the basis of the methylation status of the two DMRs and the anatomical tumor site. The methylation pattern of the H19 and SNRPN DMRs was total erasure in seminomas, mostly physiological in teratomas, and various in yolk sac tumors. There were no correlations between the methylation status of the H19 DMR and mono‐ or biallelic expression of H19 or IGF2. Furthermore, we found that yolk sac tumors had a higher number of methylated TSGs than seminomas (P < 0.001) teratomas (P = 0.004) or other childhood tumors. While TSG methylation was known to have prognostic implications in various cancers, it did not affect the outcomes of patients with yolk sac tumor, suggesting that mechanisms of TSG methylation may be different between yolk sac tumor and other cancers. (Cancer Sci 2009; 100: 698–708)
Childhood germ‐cell tumor (GCT) is a rare malignant neoplasm of the gonad or the extra‐gonadal site, occurring at an incidence of 2.4 per million children and representing approximately 1% of cancers diagnosed in persons younger than 15 years.( 1 ) In contrast, testicular GCT in men between the ages 20–40 is the most common cancer with an incidence of 8–10 per 100 000 in some European countries.( 2 ) GCT is a unique tumor, thought to arise from primordial germ cells (PGCs) that undergo epigenetic reprogramming.( 3 ) Genomic imprinting is the phenomenon that the paternal and maternal sets of chromosomes have different functionality in mammals, due to parental‐specific epigenetic modification of the genome. This modification in mouse PGCs starts with imprint erasure on embryonic day 10.5 and is completed with imprint reestablishment at gametogenesis.( 4 , 5 , 6 )
The H19 gene is located in 11p15.5 and encodes non‐coding RNA, and the gene SNRPN (small nuclear ribonucleoprotein associated polypeptide N) located in 15q11‐13 and encodes SmN, a protein involved in spliceosomes.( 7 , 8 ) In normal tissues, while paternal H19 is not expressed due to methylation of the H19 differentially methylated region (DMR), maternal H19 is expressed due to the unmethylation of H19 DMR. Likewise, while paternal SNRPN is expressed due to the unmethylation of SNRPN DMR, maternal SNRPN is not expressed due to the methylation of SNRPN DMR. It has been suggested in several studies that the pattern of H19 and SNRPN methylation in GCTs can be used as a marker of gametic development of PGC giving rise to the tumor.( 9 , 10 , 11 , 12 ) The methylation status of these two DMRs was studied in childhood GCTs, and the results suggested that the methylation status of DMR of H19 or SNRPN might reflect preservation of the physiologic imprinting erasure in PGCs.( 10 , 11 , 12 ) These studies analyzed the methylation status of either H19 DMR or SNRPN DMR in GCTs using various methods, including Southern blot with a methylation‐sensitive enzyme, combined bisulfite restriction assay (COBRA) and the methylation‐sensitive single‐nucleotide primer extension method, and did not determine the relationship of the methylation statuses of the two DMRs in the same GCTs.
Methylation of the promoter region of a tumor suppressor gene (TSG) has been regarded as an important epigenetic change in the development of various cancers.( 13 ) The methylation status of many TSGs has been studied mainly in adult testicular GCTs, and these studies showed that non‐seminomas had a higher incidence of promoter methylation in many TSGs than seminomas.( 14 , 15 ) Furthermore, Smiraglia et al. reported that seminomas show almost no CpG island methylation, in contrast to non‐seminomas that show CpG island methylation at a level similar to other solid tumors by restriction landmark genomic scanning.( 16 ) It remains unknown whether childhood non‐seminoma has a high incidence of methylated TSGs similar to adult non‐seminoma, whether dysgerminoma, histologically equivalent to seminoma developing in the ovary, share a low incidence of TSG methylation, and whether childhood yolk sac tumor has a similar incidence of TSG methylation to other childhood solid tumors.
To explore the epigenetic reprogramming process of 45 GCTs, we examined the methylation status of DMR of two imprinted genes, H19 and SNPRN, and the relationship between the DMR methylation and the expression status of H19, IGF2 and/or SNRPN. Furthermore, we evaluated the methylation status of the promoter region in 17 TSGs, and examined differences in the numbers of methylated TSGs between the two groups of tumors classified by clinical, pathological and epigenetic characteristics. We also clarified the relationship between the reprogramming pathway and the clinical, pathological and epigenetic findings.
Materials and methods
Patients and samples. Tumor tissues were obtained from 38 children and seven adults with GCT. There were 23 males and 22 females, ranging in age from 1 month to 42 years with a median age of 8 years and 7 months (Table 1). In all tumors, the diagnosis of GCT was made by pathologists at each institution according to the classification proposed by Woodward et al. (2004) and/or the Japanese Pathological Society.( 2 , 17 ) The pathologist in each institution verified that each sample for molecular genetic analysis contained 70% or more tumor cells. Tumor tissues, which were obtained at Saitama Cancer Center or sent from other hospitals, were immediately frozen after resection or on arrival at the center. The study design was approved by the ethics committee of Saitama Cancer Center. Histologically and clinically, GCTs are subdivided into seminoma (called seminoma when occurring in the testis, dysgerminoma when occurring in the ovary or dysgenetic gonads, and germinoma when occurring in the brain) and non‐seminoma, including teratoma, yolk sac tumor, embryonal carcinoma and choriocarcinoma.( 2 , 17 ) There were four seminomas, five dysgerminomas and 36 non‐seminomas, including 27 yolk sac tumors, 25 of which occurred in children, five teratomas and four immature teratomas. The representative histology was used for seven yolk sac tumors and one immature teratoma that showed more than one histological criterion (Table 1). Thirty‐five tumors originated from the gonad (ovary, 20; testis, 15), and 10 from extra‐gonadal sites (mediastinum, 5; the sacrococcygeal region, 2; retroperitoneum 1; stomach 1; and pineal body, 1). Data on outcomes were available for all 45 patients, four (Nos. 5, 9, 33 and 42) of whom died of the disease, and the others were alive at the last follow‐up.
Table 1.
Clinical characteristics and genetic and epigenetic status of H19 and SNRPN regions in 45 germ cell tumors
| Patient number | Age/sex | Primary site | Pathology | % methyl H19 DMR | 11p15 SNP | ApaI site | % methyl SNRPN DMR | 15q11 SNP | HpaII site | Epigenotype H19/SNRPN | Reprogram. pathway | Methylated TSGs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Extra‐gonadal tumors (n = 10) | ||||||||||||
| Teratomas (n = 5) | ||||||||||||
| 1 | 2 m/F | Sacrococcygeal | Teratoma | 44.7 | ND | ND | 41.8 | ND | Homo | MU/UM | Normal | 0 |
| 2 | 5 m/F | Mediastinum | Teratoma | 66.2 | ND | ND | 41.2 | ND | Hetero | MU/UM | Normal | 0 |
| 3 | 13 y/M | Mediastinum | Teratoma | 36.0 | ND | Hetero | 41.4 | ND | ND | MU/UM | Normal | 9 |
| 4 | 5 y/M | Pineral body | Immat. teratoma | 2.8 | ND | Homo | 0.4 | ND | ND | UU/UU | Abnormal | 1 |
| 5 | 14 y/M | Mediastinum | Immat. teratoma | 39.7 | ND | Hetero | 27.7 | ND | Homo | MU/UU | Abnormal | 1 |
| Yolk sac tumors (n = 5) | ||||||||||||
| 6 | 10 m/F | Sacrococcygeal | Yolk sac tumor | 65.5 | ND | Homo | 35.8 | ND | Hetero | MU/UM | Normal | 8 |
| 7 | 2 y/M | Stomach | Yolk sac tumor | 16.1 | ND | Hetero | 45.3 | ND | Hetero | UU/UM | Abnormal | 6 |
| 8 | 10 y/M | Mediastinum | Yolk sac tumor | 10.0 | ND | ND | 1.7 | ND | ND | UU/UU | Abnormal | 7 |
| 9 | 13 y/M | Mediastinum | Yolk sac tumor | 12.5 | ND | Homo | 6.7 | ND | Homo | UU/UU | Abnormal | 2 |
| 10 | 11 m/M | Retroperitoneum | Yolk sac tumor | 67.3 | ROH | Homo | 34.6 | ROH | ND | MM/UM | Abnormal | 1 |
| Gonadal tumors (n = 35) | ||||||||||||
| Teratomas (n = 4) | ||||||||||||
| 11 | 13 y/M | Testis | Immat. teratoma/choriocarcinoma | 1.7 | ND | Hetero | 1.5 | ND | Hetero | UU/UU | Normal | 2 |
| 12 | 4 y/F | Ovary | Immat. teratoma | 10.8 | UPD | Homo | 84.7 | UPD | Homo | UU/MM | Normal | 1 |
| 13 | 9 y/F | Ovary | Teratoma | 3.1 | ROH | Homo | 86.0 | ROH | Homo | UU/MM | Normal | 0 |
| 14 | 20 y/F | Ovary | Teratoma | 39.4 | ND | Hetero | 52.7 | ND | ND | MU/UM | Abnormal | 0 |
| Yolk sac tumors (n = 22) | ||||||||||||
| 15 | 18 m/M | Testis | Yolk sac tumor | 17.5 | ND | Homo | 14.7 | ND | Hetero | UU/UU | Normal | 3 |
| 16 | 18 m/M | Testis | Yolk sac tumor | 14.0 | ND | Hetero | 23.7 | ND | ND | UU/UU | Normal | 2 |
| 17 | 2 y/M | Testis | Yolk sac tumor | 15.0 | ND | Hetero | 8.6 | ND | Homo | UU/UU | Normal | 5 |
| 18 | 9 y/F | Ovary | Yolk sac tumor/dysgerminoma | 20.2 | ND | Homo | 5.4 | ND | Homo | UU/UU | Normal | 5 |
| 19 | 13 y/F | Ovary | Yolk sac tumor | 20.2 | ND | Hetero | 12.9 | ND | Hetero | UU/UU | Normal | 8 |
| 20 | 14 y/F | Ovary | Yolk sac tumor/teratoma | 29.0 | ND | Homo | 6.7 | ND | Hetero | UU/UU | Normal | 6 |
| 21 | 38 y/M | Testis | Yolk sac tumor./embryonal ca. | 7.4 | ND | Homo | 2.2 | ND | Homo | UU/UU | Normal | 0 |
| 22 | 6 y/F | Ovary | Yolk sac tumor/immat. teratoma | 12.9 | ROH | Homo | 81.4 | ROH | Homo | UU/MM | Normal | 1 |
| 23 | 11 y/F | Ovary | Yolk sac tumor | 19.1 | ROH | Homo | 70.6 | ROH | Homo | UU/MM | Normal | 7 |
| 24 | 9 y/F | Ovary | Yolk sac tumor | 15.9 | ND | Homo | 53.1 | ND | Homo | UU/UM | Abnormal | 8 |
| 25 | 6 m/M | Testis | Yolk sac tumor | 38.7 | ND | Hetero | 17.4 | ND | Homo | MU/UU | Abnormal | 7 |
| 26 | 11 m/M | Testis | Yolk sac tumor | 41.8 | ND | Hetero | 19.2 | ND | Homo | MU/UU | Abnormal | 6 |
| 27 | 18 m/M | Testis | Yolk sac tumor | 36.4 | ND | Hetero | 17.8 | ND | Hetero | MU/UU | Abnormal | 6 |
| 28 | 18 m/M | Testis | Yolk sac tumor | 42.0 | ND | Homo | 29.3 | ND | Homo | MU/UU | Abnormal | 3 |
| 29 | 2 y/M | Testis | Yolk sac tumor | 39.4 | ND | ND | 11.5 | ND | Homo | MU/UU | Abnormal | 8 |
| 30 | 1 m/M | Testis | Yolk sac tumor | 57.5 | ND | Homo | 39.4 | ND | Homo | MU/UM | Abnormal | 6 |
| 31 | 9 y/F | Ovary | Yolk sac tumor | 33.6 | ND | Hetero | 48.7 | ND | Homo | MU/UM | Abnormal | 5 |
| 32 | 11 y/F | Ovary | Yolk sac tumor | 34.1 | ND | Homo | 40.3 | ND | Homo | MU/UM | Abnormal | 3 |
| 33 | 14 y/F | Ovary | Yolk sac tumor | 43.7 | ND | Homo | 58.5 | ND | Hetero | MU/UM | Abnormal | 4 |
| 34 | 18 m/F | Ovary | Yolk sac tumor/dysgerminoma | 77.9 | ROH | Homo | 35.1 | ROH | Hetero | MM/UM | Abnormal | 7 |
| 35 | 6 y/F | Ovary | Yolk sac tumor/immat. teratoma | 72.8 | ROH | Homo | 56.3 | ROH | Homo | MM/UM | Abnormal | 9 |
| 36 | 33 y/M | Testis | Yolk sac tumor/embryonal ca. | 47.9 | ND | Homo | 37.7 | ND | Homo | MU/UM | Abnormal | 1 |
| Seminomas (n = 9) | ||||||||||||
| 37 | 5 m/F | Ovary | Dysgerminoma | 22.3 | ND | Hetero | 12.5 | ND | Hetero | UU/UU | Normal | 0 |
| 38 | 8 y/F | Ovary | Dysgerminoma | 28.2 | ND | Homo | 14.9 | ND | Homo | UU/UU | Normal | 0 |
| 39 | 8 y/F | Ovary | Dysgerminoma | 23.4 | ND | Homo | 13.4 | ND | Hetero | UU/UU | Normal | 0 |
| 40 | 11 y/F | Ovary | Dysgerminoma | 15.5 | ND | Hetero | 4.5 | ND | ND | UU/UU | Normal | 7 |
| 41 | 13 y/F | Ovary | Dysgerminoma | 9.7 | ND | Homo | 9.2 | ND | Homo | UU/UU | Normal | 0 |
| 42 | 31 y/M | Testis | Seminoma | 14.8 | ND | Homo | 5.7 | ND | Hetero | UU/UU | Normal | 0 |
| 43 | 34 y/M | Testis | Seminoma | 9.5 | ND | Hetero | 10.2 | ND | Hetero | UU/UU | Normal | 0 |
| 44 | 38 y/M | Testis | Seminoma | 12.1 | ND | Hetero | 31.8 | Hetero | ND | UU/UU | Normal | 0 |
| 45 | 42 y/M | Testis | Seminoma | 9.4 | ND | Hetero | 3.1 | ND | Hetero | UU/UU | Normal | 0 |
% methyl., % methylation; Epigenotype, see text; Reprogram., Reprogramming; TSGs, tumor suppressor genes; m, months; y, years; F, female; M, male; ND, not done; ROH, retention of heterozygosity; UPD, uniparental disomy; Homo, homozygosity; Hetero, heterozygosity; Immat., immature; ca, carcinoma; DMR, differential methylated region; SNP, single‐nucleotide polymorphism.
Bisulfite treatment and combined bisulfite restriction assay (COBRA) of the CTCF6 binding site at H19 DMR and the 5′‐untranslated region of SNRPN DMR. Genomic DNA from tumor samples was treated with sodium bisulfite, and we performed COBRA to determine the methylation status of the CCCTC‐binding factor binding site 6 (CTCF6) binding site at H19 DMR, as described previously.( 18 ) In addition, we also evaluated the methylation status of the 5′‐untranslated region of SNRPN DMR.( 19 ) MluI and CfoI were used as restriction enzymes to evaluate the methylation status of H19 DMR and SNRPN DMR, respectively. The intensity of methylated and unmethylated bands was examined by a fluoro‐image analyzer, FLA‐3000G (Fujifilm, Tokyo, Japan). Two methylated bands represented H19 DMR; 197 bp and 82 bp, and only the larger band was evaluated because the smaller band was too faint to be evaluated in all samples. Likewise, two methylated bands represented SNRPN DMR; 175 bp and 65 bp, and only the larger band was evaluated. The experiments were performed three times, and the mean value of the DNA methylation percentages was calculated.
Loss of heterozygosity (LOH) analysis of IGF2 and SNRPN. High‐resolution single‐nucleotide polymorphism (SNP) array, Affymetrix Mapping 250K‐Nsp array (Affymetrix, Santa Clara, CA, US), was used to analyze chromosomal aberrations, including 11p15.5 and 15q11‐13 where IGF2 and SNRPN reside, respectively. Genomic DNA in 7/45 tumors was assayed according to the manufacturer's protocol, and the genomic status of IGF2 and SNRPN was determined using copy number analyzer for GeneChip® (CNAG) software as previously described (Table 1).( 20 , 21 ) In addition, the ApaI polymorphic site in exon 9 of IGF2 was used to evaluate heterozygosity or homozygosity in 42 tumors, as described previously,( 19 ) and the HpaII polymorphic site in intron 1 of SNRPN was also used to evaluate heterozygosity or homozygosity in 38 tumors using the polymerase chain reaction (PCR)‐primers as follows; 5′‐GGCGACAGTGGGTATTGG‐3′ and 5′‐CAGAAATGCGGTGGAATCCTT‐3′.
Expression analysis of H19, IGF2 and SNRPN and allelic expression analysis of H19 and IGF2. Reverse transcription (RT)‐PCR analysis was performed in 12 tumors, the RNA of which was available. Primers used for H19 were 5′‐TACAACCACTGCACTACCTG‐3′ and 5′‐TGGAATGCTTGAAGGCTGCT‐3′, those for SNRPN were 5′‐CCACCAGGCATTAGAGGTCCAC‐3′ and 5′‐GCAGAATGAGGGAACAAAAAGCTC‐3′, and those for IGF2 were reported previously.( 18 , 22 , 23 ) The RsaI polymorphic site in H19 and the ApaI polymorphic site in IGF2 were used to evaluate allelic expression of respective genes. PCR products were analyzed by electrophoresis in a 2.0% agarose gel and stained with ethidium bromide (Sigma, St Louis, MO, US).
Methylation‐specific PCR (MSP) analysis of various TSGs. The methylation status of the promoter region in 17 genes was analyzed by MSP, as previously reported.( 15 , 24 ) The genes examined were RASSF1A, HOXA9, RUNX3, CASP8, SCGB3A1, SFRP2, DCR2, RASSF5, RASSF2A, BLU, SFRP5, HOXB5, p16INK4A, SFRP1, p14ARF, RIZ1 and SFRP4. The methylation status of 12 of the 17 genes was also examined in 20 Wilms tumors to compare the number of methylated TSGs between childhood yolk sac tumors and Wilms tumors. The primer sequences and their location in the original genomic sequences were reported elsewhere.( 15 , 24 ) CpG genome™ Universal Methylated DNA (Chemicon International, Temecula, CA, US) and normal lymphocyte DNA were used as controls for completely methylated and unmethylated templates, respectively. PCR products were run on 2% agarose gels and visualized after staining with ethidium bromide.
Results
Combined bisulfite restriction assay of the CTCF6 site in H19 DMR and SNRPN DMR. COBRA showed that the mean methylation percentage ± standard deviation (SD) of seven lymphocyte samples was 49.0 ± 1.6% at H19 DMR and 40.1 ± 5.7% at SNRPN DMR. Forty‐five tumors showed a mean methylation percentage of CTCF6 ranging from 1.7% to 77.9% and that of SNRPN DMR from 0.4% to 86.0%. We chose a cut‐off value of <33.3% methylation, between 33.3% and 66.6% methylation, and >66.6% methylation as definitions of hypomethylated, somatically methylated, and hypermethylated states, respectively, as proposed by Sievers et al.( 12 ) H19 DMR and SNRPN DMR were hypomethylated in 26 and 26 tumors, somatically methylated in 16 and 15 tumors, and hypermethylated in three and four tumors, respectively (Table 1 and Fig. 1).
Figure 1.
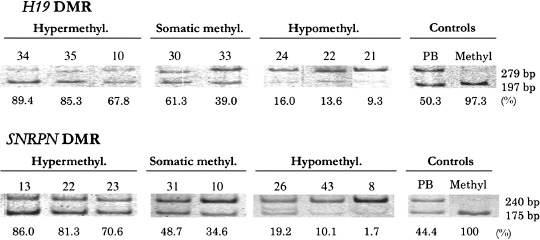
Examples of the methylation status of the CCCTC‐binding factor binding site 6 (CTCF6) binding site of H19 differential methylated region (DMR) and SNPRN DMR analyzed by a combined bisulfate restriction assay (COBRA). Bisulfate‐modified polymerase chain reaction products were digested with MluI or CfoI. Upper 279 bp and lower 197 bands in the upper lane show unmethylated and methylated fragments containing the CTCF6 binding site, respectively, and upper 240 and lower 175 bands in the lower lane show unmethylated and methylated fragments containing SNPRN DMR, respectively. PB, lymphocyte DNA showing normal somatic methylation; Methyl. control methylated DNA. Numbers above lanes indicate the tumor number. The percentage of methylated DNA calculated from the bands above is shown below each lane. The tumors were classified as hypermethylated (>66.6%), somatically methylated (33.3–66.6%), and hypomethylated (<33.3%).
Normal and abnormal reprogramming pathways defined by the methylation status of H19 DMR and SNRPN DMR and the anatomical site in germ cell tumors. Normal mouse PGCs originate from proximal epiblast cells of embryonic day 6.5 (E6.5) egg cylinder, and migrate into the genital ridge.( 4 , 5 , 6 ) PGCs before entering the genital ridge retain parental imprints (somatic methylation); that is, while the paternal and maternal H19 DMRs are methylated and unmethylated, respectively; this epigenotype is abbreviated to (MU) in the present study, and the paternal and maternal SNRPN DMRs are unmethylated and methylated, respectively (UM) (Fig. 2).( 9 , 10 , 11 , 12 ) When PGCs enter the genital ridge, parental imprints are erased (H19 DMR/SNRPN DMR; UU/UU). PGCs maintain the erased imprint in the ovary or testis, and parental imprints are re‐established when PGCs proceed to the meiosis I stage of gametogenesis in female (UU/MM) and to the premeiotic stage in male (MM/UU). Embryologic studies reveal that PGCs actively migrate along the mesentery to the posterior hindgut, in very close proximity to the coccyx, before they enter the genital ridges;( 4 , 5 , 6 ) therefore, tumors located in the extra‐gonadal sites, including the sacrococcyx, mediastinum, retroperitoneum and pineal body, are postulated to have arisen from PGCs that had yet to reach the genital ridge and to erase their imprint. Thus, we assume that GCT cells originating from the extra‐gonadal site retain parental imprints (MU/UM), whereas GCT cells originating from the gonad show an erased imprint (UU/UU) or re‐established imprint (MM/UU or UU/MM).( 3 , 4 , 5 , 6 )
Figure 2.
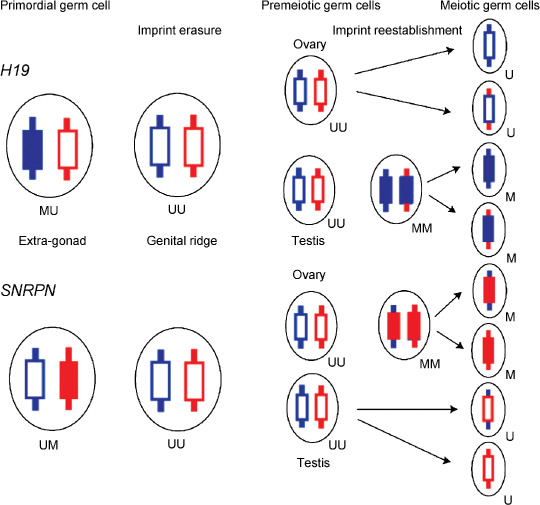
Model of H19 and SNRPN imprint resetting in the human germline. Primordial germ cells (PGCs) before entering the genital ridge retain a somatic methylation pattern showing the paternal imprint at H19 differential methylated region (DMR) and the maternal imprint at SNRPN DMR (H19 DMR/SNRPN DMR is shown as MU/UM). When PGCs have entered the genital ridge, both homologs are unmethylated and the imprint is erased (UU/UU). Imprint is re‐established when both homologs of H19 DMR in male PGCs are methylated (MM) at premeiotic stage in the testis and both homologs of SNRPN DMR in female PGCs are methylated (MM) at meiosis in the ovary. M, methylated DMR; U, unmethylated DMR.
Of 10 tumors originating from the extra‐gonadal site, four showed a normal somatic imprint status, MU/UM, and the other six showed various abnormal imprint statuses. Likewise, of 35 gonadal tumors, 21 showed the normal imprint status of UU/UU (17 tumors with imprint erasure) or UU/MM (4 ovarian tumors with re‐establishment of the maternal imprint), and 14 showed various abnormal imprint statuses (Table 2).
Table 2.
Epigenotype and normal and abnormal reprogramming pathways in extra‐gonadal and gonadal germ cell tumors
| EpigenotypeH19 DMR/SNRPN DMR | Extra‐gonad (n = 10) | Gonad (n = 35) |
|---|---|---|
| Normal reprogramming pathway (n = 25) | ||
| MU/UM | 4 (M 1: Tera 1; F 3: Tera 2, YST 1) | |
| UU/UU | 17 (M 9: YST 4, Sem 4, Tera 1; F 8: Dys 5, YST 3) | |
| UU/MM | 4 (F 4: Tera 2, YST 2) | |
| Abnormal reprogramming pathway (n = 20) | ||
| MM/UM | 1 (M 1: YST 1) | 2 (F 2: YST 2) |
| UU/UU | 3 (M 3: YST 2, Tera 1) | |
| MU/UM | 6 (M 1: YST 1; F 5: YST 4, Tera 1) | |
| MU/UU | 1 (M 1: Tera 1) | 5 (M 5: YST 5) |
| UU/UM | 1 (M 1: YST 1) | 1 (F 1: YST 1) |
MU/UM represents methylated paternal H19 allele and unmethylated maternal H19 allele/unmethylated paternal SNRPN allele and methylated maternal SNRPN allele. Please also see text and Fig. 2.
M, male; F, female; Tera, teratoma; YST, yolk sac tumor; Sem, seminoma; Dys, dysgerminoma; DMR, differential methylated region.
LOH analysis of seven tumors with hypermethylated H19 DMR or SNRPN DMR. SNP array analysis was performed in three tumors with hypermethylated H19 DMR and four with hypermethylated SNRPN DMR. Of the three tumors, one developed from the extra‐gonadal site of a boy and two from the gonadal site of two girls showing heterozygous H19‐IGF2 alleles, indicating aberrant methylation of the maternal allele (Table 1). Of the remaining four tumors, all developed from the ovary, three showed heterozygous SNRPN alleles, indicating physiological methylation of the maternal allele, SNRPN MM (Fig. 2), and one showed uniparental duplication of the maternal SNRPN allele. ApaI polymorphism analysis showed heterozygosity of the IGF2 locus in 18 tumors and homozygosity in 24 tumors. Of the 18 tumors, eight had the MU epigenotype in H19 DMR, and 10 had the UU epigenotype, indicating that the UU genotype was caused by demethylation of the paternal H19 DMR but not by duplication of the maternal H19 DMR. Likewise, HpaII polymorphism analysis showed heterozygosity of the SNRPN locus in 16 tumors and homozygosity in 22 tumors. Of the 16 tumors, five had the UM epigenotype and 11 had the UU epigenotype, indicating that the UU epigenotype was caused by demethylation of maternal SNRPN DMR but not by duplication of paternal SNRPN DMR. Thus, it was proved that uniparental disomy was not the cause of the MM/UM, UU/MM, or UU/UU epigenotype in the majority of GCTs. SNP analysis demonstrated that one immature teratoma (No. 12) with the UU/MM epigenotype had uniparental disomy of both IGF2‐H19 and SNRPN regions, indicating that duplication of the uniparental H19 DMR or SNRPN DMR may cause the UU/MM epigenotype in a minority of GCTs.
Expression analysis of H19, IGF2 and SNRPN and allelic expression analysis of H19 and IGF2. The results of RT‐PCR analysis are shown in Table 3 and Fig. 3. The expression status of each gene was defined as 1+ or 2+. Tumors with the H19 MU epigenotype expressed 2+ IGF2 mRNA more frequently than tumors with the H19 UU epigenotype (P = 0.0133), whereas tumors with the H19 MU epigenotype and those with the H19 UU epigenotype expressed 2+ H19 mRNA with a similar frequency (P = 0.1982). Tumors with the SNRPN UU epigenotype tended to express 2+ SNRPN mRNA more frequently than tumors with the UM or MM SNRPN epigenotype (P = 0.0668). The reasons of the correlation between the H19 MU DMR and IGF2 2+ expression is presently unknown.
Table 3.
Methylation status of H19 and SNRPN DMRs and total and allelic expression of H19, IGF2 and SNRPN
| PatientNumber | Pathology | H19 DMR | H19 mRNA | RsaI site | H19 allelic expression | IGF2 mRNA | ApaI site | IGF2 allelic expression | SNRPN DMR | SNRPN mRNA |
|---|---|---|---|---|---|---|---|---|---|---|
| 13 | Teratoma | UU | 2+ | Homo | Not done | 2+ | Homo | Not done | MM | 1+ |
| 15 | Yolk sac tumor | UU | 2+ | Homo | Not done | 2+ | Homo | Not done | UU | 2+ |
| 21 | Yolk sac tumor/embryonal Ca | UU | 1+ | Hetero | Biallelic | 1+ | Homo | Not done | UU | 2+ |
| 24 | Yolk sac tumor | UU | 2+ | Homo | Not done | 1+ | Homo | Not done | UM | 1+ |
| 26 | Yolk sac tumor | MU | 2+ | Homo | Not done | 2+ | Hetero | Biallelic | UU | 2+ |
| 27 | Yolk sac tumor | MU | 2+ | Homo | Not done | 2+ | Hetero | Monoallelic † | UU | 2+ |
| 30 | Yolk sac tumor | MU | 2+ | Homo | Not done | 2+ | Homo | Not done | UM | 2+ |
| 31 | Yolk sac tumor | MU | 2+ | Hetero | Monoallelic | 2+ | Hetero | Biallelic | UM | 2+ |
| 36 | Yolk sac tumor/embryonal Ca | MU | 2+ | Homo | Not done | 2+ | Homo | Not done | UM | 2+ |
| 43 | Seminoma | UU | 1+ | Hetero | Monoallelic | 1+ | Hetero | Monoallelic † | UU | 2+ |
| 44 | Seminoma | UU | 1+ | Hetero | Monoallelic | 1+ | Hetero | Biallelic | UU | 2+ |
| 45 | Seminoma | UU | 1+ | Homo | Not done | 1+ | Hetero | Monoallelic † | UU | 2+ |
UU/MM/MU/UM, see the legend for Table 2; Homo, homozygosity; Hetero, heterozygosity; Ca, carcinoma; DMR, differential methylated region; †Monoallelic, a faint opposite band is visible.
Figure 3.
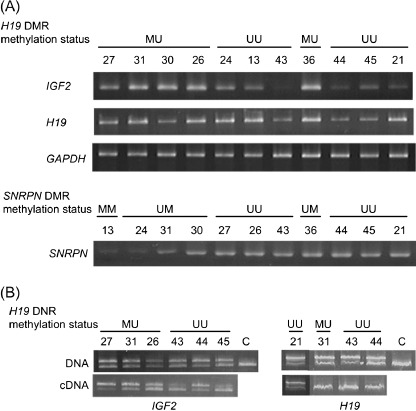
(A) The upper lanes show examples of IGF2 and H19 expression in germ cell tumors with various methylation statuses of H19 differential methylated region (DMR) (CCCTC‐binding factor binding site 6 [CTCF6]). The lower lanes show examples of SNRPN expression in germ cell tumors with various methylation statuses of SNRPN DMR. (B) Allelic expression analysis of IGF2 and H19 mRNA. Electrophoretic patterns of products of genomic DNA polymerase chain reaction (PCR) or reverse transcritpion‐PCR after ApaI digestion for IGF2, and after RsaI digestion for H19. Biallelic expression of IGF2 was found in Nos. 31, 26 and 44, and monoallelic expression of IGF2 with a faintly stained opposite band was found in Nos. 27, 43 and 45. Three tumors (Nos. 31, 43 and 44) showed monoallelic expression of H19, and one (No. 21) showed biallelic expression of H19. c, control.
Of four tumors with the heterozygous RsaI site, three and one showed monoallelic and biallelic expression, respectively, of H19. Two of three tumors with the H19 UU DMR and one with H19 MU DMR showed the monoallelic expression. Of six tumors with the heterozygous ApaI site, three and three showed monoallelic and biallelic expression, respectively, of IGF2. All three tumors (Nos. 27, 43 and 45) with the monoallelic expression simultaneously showed a faintly stained opposite band. Two tumors with the monoallelic expression and one with the biallelic expression had the H19 UU DMR, whereas one tumor with the monoallelic expression and two with the biallelic expression had the H19 MU DMR. Thus, there seems to be no correlation between the methylation status of H19 DMR and the allelic expression status of H19 and IGF2.
MSP analysis of various TSGs in germ cell tumors. We examined the methylation status of 17 TSGs in 45 GCT samples by MSP, and found hypermethylation of RASSF1A in 25 (55.6%), HOXA9 in 24 (53.3%), RUNX3 in 22 (48.9%), CASP8 in 16 (35.6%), SCGB3A1 in 16 (35.6%), SFRP2 in 16 (35.6%), DCR2 in 15 (33.3%), RASSF5 in eight (17.8%), RASSF2A in five (11.1%), BLU in three (6.7%), SFRP5 in two (4.4%), HOXB5 in one (2.2%), p16INK4A in one (2.2%) and SFRP1 in one (2.2%) (Table 4 and Fig. 4). No tumors showed hypermethylation of p14ARF, RIZ1 and SFRP4.
Table 4.
Tumor suppressor genes and the incidence of methylated seminoma, non‐seminoma, childhood yolk sac tumor, hepatoblastoma, Wilms tumor and neuroblastoma examined by methylation‐specific polymerase chain reaction
| Gene | Seminoma | Non‐seminoma | Childhood solid tumors | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yolk sac tumor (n = 25) | Hepatoblastoma | Wilms tumor | Neuroblastoma | |||||||||
| Present study (n = 9) | Lind et al. 2006 15 (n = 20) | Present study (n = 36) | Lind et al. 2006 15 (n = 35) | P‐value | (n = 20 † , 39 ‡ , 97 § ) | P‐value | (n = 20 ¶ , 39 †† , 40 ‡‡ ) | P‐value | (n = 56 §§ , 70 ¶¶ ) | P‐value | ||
| RASSF1A | 1 (11%) | 0 (0%) | 24 (67%) | 10 (29%) | 0.001 | 21 (84%) | 43 § (44%) | 0.0020 | 21 ‡ (54%) | 0.0272 | 39 † (70%) | 0.2768 |
| HOXA9 | 1 (11%) | 0 (0%) | 23 (64%) | 9 (26%) | 0.003 | 22 (88%) | 0 † (0%) | <0.0001 | 0 † (0%) | < 0.0001 | 0 † (0%) | <0.0001 |
| RUNX3 | 1 (11%) | 0 (0%) | 21 (58%) | 1 (3%) | <0.001 | 20 (80%) | 0 † (0%) | <0.0001 | 4 † (20%) | 0.0002 | 0/45 | |
| CASP8 | 0 (0%) | 16 (44%) | 14 (56%) | 15 § (16%) | <0.0001 | 17 § (43%) | 0.2891 | 39 ‡ (56%) | 0.9803 | |||
| SCGB3A1 | 1 (11%) | 0 (0%) | 15 (42%) | 19 (54%) | 0.287 | 13 (52%) | 0 ‡ (0%) | <0.0001 | 5 † (25%) | 0.0777 | 15 ‡ (21%) | 0.0040 |
| SFRP2 | 1 (11%) | 15 (42%) | 13 (52%) | 0 ‡ (0%) | <0.0001 | 2 † (10%) | 0.0040 | |||||
| DCR2 | 1 (11%) | 14 (39%) | 11 (44%) | 0 ‡ (0%) | <0.0001 | 6 † (30%) | 0.5137 | 31 ‡ (44%) | 0.9803 | |||
| RASSF5 | 1 (11%) | 7 (19%) | 7 (28%) | 0 † (0%) | 0.012 | 6 § (15%) | 0.3390 | 0/45 | ||||
| RASSF2A | 0 (0%) | 5 (14%) | 5 (20%) | 0 † (0%) | 0.0561 | 2 † (10%) | 0.4367 | 0/45 | ||||
| BLU | 0 (0%) | 3 (8%) | 3 (12%) | 0 † (0%) | 0.2424 | 0 † (0%) | 0.2424 | 5/45 | ||||
| SFRP5 | 0 (0%) | 2 (6%) | 1 (4%) | 0 ‡ (0%) | 0.3906 | 0 † (0%) | 0/45 | |||||
| HOXB5 | 0 (0%) | 0 (0%) | 1 (3%) | 5 (13%) | 0.107 | 1 (4%) | 0 † (0%) | 1 | 0 † (0%) | 1 | 0/45 | |
| P161NK4A | 0 (0%) | 1 (3%) | 0 (0%) | 0 † (0%) | 1 | 4 § (10%) | 0.2706 | 0/45 | ||||
| SFRP1 | 0 (0%) | 1 (3%) | 1 (4%) | 0 † (0%) | 1 | 0 † (0%) | 1 | 0/45 | ||||
| P14ARF | 0 (0%) | 0 (0%) | 0 (0%) | 0 † (0%) | 1 | 6 § (15%) | 0.0743 | 0/45 | ||||
| RIZ1 | 0 (0%) | 0 (0%) | 0 (0%) | 0 † (0%) | 1 | 0 † (0%) | 1 | 0/45 | ||||
| SFRP4 | 0 (0%) | 0 (0%) | 0 (0%) | 0 † (0%) | 1 | 0 † (0%) | 1 | 0/45 | ||||
Figure 4.
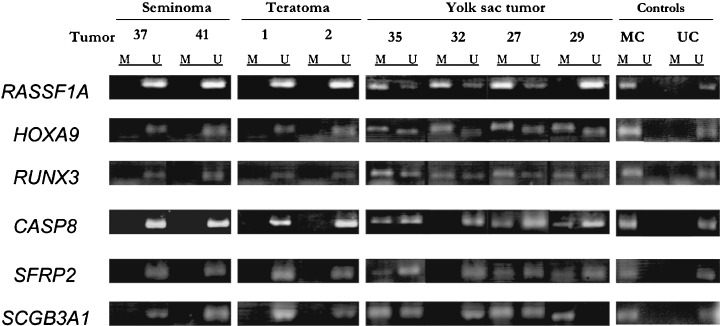
Examples of methylation status using methylation‐specific polymerase chain reaction (PCR). PCR products of methylated or unmethylated RASSF1A, HOXA9, RUNX3, CASP8, SFRP2 and SCGB3A1 from germ cell tumors are shown. M, methylated products; U, unmethylated products.
We compared the numbers of methylated TSGs between childhood yolk sac tumors and other childhood solid tumors (Table 4).( 25 , 26 , 27 , 28 ) The incidences of CASP8 and DCR2 methylation were similarly high in childhood yolk sac tumors, Wilms tumors and neuroblastomas, although the incidences were lower in hepatoblastomas. The incidences of RASSF1A, RUNX3 and SFRP2 methylation were higher in childhood yolk sac tumors than in hepatoblastomas (P = 0.0020, <0.0001 and < 0.0001) and Wilms tumors (P = 0.0272, < 0.0001 and 0.0040). HOXA9 and SGGB3A1 methylation was more frequent in yolk sac tumors than in hepatoblastomas (P < 0.0001 and <0.0001), Wilms tumors (P < 0.0001 and 0.0777) and neuroblastomas (P < 0.0001 and P = 0.0040). In summary, both the number of methylated TSGs and the incidence of TSG methylated tumors were higher in childhood yolk sac tumor than in other childhood solid tumors.
Association between epigenetic reprogramming pathways and clinical, pathological, and epigenetic findings ( Table 5 ). There were no differences in age, sex and the primary site between tumors in the normal pathway and those in the abnormal pathway by chi‐square test. In contrast, seminomas and teratomas were more frequent in the normal pathway than yolk sac tumors (P = 0.013). Furthermore, tumors with the UU/UU epigenotype and tumors with no methylated TSG were more frequent in the normal pathway than tumors with the other epigenotypes (P = 0.0007) and tumors with 1–9 methylated TSGs (P = 0.0009), respectively.
Table 5.
Association between two or three groups of tumors classified by clinical, pathological and epigenetic factors and reprogramming pathways
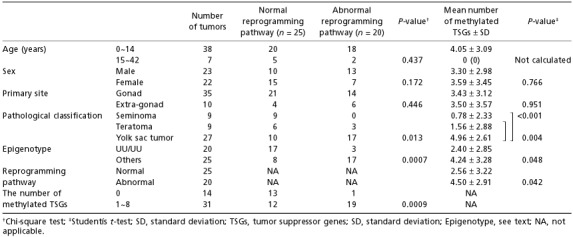
Differences in the number of methylated TSGs between two groups of tumors classified by clinical, pathological and epigenetic characteristics ( Table 5 ). All seven adult tumors had no methylated TSGs. There were no differences in the numbers of methylated TSGs between tumors in males and females, and between tumors developed in the gonad and in the extra‐gonadal site. In contrast, yolk sac tumors had a higher number of methylated TSGs than seminomas (P < 0.001) or teratomas (P = 0.004) (Table 5 and Fig. 5) by Student's t‐test. Tumors with the UU/UU epigenotype and tumors in the normal pathway had a lower number of methylated TSGs than tumors with other epigenotypes (P = 0.048) and tumors in the abnormal pathway, respectively (P = 0.042).
Figure 5.
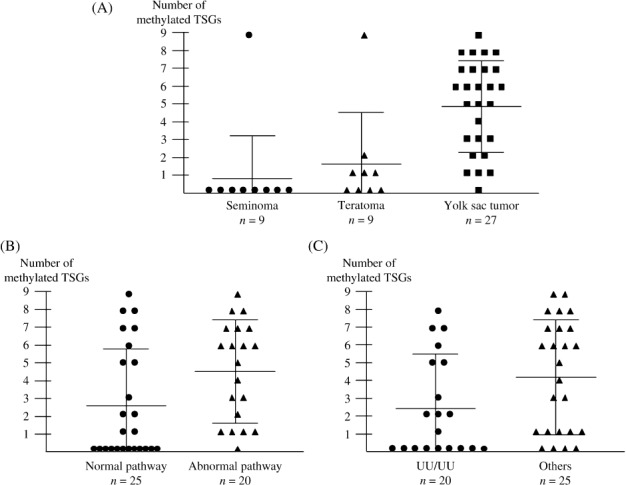
Numbers of methylated tumor suppressor genes (TSGs) in 45 germ cell tumors. The large and small horizontal bars indicate the mean number of methylated TSGs and the mean number ± standard deviation (SD), respectively. (A) The number of methylated TSGs was higher in yolk sac tumors than in seminomas (P < 0.001) or teratomas (P = 0.004) by Student's t‐test. (B, C) The number of methylated TSGs was lower in tumors in the normal reprogramming pathway than in tumors in the abnormal pathway (P = 0.042), and in tumors with the UU/UU epigenotype than in tumors with other epigenotypes (P = 0.048).
In 27 yolk sac tumors, there were no significant differences in the number of methylated TSGs between 22 gonadal tumors and five extra‐gonadal tumors, between 10 tumors in the normal reprogramming pathway and 17 tumors in the abnormal pathway, and between nine tumors with the UU/UU epigenotype and 18 tumors with other epigenotypes.
On the basis of the methylation status of the H19 and SNRPN DMRs and the numbers of methylated TSGs, we epigenetically characterized GCTs into three groups; seminomas with total erasure of H19 and SNRPN DMR methylation and few methylated TSGs; teratomas with mostly physiological methylation and demethylation of the H19 and SNRPN DMRs and few methylated TSGs, and yolk sac tumors with various methylation statuses of the H19 and SNRPN DMRs and a high incidence of methylated TSGs.
1p36 deletion and the methylation status of RUNX3 and RIZ1. SNP array analysis showed hemizygous deletion of the 1p36 chromosomal region in 3/7 tumors; the deletion region of 1p36 was 15.5–37.2 Mb in No. 23, 0–31.7 Mb in No. 34 and 0–35.6 Mb in No. 35 (National centre for Biotechnology Information [NCBI] v36.2). Because RUNX3 and RIZ1 are located in the 25.1 Mb and 13.9 Mb regions (NCBI v36.2), respectively, all three tumors lost the RUNX3 locus and 2/3 lost the RIZ1 locus. All three tumors with 1p36 deletion had RUNX3 methylation but no RIZ1 methylation, whereas the other four tumors without 1p36 deletion had neither RUNX3 methylation nor RIZ1 methylation.
Methylation status of TSGs and the outcome of patients. Of 45 patients who were followed from 3 to 278 months with a median of 97 months, only four died of the disease. Of these four patients, two children with yolk sac tumor had HOXA9 and CASP8 methylation; one of the two had RASSF1A methylation, one child with immature teratoma had DCR2 methylation and one adult with seminoma had no methylation of 17 TSGs. Conversely, 24 of 25 patients with RASSF1A methylation, 14 of 16 patients with CASP8 methylation, and 14 of 15 patients with DCR2 methylation were alive. Thus, there was no correlation between TSG methylation and the outcome in patients with GCT in the present study, although studies including a large number of deceased patients may be necessary to get a definite conclusion.
Discussion
Primordial germ cells undergo erasure of the somatic‐like epigenetic pattern, and are transformed into the sex‐specific pattern of mature germ cells during the epigenetic reprogramming process.( 4 , 5 , 6 ) Previous studies suggested that GCTs, thought to originate from PGCs, show epigenetic patterns reflecting the developmental process of PGCs.( 9 , 10 , 11 , 12 ) In the present series of 45 childhood and adult GCTs, 25 and 20 tumors showed the epigenetic patterns thought to reflect normal and abnormal reprogramming pathways, respectively (Table 2).
Kawakami et al.( 29 ) showed that the patterns of methylation of the promoter and CTCF‐binding site upstream of H19 were thoroughly unmethylated in testicular GCTs, and proposed that the patterns of methylation of H19 in testicular GCTs showed erasure of methylation. The present study, which showed the UU/UU epigenotype in all four seminomas and in all five dysgerminomas, confirmed the findings reported by Kawakami et al. ( 29 ) and furthermore indicated a novel evidence that dysgerminoma, that is, the counterpart of seminoma in females, also retained unmethylated H19 and SNRPN DMRs. Thus, we present data that seminoma developing in the testis and dysgerminoma developing in the ovary share not only the same morphological features but also the same epigenotype, indicating that both may be derived from PGCs in the same reprogramming process.
All four tumors with hypermethylated SNRPN DMR (MM) originated from the ovary, and 3/4 retained heterozygosity in SNRPN alleles. These findings suggest that PGCs that entered into female gametogenesis and underwent biparental methylation of SNRPN may have transformed to teratoma and yolk sac tumor (Fig. 2). Bussey et al. stated that SNRPN methylation pattern in germ cell tumors reflects primordial germ cell development.( 11 ) Other investigators also reported hypermethylation of SNRPN in some benign and immature ovarian teratomas.( 9 , 10 ) While three extra‐gonadal tumors showed the UU/UU epigenotype, which is the normal imprint status of gonadal tumors, suggesting that aberrant imprint erasure may have occurred in PGCs before entering the genital ridge, and five ovarian and one testicular tumors showed MU/UM, which is the normal imprint status of extra‐gonadal tumors, suggesting that incomplete imprint erasure may have occurred in PGCs in the ovary. Thus, the present study showed that half of PGCs giving rise to GCT underwent developmentally regulated erasure and resetting of imprinted genes, and that the other half of PGCs underwent inappropriate timing of imprint erasure or aberrant retention of the imprint. In regard to histology, while all seminomas and most teratomas were in the normal reprogramming pathway, the majority of yolk sac tumors were in the abnormal pathway.
In regard to the relationship between the methylation status of H19 DMR and H19 expression, tumors with somatically methylated H19 DMR (MU) showed a higher amount of IGF2 mRNA than tumors with hypomethylated H19 DMR (UU). Furthermore, the methylation status of H19 DMR was not correlated with monoallelic or biallelic expression of H19 or IGF2 (Table 3). These findings are not consistent with the enhancer completion model in somatic cells, that is, IGF2 and H19 promoters compete on the same chromosome for a shared enhancer, and access of the maternal IGF2 allele to this enhancer is blocked by H19 DMR when unmethylated, because of the insulator activity of CTCF binding to unmethylated H19 DMR.( 30 , 31 ) Jaenisch suggested that DNA methylation has no role in non‐somatic lineages such as embryonic stem (ES) cells, PGCs and cleavage embryo.( 32 ) The present and previous studies suggest that the hypothesis is applicable to GCT.( 29 )
In a study of 55 primary adult testicular GCTs, Lind et al.( 15 ) reported that non‐seminomas were significantly more often methylated than seminomas, and that the three most frequently methylated genes among this subtype were SCGB3A1 (54%), RASSF1A (29%), and HOXA9 (26%). Our data not only confirmed the higher incidence of TSG methylation in non‐seminomas than in seminomas but also revealed that the incidences of RASSF1A (67%), HOXA9 (64%) and RUNX3 (58%) methylation were much higher in the present series of non‐seminomas, mostly occurring in children, than in the previous series of adult testicular non‐seminomas, although there was no difference in the incidence of SCGB3A1 methylation between the two series of non‐seminomas (Table 4). In addition, a high incidence of RUNX3 methylation in infant testicular yolk sac tumors and absence of RUNX3 methylation in adult testicular non‐seminomas were reported previously.( 33 ) The present findings that all three childhood yolk sac tumors with 1p36 loss had RUNX3 methylation but no RIZ1 methylation, and that all four yolk sac tumors or teratomas with no 1p36 loss had neither RUNX3 nor RIZ1 methylation. The biological significance of RUNX3 methylation should be elucidated.
Recently, Calvanese et al. reported that some of the genes frequently hypermethylated in cancer are also frequently hypermethylated in human embryonic stem cells, and proposed a hypothesis that the aberrant methylation of some TSGs in cancer should be understood as a defect in establishing an unmethylated promoter during differentiation, rather than as an anomalous process of de novo hypermethylation.( 34 ) Yolk sac tumors had more frequent TSG methylation and were more frequent in the abnormal reprogramming pathway than seminomas or teratomas. Furthermore, the incidence of methylated TSGs was higher in childhood yolk sac tumors than in other childhood tumors. Nevertheless, methylation of TSGs did not affect the outcome of children with yolk sac tumors, although prognostic implication of methylated TSGs has been reported in many childhood and adult cancers.( 13 , 25 , 28 ) The number of methylated TSGs seems to be not correlated with specific methylation patterns of H19 and SNRPN DMRs in yolk sac tumors, although the number of the tumors with specific methylation patterns of the two DMRs may be too small to draw a conclusion. The high incidence but no prognostic significance of methylated TSGs in childhood yolk sac tumor may be explained by a hypothesis, that is, hypermethylation of TSGs may reflect epigenetic characteristics of PGCs giving rise to yolk sac tumor, but not the aberrant promoter methylation occurring in the tumorigenic process of various other cancers.( 34 ) The present findings are also consistent with the hypothesis proposed by Jaenisch who suggested that DNA methylation has no role in PGCs.( 32 ) In contrast, the low incidence of methylated TSGs in seminoma and teratoma suggests that TSGs in these tumors may be repressed by the Polycomb group of proteins but not by the promoter DNA methylation. In conclusion, the present study of epigenetic reprogramming pathways and TSG methylation in seminoma, teratoma and yolk sac tumors disclosed distinct epigenetic characteristics in each of the three histological subtypes of GCT.
Acknowledgments
This work was supported by the Ministry of Health, Labor and Welfare, Japan for Third‐term Comprehensive Control Research for Cancer (Y. Kaneko). We are grateful to the physicians who provided samples and clinical data.
References
- 1. Cushing B, Perlman E, Marina NM et al . Germ cell tumors. In: Pizo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology, 5th edn. Philadelphia: Lippincott‐Raven, 2006: 1116–38. [Google Scholar]
- 2. Woodward PJ, Heidenreich A, Looijenga LHJ et al . Germ cell tumours. In: Eble JN, Suter G, Epstein JI, Sesterhenn IA, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon: IARC Press, 2004: 221–49. [Google Scholar]
- 3. Oosterhuis JW, Looijenga LH. Testicular germ‐cell tumours in a broader perspective. Nat Rev Cancer 2005; 5: 210–22. [DOI] [PubMed] [Google Scholar]
- 4. Wylie C. Germ cells. Cell 1999; 96: 165–74. [DOI] [PubMed] [Google Scholar]
- 5. Surani MA. Reprogramming of genome function through epigenetic inheritance. Nature 2001; 414: 122–8. [DOI] [PubMed] [Google Scholar]
- 6. Hajkova P, Ancelin K, Waldmann T et al . Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 2008; 452: 877–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gabory A, Ripoche MA, Yoshimizu T, Dandolo L. The H19 gene: regulation and function of a non‐coding RNA. Cytogenet Genome Res 2006; 113: 188–93. [DOI] [PubMed] [Google Scholar]
- 8. Horsthemke B, Buiting K. Imprinting defects on human chromosome 15. Cytogenet Genome Res 2006; 113: 292–9. [DOI] [PubMed] [Google Scholar]
- 9. Miura K, Obama M, Yun K et al . Methylation imprinting of H19 and SNRPN genes in human benign ovarian teratomas. Am J Hum Genet 1999; 65: 1359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schneider DT, Schuster AE, Fritsch MK et al . Multipoint imprinting analysis indicates a common precursor cell for gonadal and nongonadal pediatric germ cell tumors. Cancer Res 2001; 61: 7268–76. [PubMed] [Google Scholar]
- 11. Bussey KJ, Lawce HJ, Himoe E et al . SNRPN methylation patterns in germ cell tumors as a reflection of primordial germ cell development. Genes Chromosomes Cancer 2001; 32: 342–52. [DOI] [PubMed] [Google Scholar]
- 12. Sievers S, Alemazkour K, Zahn S et al . IGF2/H19 imprinting analysis of human germ cell tumors (GCTs) using the methylation‐sensitive single‐nucleotide primer extension method reflects the origin of GCTs in different stages of primordial germ cell development. Genes Chromosomes Cancer 2005; 44: 256–64. [DOI] [PubMed] [Google Scholar]
- 13. Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358: 1148–59. [DOI] [PubMed] [Google Scholar]
- 14. Koul S, Houldsworth J, Mansukhani MM et al . Characteristic promoter hypermethylation signatures in male germ cell tumors. Mol Cancer 2002; 1: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lind GE, Skotheim RI, Fraga MF, Abeler VM, Esteller M, Lothe RA. Novel epigenetically deregulated genes in testicular cancer include homeobox genes and SCGB3A1 (HIN‐1 ). J Pathol 2006; 210: 441–9. [DOI] [PubMed] [Google Scholar]
- 16. Smiraglia DJ, Szymanska J, Kraggerud SM, Lothe RA, Peltomäki P, Plass C. Distinct epigenetic phenotypes in seminomatous and nonseminomatous testicular germ cell tumors. Oncogene 2002; 21: 3909–16. [DOI] [PubMed] [Google Scholar]
- 17. Japanese Pathological Society . Germ cell tumors and sex cord/stromal tumors in childhood. Committee on Histological Classification of Childhood Tumors. Tokyo: Kanahara Shuppan, 1999. [Google Scholar]
- 18. Watanabe N, Haruta M, Soejima H et al . Duplication of the paternal IGF2 allele in trisomy 11 and elevated expression levels of IGF2 mRNA in congenital mesoblastic nephroma of the cellular or mixed type. Genes Chromosomes Cancer, 2007; 46: 929–35. [DOI] [PubMed] [Google Scholar]
- 19. Kobayashi H, Sato A, Otsu E et al . Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum Mol Genet 2007; 16: 2542–51. [DOI] [PubMed] [Google Scholar]
- 20. Yamamoto G, Nannya Y, Kato M et al . Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of Affymetrix single‐nucleotide‐polymorphism genotyping microarrays. Am J Hum Genet 2007; 81: 114–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haruta M, Arai Y, Sugawara W et al . Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chromosomes Cancer 2008; 47: 712–27. [DOI] [PubMed] [Google Scholar]
- 22. Tanaka K, Shiota G, Meguro M et al . Loss of imprinting of long QT intronic transcript 1 in colorectal cancer. Oncology 2001; 60: 268–73. [DOI] [PubMed] [Google Scholar]
- 23. Reed ML, Leff SE. Maternal imprinting of human SNRPN, a gene deleted in Prader–Willi syndrome. Nat Genet 1994; 6: 163–7. [DOI] [PubMed] [Google Scholar]
- 24. Honda S, Haruta M, Sugawara W et al . The methylation status of RASSF1A promoter predicts responsiveness to chemotherapy and eventual cure in hepatoblastoma patients. Int J Cancer 2008; 123: 1117–25. [DOI] [PubMed] [Google Scholar]
- 25. Yang Q, Zage P, Kagan D et al . Association of epigenetic inactivation of RASSF1A with poor outcome in human neuroblastoma. Clin Cancer Res 2004; 10: 8493–500. [DOI] [PubMed] [Google Scholar]
- 26. Morris MR, Hesson LB, Wagner KJ et al . Multigene methylation analysis of Wilms’ tumour and adult renal cell carcinoma. Oncogene 2003; 22: 6794–801. [DOI] [PubMed] [Google Scholar]
- 27. Astuti D, Da Silva NF, Dallol A et al . SLIT2 promoter methylation analysis in neuroblastoma, Wilms’ tumour and renal cell carcinoma. Br J Cancer 2004; 90: 515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang Q, Kiernan CM, Tian Y et al . Methylation of CASP8, DCR2, and HIN‐1 in neuroblastoma is associated with poor outcome. Clin Cancer Res 2007; 13: 3191–7. [DOI] [PubMed] [Google Scholar]
- 29. Kawakami T, Zhang C, Okada Y, Okamoto K. Erasure of methylation imprint at the promoter and CTCF‐binding site upstream of H19 in human testicular germ cell tumors of adolescents indicate their fetal germ cell origin. Oncogene 2006; 25: 3225–36. [DOI] [PubMed] [Google Scholar]
- 30. Bell AC, Felsenfeld G. Methylation of a CTCF‐dependent boundary controls imprinted expression of the Igf2 gene. Narture 2000; 405: 482–5. [DOI] [PubMed] [Google Scholar]
- 31. Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation‐sensitive enhancer‐blocking activity at the H19/Igf2 locus. Nature 2000; 405: 486–9. [DOI] [PubMed] [Google Scholar]
- 32. Jaenisch R. DNA methylation and imprinting: why bother? Trend Genet 1997; 13: 323–9. [DOI] [PubMed] [Google Scholar]
- 33. Kato N, Tamura G, Fukase M, Shibuya H, Motoyama T. Hypermethylation of the RUNX3 gene promoter in testicular yolk sac tumor of infants. Am J Pathol 2003; 163: 387–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Calvanese V, Horrillo A, Hmadcha A et al . Cancer genes hypermethylated in human embryonic stem cells. Plos ONE 2008; 3: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]