

Abstract
Obesity and related metabolic abnormalities, including insulin resistance, are risk factors for hepatocellular carcinoma in non‐alcoholic steatohepatitis as well as in chronic viral hepatitis. Branched‐chain amino acids (BCAA), which improve insulin resistance, inhibited obesity‐related colon carcinogenesis in a rodent model, and also reduced the incidence of hepatocellular carcinoma in obese patients with liver cirrhosis. In the present study, we determined the effects of BCAA on the development of diethylnitrosamine (DEN)‐induced liver tumorigenesis in obese C57BL/KsJ‐db/db (db/db) mice with diabetes mellitus. Male db/db mice were given tap water containing 40 ppm DEN for an initial 2 weeks and thereafter they received a basal diet containing 3.0% of BCAA or casein, which served as a nitrogen content‐matched control of BCAA, throughout the experiment. Supplementation with BCAA significantly reduced the total number of foci of cellular alteration, a premalignant lesion of the liver, and the expression of insulin‐like growth factor (IGF)‐1, IGF‐2, and IGF‐1 receptor in the liver when compared to the casein supplementation. BCAA supplementation for 34 weeks also significantly inhibited both the development of hepatocellular neoplasms and the proliferation of hepatocytes in comparison to the basal diet or casein‐fed groups. Supplementation with BCAA improved liver steatosis and fibrosis and inhibited the expression of α‐smooth muscle actin in the DEN‐treated db/db mice. The serum levels of glucose and leptin decreased by dietary BCAA, whereas the value of the quantitative insulin sensitivity check index increased by this agent, indicating the improvement of insulin resistance and hyperleptinemia. In conclusion, oral BCAA supplementation improves insulin resistance and prevents the development of liver tumorigenesis in obese and diabetic mice. (Cancer Sci 2009)
Hepatocellular carcinoma is a major health problem worldwide. The development of HCC is frequently associated with chronic inflammation of the liver induced by a persistent infection with the hepatitis B virus or hepatitis C virus.( 1 ) The risk of HCC is also elevated in those with metabolic syndrome, also called insulin resistance syndrome, which is commonly associated with obesity and impaired glucose tolerance.( 1 , 2 , 3 , 4 ) Non‐alcoholic fatty liver disease is known to be a hepatic manifestation of the metabolic syndrome. Diabetes mellitus, a condition associated with hyperinsulinemia, has been proposed as a risk factor for both chronic liver disease and HCC through the development of NASH, which is observed in a subset of patients with non‐alcoholic fatty liver disease and involves inflammation, cell damage, and/or fibrosis in the liver.( 5 , 6 , 7 ) In 1998, Day and James proposed, in their “two hit theory,” that insulin resistance is regarded as a critical factor in the etiology of NASH.( 8 )
C57BL/KsJ‐db/db (db/db) mice are a genetically altered animal model with phenotypes of obesity and diabetes mellitus. A functional defect in the long‐form leptin receptor, which plays a significant role in the regulation of food intake and the control of body weight, leads to hyperleptinemia in these mice.( 9 ) Because of such obesity, hyperinsulinemia, and hyperleptinemia, the db/db mice represent a suitable animal model that uniquely mimics the metabolic syndrome in humans.( 10 ) It is also reported that the db/db mice are susceptible to chemically induced carcinogenesis in certain tissues. For instance, the db/db mice are sensitive to AOM‐induced colon carcinogenesis; putative precursor lesions for colorectal cancer are greatly enhanced in db/db mice compared to db/+ or +/+ mice.( 11 ) With respect to the liver, feeding db/db mice with a methionine and choline‐deficient diet developed accelerated hepatic inflammation and fibrosis, very similar to those seen in human NASH.( 12 )
An improvement of insulin resistance by nutritional or pharmaceutical intervention might therefore be an effective and attractive strategy to inhibit the obesity‐related carcinogenesis, as already reported experimentally for the colon.( 13 ) Candidate modalities include dietary supplementation with BCAA (leucine, isoleucine, and valine) because BCAA prevents progressive hepatic failure and improves the event‐free survival in patients with chronic liver diseases, at least in part, by improving insulin resistance.( 14 , 15 , 16 ) In addition, BCAA supplementation has been shown to prevent obesity‐related colon carcinogenesis initiated with AOM( 17 ) and, furthermore, to reduce the risk of HCC in obese patients with chronic viral liver disease.( 18 ) In an obesity‐related colon cancer model, the effects of BCAA in inhibiting the development of colonic premalignancies might be associated with improvement of insulin resistance.( 17 ) However, whether BCAA prevents obesity‐related liver carcinogenesis, and the precise mechanisms of that prevention, have not been explored.
In the present study, we examined the effects of BCAA supplementation on the development of HCC, liver cell adenoma, and FCA in obese and diabetic db/db mice initiated with DEN by focusing on the improvement of insulin resistance, liver steatosis, and fibrosis. We also examined whether BCAA supplementation in the diet alters the expression of IGF‐1, IGF‐2, and IGF‐1R in the liver of DEN‐treated db/db mice. The IGF/IGF‐1R axis is closely associated with the development of HCC and might be regarded as a critical target for both HCC treatment and chemoprevention.( 19 , 20 )
Materials and Methods
Animals, chemicals, and diets. Four‐week‐old male db/db mice were obtained from Japan SLC (Shizuoka, Japan). All mice received humane care and were maintained at Gifu University Life Science Research Center (Gifu, Japan), according to the Institutional Animal Care Guidelines. DEN was purchased from Sigma Chemical Co. (St. Louis, MO, USA). BCAA and casein were obtained from Ajinomoto Co. (Tokyo, Japan). The BCAA composition (2:1:1.2 = leucine:isoleucine:valine) was set at the clinical dosage that is used for the treatment of decompensated liver cirrhosis in Japan.( 16 , 18 ) The basal diet, CRF‐1 (Oriental Yeast Co., Tokyo, Japan), contained 22.4 g of protein (1.65 g leucine, 0.83 g isoleucine, and 1.03 g valine) per 100 g of total volume.
Experimental procedure. The experimental protocol was approved by the Institutional Committee of Animal Experiments of Gifu University. At 5 weeks of age, a total of 41 db/db mice were divided into three groups. All the mice in Group 1 (n = 11), Group 2 (n = 15), and Group 3 (n = 15) were given tap water containing 40 ppm DEN for the initial 2 weeks. After treatment with DEN, Group 3 was given the CRF‐1 supplemented with 3.0% BCAA (w/w) through to the end of experiment, whereas mice in Group 2 were given the basal diet supplemented with 3.0% casein (w/w) and served as a nitrogen content‐matched control for the BCAA‐treated group. Group 1 was given the CRF‐1 diet throughout the experiment. In order to examine the effect of BCAA on the development of FCA in early phase, four mice each in groups 2 and 3 were starved for 6 h and killed by CO2 asphyxiation at 23 weeks of age (after 16 weeks supplementation with BCAA or casein). At 41 weeks of age (after 34 weeks supplementation with the experimental diet), all remaining animals (total 33 mice) were killed to determine the development of HCC, liver cell adenoma, and FCA.
Histopathology and immunohistochemical analyses for α‐SMA and PCNA. After the mice were killed, the livers were immediately removed and macroscopically inspected for the presence of neoplasms. Maximum sagittal sections of each lobe (six lobes) were used for histological examination. The tissue specimens were fixed in 10% buffered formaldehyde then embedded in paraffin. Serial sections (3–4 μm thick) were cut from the tissue blocks and stained with H&E for histopathology or Azan stain to observe liver fibrosis. The liver neoplasms (HCC and liver cell adenoma) and FCA were diagnosed according to criteria described previously.( 21 ) The multiplicity of FCA was assessed on the per area basis (per cm2).
Immunohistochemistry of α‐SMA, an indicator of HSC activation, was carried out using a primary anti‐α‐SMA antibody (Dako, Glostrup, Denmark).( 22 ) Immunohistochemistry of PCNA, a G1‐to‐S phase marker, was carried out to estimate the cell proliferative activity of the hepatocyte using a primary anti‐PCNA antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA).( 23 ) PCNA‐positive nuclei in the hepatocytes were counted and expressed as the percentage of the total number of hepatocyte nuclei. The PCNA‐labeling index (%) was determined by counting at least 500 hepatocytes in each section (total of 3000 hepatocytes per mouse).
Clinical chemistry. After the mice were killed, blood samples were collected from inferior vena cava to determine the serum concentrations of ALT, glucose, insulin, leptin, and BCAA. The levels of serum glucose, insulin, and BCAA were assayed as described previously.( 24 , 25 ) The serum leptin level was determined by ELISA (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol.( 17 ) The serum ALT activity was measured with a standard clinical automatic analyzer (type 726; Hitachi, Tokyo, Japan). Insulin resistance was estimated by QUICKI as follows: QUICKI = 1/[log(I0) + log(G0)], where I0 is the fasting insulin and G0 is the fasting glucose, which correlates with the glucose clamp method.( 26 )
Hepatic lipid analysis. To visualize intrahepatic lipids, Sudan III stain was carried out with frozen sections using the standard procedure. The hepatic lipids were also extracted from the frozen livers. Approximately 200 mg of liver was homogenized and the lipids were then extracted using chloroform:methanol (2:1 v/v) solution, as described by Folch.( 27 ) The levels of triglyceride in the liver were measured using the triglyceride E‐test kit (Wako Pure Chemical Co., Osaka, Japan) according to the manufacturer’s protocol.
Hepatic hydroxyproline analysis. Hepatic hydroxyproline content was quantified colorimetrically in duplicate samples from approximately 200 mg wet‐weight of liver tissue, as previously described.( 22 ) The hydroxyproline contents were expressed as μmol/g wet liver.
Protein extraction and Western blot analysis. Equivalent amounts of protein lysates (30 μg/lane) from the liver of experimental mice were subjected to a Western blot analysis of α‐SMA (Dako), as described previously.( 22 , 23 ) An antibody to GAPDH (Chemicon International, Temecula, CA, USA) served as a loading control. The intensities of the blots were quantified with NIH image software version 1.62.
RNA extraction and quantitative real‐time RT‐PCR analysis. A quantitative real‐time RT‐PCR analysis was carried out as described previously.( 28 ) Total RNA was isolated from the liver of the mice using the RNAqueous‐4PCR kit (Ambion Applied Biosystems, Austin, TX, USA). The cDNA was synthesized from 0.2 μg total RNA using the SuperScript III First‐Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). The primers used for the amplification of IGF‐1, IGF‐2, and IGF‐1R specific genes were as follows: IGF‐1 forward, 5′‐CTGGACCAGAGACCCTTTGC‐3′ and reverse, 5′‐GGACGGGGACTTCTGAGTCTT‐3′; IGF‐2 forward, 5′‐GTGCTGCATCGCTGC‐TTAC‐3′ and reverse, 5′‐ACGTCCCTCTCGGACTTGG‐3′; and IGF‐1R forward, 5′‐GTGGGGGCTCGTGTTTCTC‐3′ and reverse, 5′‐GATCACCGTGCAGTTTTCCA‐3′. Real‐time PCR was done in a LightCycler (Roche Diagnostics Co., Indianapolis, IN, USA) with SYBR Premix Ex Taq (TaKaRa Bio, Shiga, Japan). The expression levels of the IGF‐1, IGF‐2, and IGF‐1R genes were normalized to the β‐actin gene expression level.( 28 )
Statistical analysis. The results are presented as the mean ± SD, and they were analyzed using the GraphPad Instat software program version 3.05 (GraphPad Software, San Diego, CA, USA) for Macintosh. Differences among the groups were analyzed by either one‐way ANOVA or, as required, by two‐way ANOVA. When ANOVA showed a statistically significant effect (P < 0.05), comparisons of each experimental group with the control group were then made using Dunnett’s test, which corrects for multiple comparisons. The differences were considered to be significant when the two‐sided P value was <0.05.
Results
General observations. As shown in Table 1, there were no significant differences in the body, liver, kidney, or fat (white adipose tissue of the periorchis and retroperitoneum) weights among the groups at the end of the study. Male db/db mice well‐tolerated the treatment with DEN together with casein or BCAA. The body weight gains did not differ significantly among the groups during the experiment (data not shown). A histopathological examination suggested the absence of toxicity of BCAA in important organs, including liver, kidney, and spleen (data not shown). In addition, no clinical signs indicating the toxicity of BCAA were observed in the mice during the experiment.
Table 1.
Body, liver, kidney, spleen, and fat weights of the experimental mice
| Group no. | Diet | No. of mice | Weight (g) (mean ± SD) | |||
|---|---|---|---|---|---|---|
| Body | Liver | Kidney | Fat† | |||
| 1 | CRF‐1 | 11 | 73.0 ± 9.2 | 4.4 ± 0.9 | 0.9 ± 1.0 | 7.8 ± 2.2 |
| 2 | Casein | 11 | 66.0 ± 12.0 | 3.8 ± 1.2 | 0.6 ± 0.2 | 5.4 ± 1.0 |
| 3 | BCAA | 11 | 68.2 ± 12.4 | 3.4 ± 1.3 | 0.6 ± 0.1 | 6.2 ± 1.4 |
†White adipose tissue of the periorchis and retroperitoneum.
Incidence and multiplicity of DEN‐induced liver neoplasms and FCA in db/db mice. Macroscopically, nodular lesions (Fig. 1a) were observed in the livers of experimental mice at the termination of the study (41 weeks of age). Histopathologically, these lesions were liver cell adenoma (Fig. 1b) or HCC (Fig. 1c). FCA (Fig. 1d) also developed in the liver of experimental mice. Simultaneously, we put supplemental groups to support that db/db mice are actually susceptible to DEN‐induced liver tumorigenesis (data not shown; see Supporting Information Table S1) and found no neoplasms in C57B6 or C57BL/KsJ‐+/+ mice, genetic controls for db/db mice, regardless of DEN treatment. No tumors developed in the CRF‐1‐fed and DEN‐untreated db/db mice.
Figure 1.
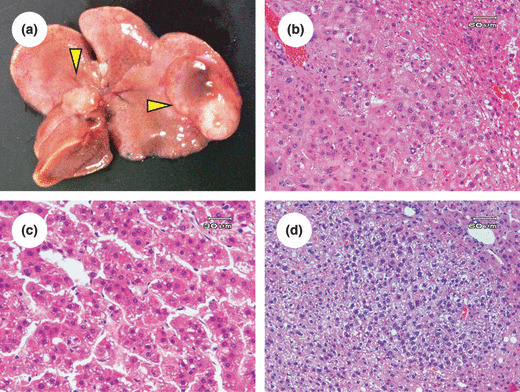
Macroscopic (a) and microscopic (b–d) analyses of liver neoplasms in diethylnitrosamine‐treated db/db mice. (a) Macroscopically, white tumors (hepatocellular carcinoma; indicated by arrowheads) were detected in the liver of diethylnitrosamine‐treated C57BL/KsJ‐db/db mice. (b–d) Paraffin‐embedded sections were stained with H&E. Representative photomicrographs show adenoma (b), hepatocellular carcinoma (c), and foci of cellular alteration (d) in liver of experimental mice.
Effects of BCAA supplementation on DEN‐induced liver tumorigenesis in db/db mice. The incidence and multiplicity of liver neoplasms (adenoma plus HCC) and FCA at 41 weeks of age are summarized in Table 2. Compared with the CRF‐1‐fed mice (Group 1), dietary supplementation with BCAA (Group 3) significantly inhibited the incidence (P < 0.05) of adenoma. BCAA supplementation also reduced the incidence (P < 0.05) and multiplicity of adenoma (P < 0.01) compared to the casein‐supplementation mice (Group 2). HCC was developed in the CRF‐1‐fed (9%) and casein‐supplementation mice (27%), but not in the mice supplemented with BCAA (0%), and the multiplicity of total liver neoplasms was significantly inhibited by supplementation with BCAA when compared to CRF‐1‐fed (P < 0.05) or Casein‐supplementation mice (P < 0.01), respectively. The number of FCA, which were developed in all experimental mice, was also significantly decreased by supplementation with BCAA when compared to CRF‐1‐fed (P < 0.05) or casein‐supplementation mice (P < 0.001), respectively.
Table 2.
Incidence and multiplicity of hepatic neoplasms and foci of cellular alteration (FCA) in obese diabetic C57BL/KsJ‐db/db mice fed basal (CRF‐1), casein‐supplemented, or branched‐chain amino acid (BCAA)‐supplemented diets
| Group no. | Diet | No. of mice | Incidence (%) | Multiplicity (no. of neoplasms/mouse) (mean ± SD) | FCA (No./cm2) (mean ± SD) | |||
|---|---|---|---|---|---|---|---|---|
| Adenoma | HCC | Total | Adenoma | HCC | ||||
| 1 | CRF‐1 | 11 | 7/11 (64) | 1/11 (9) | 1.0 ± 1.1 | 0.9 ± 1.1 | 0.1 ± 0.3 | 14.4 ± 4.4 |
| 2 | Casein | 11 | 8/11 (73) | 3/11 (27) | 1.7 ± 1.3 | 1.5 ± 1.1 | 0.3 ± 0.5 | 19.1 ± 5.7 |
| 3 | BCAA | 11 | 2/11 (18)*,** | 0/11 (0) | 0.2 ± 0.4*,*** | 0.2 ± 0.4*** | 0 | 9.6 ± 5.1*,**** |
*P < 0.05, significantly different from Group 1; **P < 0.05, significantly different from Group 2; ***P < 0.01, significantly different from Group 2; ****P < 0.001, significantly different from Group 2. HCC, hepatocellular carcinoma.
Effects of BCAA supplementation on the expression levels of IGF‐1, IGF‐2, and IGF‐1R mRNAs in the liver of DEN‐treated db/db mice. When the mice were killed at 23 weeks of age, the development of FCA was also significantly inhibited by dietary supplementation with BCAA compared with casein‐supplemented mice (P < 0.01) (Fig. 2a). In addition, semiquantitative RT‐PCR analyses showed that there was a significant decrease in the expression level of IGF‐1 (P < 0.05), IGF‐2 (P < 0.05), and IGF‐1R mRNAs (P < 0.05) in the livers of the mice supplemented with BCAA when compared to that of the livers in casein‐supplemented mice (Figs 2b–d). These findings suggest that BCAA supplementation prevents the development of FCA, at least in part, by inhibiting the expression of the IGF/IGF‐1R axis.
Figure 2.
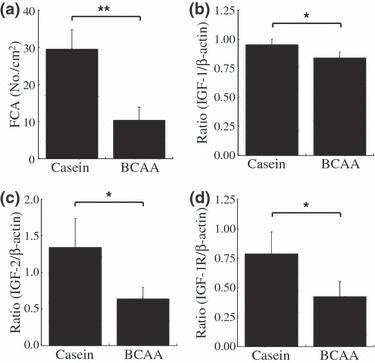
Effect of branched‐chain amino acid (BCAA) supplementation on the development of foci of cellular alteration (FCA) and on the expression of insulin‐like growth factor (IGF)‐1, IGF‐2 and IGF‐1 receptor (IGF‐1R) mRNAs in the liver of diethylnitrosamine‐treated C57BL/KsJ‐db/db mice. Livers were excised from treated mice supplemented with casein or BCAA for 16 weeks. (a) Paraffin‐embedded liver sections were stained with H&E and the total numbers of FCA were counted. Values are the means ± SD (n = 4). (b–d) Total RNA was isolated from the removed liver and the expression of IGF‐1 (b), IGF‐2 (c), and IGF‐1R (d) genes were examined by quantitative real‐time RT‐PCR. The expression of each gene was normalized to β‐actin expression. Each experiment was done in triplicate. *P < 0.05; **P < 0.01.
Effects of BCAA supplementation on serum levels of BCAA, ALT, and leptin in DEN‐treated db/db mice. BCAA supplementation caused a significant increase in the serum levels of BCAA compared to the CRF‐1‐fed (P < 0.05) and casein‐supplemented mice (P < 0.05) (Fig. 3a). These findings suggest that supplementation with 3.0% BCAA is sufficient to raise the serum concentration of BCAA. The serum ALT levels markedly increased in the db/db mice when compared to the genetic control mice (data not shown; see Supporting Information Table S1). However, BCAA supplementation significantly decreased this value in comparison to the CRF‐1‐fed (P < 0.01) and casein‐supplemented mice (P < 0.001) (Fig. 3b), thus indicating an improvement of liver damage. In addition, the mice supplemented with BCAA showed a decrease in the serum levels of leptin compared with the CRF‐1‐fed (P < 0.001) and casein‐supplemented mice (P < 0.001) (Fig. 3c).
Figure 3.
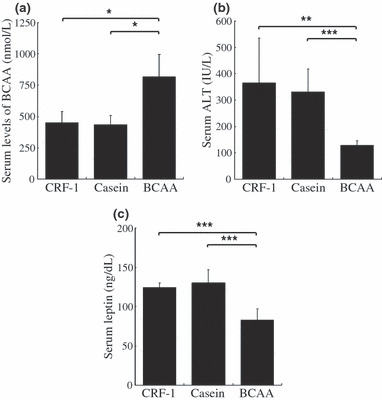
Effect of branched‐chain amino acid (BCAA) supplementation on the serum levels of BCAA, alanine aminotransferase (ALT), and leptin in diethylnitrosamine‐treated C57BL/KsJ‐db/db mice. After mice were killed, blood samples were collected and the serum levels of BCAA (a), ALT (b), and leptin (c) were then assayed. Values are the means ± SD (n = 8). *P < 0.05; **P < 0.01; ***P < 0.001.
Effects of BCAA supplementation on the hepatic steatosis in DEN‐treated db/db mice. Examination of Sudan III stained sections revealed that there was a marked macrovesicular steatosis in the DEN‐treated db/db mice, which were fed CRF‐1 or casein, but BCAA supplementation significantly improved the accumulation of the lipid in the liver (Fig. 4a). The histological findings were consistent with the results of the measurement of liver triglyceride contents; the levels of triglyceride in the liver of DEN‐treated db/db mice were significantly decreased by the supplementation with BCAA compared to those in the CRF‐1‐fed (P < 0.001) and casein‐supplementation groups (P < 0.01) (Fig. 4b).
Figure 4.
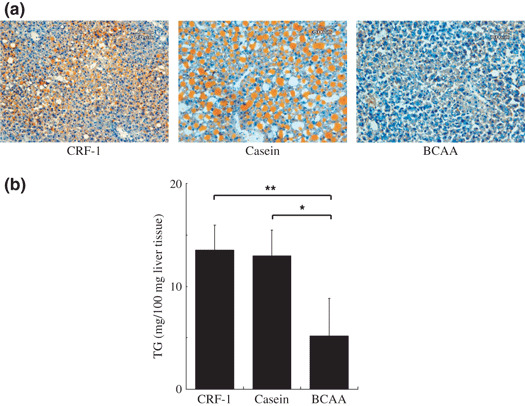
Effect of branched‐chain amino acid (BCAA) supplementation on hepatic steatosis in diethylnitrosamine‐treated C57BL/KsJ‐db/db mice. (a) Frozen sections of basal diet (CRF‐1)‐fed, casein‐supplemented, or BCAA‐supplemented treated mice were stained with Sudan III stain to show steatosis. (b) Hepatic lipids were extracted from the frozen livers and the levels of triglyceride were then measured. Values are the means ± SD (n = 8). *P < 0.01; **P < 0.001.
Effects of BCAA supplementation on liver fibrosis in DEN‐treated db/db mice. As shown in Figure 5a, examination of Azan‐stained sections indicated that DEN‐treated db/db mice of CRF‐1‐fed and casein‐supplemented groups showed the development of peri‐central venous and peri‐cellular fibrosis. However, supplementation with BCAA yielded an improvement in liver fibrosis (Fig. 5a). Similar findings were also observed in the measurement of liver hydroxyproline contents, a useful marker of hepatic fibrosis;( 22 ) when compared to CRF‐1 feeding (P < 0.05) and casein supplementation (P < 0.01), BCAA supplementation caused a significant decrease in the amounts of hydroxyproline in the liver of DEN‐treated db/db mice (Fig. 5b). In addition, both the immunohistochemical (Fig. 6a) and Western blot analyses (Fig. 6b) showed the expression levels of α‐SMA in the liver to be elevated in the CRF‐1‐fed and casein‐supplemented mice, whereas supplementation with BCAA significantly decreased the expression of this protein (P < 0.05 and P < 0.01, respectively).
Figure 5.
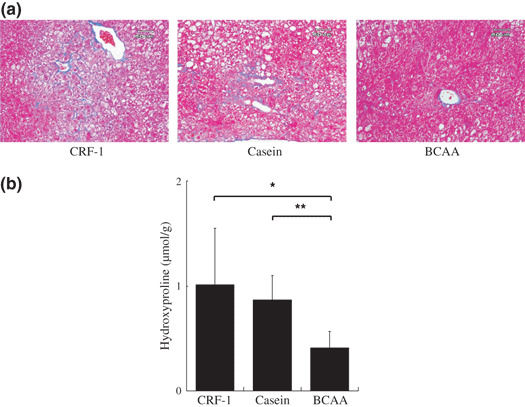
Effect of branched‐chain amino acid (BCAA) supplementation on hepatic fibrosis in diethylnitrosamine‐treated C57BL/KsJ‐db/db mice. (a) Paraffin‐embedded sections of basal diet (CRF‐1)‐fed, casein‐supplemented, or BCAA‐supplemented treated mice were stained with Azan stain to show fibrosis. (b) Hepatic hydroxyproline contents were quantified colorimetrically. Values are the means ± SD (n = 8). *P < 0.05; **P < 0.01.
Figure 6.
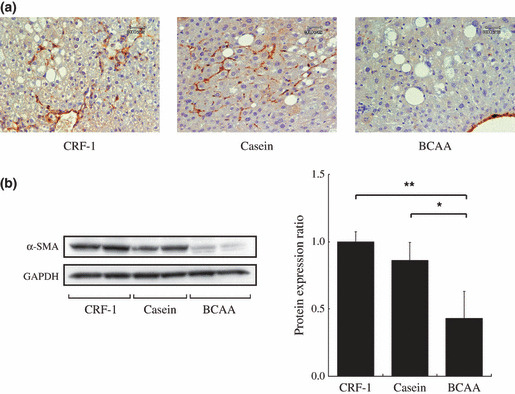
Effect of branched‐chain amino acid (BCAA) supplementation on the expression of α‐smooth muscle actin (α‐SMA) in diethylnitrosamine‐treated C57BL/KsJ‐db/db mice. (a) Immunohistochemical expression of α‐SMA in the liver of basal diet (CRF‐1)‐fed, casein‐supplemented, or BCAA‐supplemented treated mice. (b) Total protein was extracted from the liver of experimental mice and the expression of α‐SMA protein was examined by Western blot analysis. An antibody to GAPDH served as a loading control. Repeat Western blots gave similar results. The results obtained were quantitated by densitometry and are shown in the right‐hand panels. Values are the means ± SD (n = 5). *P < 0.05; **P < 0.01.
Effects of BCAA supplementation on insulin resistance and serum level of glucose in DEN‐treated db/dbdb mice. Insulin resistance plays a critical role in obesity‐related HCC development.( 1 , 2 , 3 , 4 ) Therefore, the effects of BCAA supplementation on the value of QUICKI and the serum levels of glucose were examined in DEN‐treated db/db mice. As shown in Figure 7a, supplementation with BCAA caused a significant increase in the value of QUICKI compared to the CRF‐1‐fed (P < 0.01) and casein‐supplemented mice (P < 0.01), thus indicating an improvement of insulin resistance. The serum glucose level also decreased after the supplementation with BCAA compared to CRF‐1‐fed (P < 0.001) and casein‐supplementation (P < 0.01) (Fig. 7b).
Figure 7.
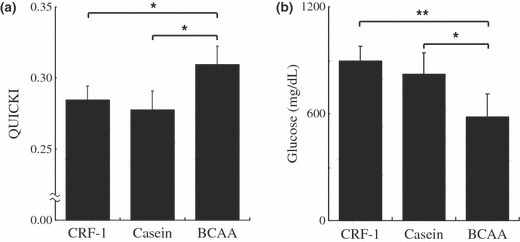
Effect of branched‐chain amino acid (BCAA) supplementation on insulin sensitivity and the serum level of glucose in diethylnitrosamine‐treated C57BL/KsJ‐db/db mice fed basal diet (CRF‐1), or supplemented with casein or BCAA. (a) The value of the quantitative insulin sensitivity check index (QUICKI), was calculated to evaluate the insulin sensitivity. (b) The serum concentration of glucose was measured by the hexokinase method. Values are the means ± SD (n = 8). *P < 0.01; **P < 0.001.
Effects of BCAA supplementation on cell proliferative activity in liver of DEN‐treated db/db mice. The PCNA‐labeling index of non‐lesional hepatocytes in DEN‐treated db/db mice was determined based on the findings of PCNA‐immunohistochemical sections (Fig. 8a). As illustrated in Figure 8b, the mean PCNA‐labeling index in the BCAA‐supplemented mice was significantly lower than that of the CRF‐1‐fed (P < 0.01) and casein‐supplemented mice (P < 0.05), thus indicating that BCAA supplementation significantly inhibited cell proliferation in the liver of DEN‐treated db/db mice.
Figure 8.
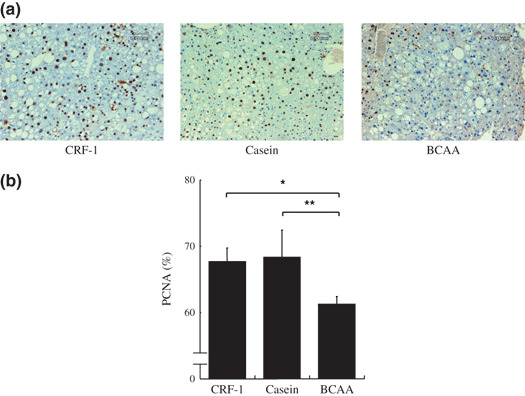
Effect of branched‐chain amino acid (BCAA) supplementation on hepatic cell proliferation in diethylnitrosamine‐treated C57BL/KsJ‐db/db mice. (a) Immunohistochemical expression of proliferating cell nuclear antigen (PCNA) in the liver of basal diet (CRF‐1)‐fed, or casein‐ or BCAA‐supplemented treated mice. (b) PCNA‐labeling index in non‐lesional hepatocytes was determined by counting the PCNA‐positive nuclei in the hepatocytes. *P < 0.05; **P < 0.01.
Discussion
Recent studies have shown that obesity and diabetes mellitus are risk factors for HCC through the development of NASH.( 5 , 6 , 7 ) The present study clearly indicated that db/db mice, which develop obesity and severe diabetes mellitus, easily developed steatosis‐related liver neoplasms by treatment with liver carcinogen DEN (Table 2 and Fig. 1), whereas background C57B6 or C57BL/KsJ‐+/+ mice did not. Furthermore, this study showed that dietary supplementation with BCAA effectively decreased the serum levels of ALT (Fig. 3b), which increase due to severe steatosis (Fig. 4a) and fibrosis (Fig. 5a), and inhibited the development of liver neoplasms (Table 2) in DEN‐treated db/db mice. A clinical trial recently indicated that dietary supplementation with BCAA can reduce the risk of HCC in cirrhotic patients who are obese.( 18 ) How can BCAA exert chemopreventive effects on obesity‐related HCC? Presumably, the improvement of insulin resistance by BCAA (Fig. 7a) plays a critical role in this beneficial effect because, in addition to the role of insulin in glucose uptake and glycogen biosynthesis in liver and skeletal muscle, insulin has oncogenic properties on HCC cells, including the stimulation of cell growth and induction of anti‐apoptotic activity.( 29 , 30 ) These reports, therefore, suggest the possibility that BCAA inhibits the excessive cell proliferation in the whole liver of DEN‐treated db/db mice (Fig. 8) by improving insulin resistance (Fig. 7a). Recent studies have also revealed that BCAA improves glucose tolerance by modulating the insulin‐independent glucose uptake into skeletal muscle.( 31 , 32 ) Isoleucine increased muscle glucose uptake and depressed gluconeogenesis in the liver without causing significant elevation of the plasma insulin level, thereby leading to the hypoglycemic effect in a rodent model.( 33 ) Both improved insulin resistance and glucose tolerance by BCAA have also been indicated in clinical trials.( 14 , 34 )
In addition to the improvement of insulin resistance (Fig. 7a), the present study also indicated that dietary supplementation with BCAA significantly decreased the expression levels of IGF‐1, IGF‐2, and IGF‐1R mRNAs in the liver of DEN‐treated db/db mice (Figs 2b–d). These findings seem to be significant because abnormal activation of the IGF/IGF‐1R axis, which is caused by insulin resistance, is involved in the development of HCC and, therefore, might be a critical target to prevent this malignancy.( 19 , 20 ) These findings are also consistent with those of a previous report that showed BCAA supplementation decreased the serum levels of both IGF‐1 and IGF‐2 while also inhibiting the expression of IGF‐1R on the colonic mucosa, thereby preventing the development of AOM‐induced colonic neoplastic lesions in db/db mice.( 17 ) This previous report,( 17 ) together with our present findings (Fig. 2), suggest the possibility that the inhibition of IGF/IGF‐1R activation is one of the critical mechanisms to suppress obesity‐related tumorigenesis in specific organs, such as the colon and liver, and BCAA might be able to exert its chemopreventive effect on obesity‐associated carcinogenesis by targeting this axis.
Insulin stimulates glucose uptake and triglyceride biosynthesis, which are stored in adipose tissue. The improvement of insulin resistance by BCAA, therefore, inhibits the release of free fatty acid from adipose tissue, improves hypertriglyceridemia, and thus resulted in improvement of hepatic steatosis in the present study (Fig. 4). Ectopic triglyceride accumulation in the liver is directly responsible for the development of insulin resistance.( 35 ) In addition, several studies support the concept that hepatic steatosis promotes the development of HCC.( 36 ) For instance, HCV core protein gene transgenic mice, a model for HCV‐related hepatocarcinogenesis,( 37 ) show marked hepatic steatosis and insulin resistance.( 38 , 39 ) Hepatic steatosis is a major accelerating factor of hepatocarcinogenesis in chronic HCV infected patients.( 40 ) In addition, a significant relationship has also been reported between steatosis and hepatic fibrosis, a potent risk factor for HCC development.( 36 ) Therefore, the reduction of hepatic lipid accumulation might be an effective strategy for HCC chemoprevention. The improvement of hepatic steatosis (Fig. 4) and fibrosis (Fig. 5) by BCAA is thus considered to be advantageous to accomplish this objective.
There are two study limitations that might suggest additional investigations. The first is that the incidence of HCC itself was not very high in the present study (Table 2) because the duration of the experiments (41 weeks) might have been sufficient to develop adenoma but not HCC. Therefore, future study should recruit longer‐term experiments to see that DEN‐treated db/db mice develop HCC more frequently. The second is that, although our model seems to be useful to elucidate the pathogenesis underlying NASH‐associated HCC, there is one difference between the liver of db/db mice and human NASH, as hepatic fibrosis was severe but did not reach liver cirrhosis at the end point of this experiment (Fig. 5a). This might be explained by a functional defect in the long‐form leptin receptor because leptin exerts a pro‐fibrogenic activity in the injured liver.( 41 , 42 ) However, BCAA supplementation significantly decreased the serum levels of leptin (Fig. 3c), inhibited the development of liver fibrosis (Fig. 5), and suppressed the expression of α‐SMA (Fig. 6), thus indicating the inhibition of HSC activation. These findings seem to be significant because activated HSCs are a major cellular source of collagen in the injured liver and thus may be a critical target for inhibiting the development of liver fibrosis.( 43 ) Therefore, BCAA supplementation prevents the development of hepatic fibrosis, at least in part, by inhibiting the HSC activation (Fig. 6). In addition, a previous study also indicated that supplementation with BCAA effectively suppressed the hyperleptinemia in db/db mice with colonic carcinogenesis model.( 17 ) These findings suggest that leptin is also one of the critical targets of BCAA in obese mice. Future studies would be important to evaluate whether BCAA could also prevent the development of liver fibrosis using a more aggressive fibrotic model, such as methionine and choline‐deficient diet‐fed db/db mice, known to be a good model of progressive NASH.( 44 )
Finally, it should be emphasized again that, in a recent study, BCAA supplementation in the basal diet was shown to improve insulin resistance, thereby preventing the development of colonic premalignancies in an obesity‐related colon cancer model.( 17 ) Both obesity and insulin resistance are strongly associated with the development of not only HCC, but also colorectal cancer.( 45 ) These previous reports, therefore, further strengthen our conclusion that the prevention of HCC by targeting the dysregulation of energy homeostasis, particularly an increased insulin resistance, might be a promising strategy for obese people who are at increased risk for developing HCC. BCAA appears to be a potentially effective and critical candidate for this purpose because it can improve insulin resistance (Fig. 7a), hepatic steatosis (Fig. 4), and fibrosis (Fig. 5) in obese and diabetic db/db mice.
In conclusion, BCAA might therefore represent a new effective strategy for chemoprevention against HCC, especially in obese people. Among the beneficial effects of BCAA shown in this study, the improvement of insulin resistance might play a crucial role to prevent the development of obesity‐related liver tumorigenesis because the state of insulin resistance is closely associated with the activation of the IGF/IGF‐1R axis, the development of hepatic steatosis and fibrosis.( 5 , 6 , 7 ) In addition, a recent study revealed that BCAA supplementation also suppressed hepatic neovascularization in insulin‐resistance‐based hepatocarcinogenesis in obese rats.( 46 )
Abbreviations
- α‐SMA
α‐smooth muscle actin
- ALT
alanine aminotransferase
- AOM
azoxymethane
- BCAA
branched‐chain amino acids
- DEN
diethylnitrosamine
- FCA
foci of cellular alteration
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HSC
hepatic stellate cell
- IGF
insulin‐like growth factor
- IGF‐1R
insulin‐like growth factor‐1 receptor
- NASH
non‐alcoholic steatohepatitis
- PCNA
proliferating cell nuclear antigen
- QUICKI
quantitative insulin sensitivity check index
Supporting information
Table S1. Incidence and multiplicity of hepatic neoplasms and FCA and serum levels of ALT in db/db, +/+ and B6 mice.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
We thank Ms. Yukari Nomura for her excellent technical assistance. This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Science, Sports and Culture of Japan (Grant No. 18790457 to M.S. and Grant No. 17015016 to H.M.).
References
- 1. El‐Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132: 2557–76. [DOI] [PubMed] [Google Scholar]
- 2. Calle EE, Rodriguez C, Walker‐Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003; 348: 1625–38. [DOI] [PubMed] [Google Scholar]
- 3. El‐Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol 2006; 4: 369–80. [DOI] [PubMed] [Google Scholar]
- 4. El‐Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004; 126: 460–8. [DOI] [PubMed] [Google Scholar]
- 5. Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non‐alcoholic steatohepatitis. Trends Mol Med 2008; 14: 72–81. [DOI] [PubMed] [Google Scholar]
- 6. Saadeh S. Nonalcoholic Fatty liver disease and obesity. Nutr Clin Pract 2007; 22: 1–10. [DOI] [PubMed] [Google Scholar]
- 7. Smedile A, Bugianesi E. Steatosis and hepatocellular carcinoma risk. Eur Rev Med Pharmacol Sci 2005; 9: 291–3. [PubMed] [Google Scholar]
- 8. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology 1998; 114: 842–5. [DOI] [PubMed] [Google Scholar]
- 9. Lee GH, Proenca R, Montez JM et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996; 379: 632–5. [DOI] [PubMed] [Google Scholar]
- 10. Scheen AJ, Luyckx FH. Obesity and liver disease. Best Pract Res Clin Endocrinol Metab 2002; 16: 703–16. [DOI] [PubMed] [Google Scholar]
- 11. Hirose Y, Hata K, Kuno T et al. Enhancement of development of azoxymethane‐induced colonic premalignant lesions in C57BL/KsJ‐db/db mice. Carcinogenesis 2004; 25: 821–5. [DOI] [PubMed] [Google Scholar]
- 12. Sahai A, Malladi P, Pan X et al. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short‐form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol 2004; 287: G1035–43. [DOI] [PubMed] [Google Scholar]
- 13. Shimizu M, Shirakami Y, Sakai H et al. (−)‐Epigallocatechin gallate suppresses azoxymethane‐induced colonic premalignant lesions in male C57BL/KsJ‐db/db mice. Cancer Prev Res 2008; 1: 298–304. [DOI] [PubMed] [Google Scholar]
- 14. Kawaguchi T, Nagao Y, Matsuoka H, Ide T, Sata M. Branched‐chain amino acid‐enriched supplementation improves insulin resistance in patients with chronic liver disease. Int J Mol Med 2008; 22: 105–12. [PubMed] [Google Scholar]
- 15. Marchesini G, Bianchi G, Merli M et al. Nutritional supplementation with branched‐chain amino acids in advanced cirrhosis: a double‐blind, randomized trial. Gastroenterology 2003; 124: 1792–801. [DOI] [PubMed] [Google Scholar]
- 16. Muto Y, Sato S, Watanabe A et al. Effects of oral branched‐chain amino acid granules on event‐free survival in patients with liver cirrhosis. Clin Gastroenterol Hepatol 2005; 3: 705–13. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu M, Shirakami Y, Iwasa J et al. Supplementation with branched‐chain amino acids inhibits azoxymethane‐induced colonic preneoplastic lesions in male C57BL/KsJ‐db/db mice. Clin Cancer Res 2009; 15: 3068–75. [DOI] [PubMed] [Google Scholar]
- 18. Muto Y, Sato S, Watanabe A et al. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long‐term oral supplementation with branched‐chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol Res 2006; 35: 204–14. [DOI] [PubMed] [Google Scholar]
- 19. Alexia C, Fallot G, Lasfer M, Schweizer‐Groyer G, Groyer A. An evaluation of the role of insulin‐like growth factors (IGF) and of type‐I IGF receptor signalling in hepatocarcinogenesis and in the resistance of hepatocarcinoma cells against drug‐induced apoptosis. Biochem Pharmacol 2004; 68: 1003–15. [DOI] [PubMed] [Google Scholar]
- 20. Shimizu M, Shirakami Y, Sakai H et al. EGCG inhibits activation of the insulin‐like growth factor (IGF)/IGF‐1 receptor axis in human hepatocellular carcinoma cells. Cancer Lett 2008; 262: 10–8. [DOI] [PubMed] [Google Scholar]
- 21. Frith CH, Ward JM, Turusov VS. Tumours of the liver. In: Turusor VS, Mohr U eds. Pathology of Tumors in Laboratory Animals. Vol 2. Lyon: IARC Scientific Publications, 1994; 223–70. [PubMed] [Google Scholar]
- 22. Yasuda Y, Shimizu M, Sakai H et al. (−)‐Epigallocatechin gallate prevents carbon tetrachloride‐induced rat hepatic fibrosis by inhibiting the expression of the PDGFR and IGF‐1R. Chem Biol Interact 2009; 182: 159–64. [DOI] [PubMed] [Google Scholar]
- 23. Shimizu M, Hara A, Okuno M et al. Mechanism of retarded liver regeneration in plasminogen activator‐deficient mice: impaired activation of hepatocyte growth factor after Fas‐mediated massive hepatic apoptosis. Hepatology 2001; 33: 569–76. [DOI] [PubMed] [Google Scholar]
- 24. Shiraki M, Shimomura Y, Miwa Y et al. Activation of hepatic branched‐chain alpha‐keto acid dehydrogenase complex by tumor necrosis factor‐alpha in rats. Biochem Biophys Res Commun 2005; 328: 973–8. [DOI] [PubMed] [Google Scholar]
- 25. Suzuki R, Kohno H, Yasui Y et al. Diet supplemented with citrus unshiu segment membrane suppresses chemically induced colonic preneoplastic lesions and fatty liver in male db/db mice. Int J Cancer 2007; 120: 252–8. [DOI] [PubMed] [Google Scholar]
- 26. Katz A, Nambi SS, Mather K et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 2000; 85: 2402–10. [DOI] [PubMed] [Google Scholar]
- 27. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957; 226: 497–509. [PubMed] [Google Scholar]
- 28. Tomita H, Yamada Y, Oyama T et al. Development of gastric tumors in Apc(Min/+) mice by the activation of the beta‐catenin/Tcf signaling pathway. Cancer Res 2007; 67: 4079–87. [DOI] [PubMed] [Google Scholar]
- 29. Kang S, Song J, Kang H, Kim S, Lee Y, Park D. Insulin can block apoptosis by decreasing oxidative stress via phosphatidylinositol 3‐kinase‐ and extracellular signal‐regulated protein kinase‐dependent signaling pathways in HepG2 cells. Eur J Endocrinol 2003; 148: 147–55. [DOI] [PubMed] [Google Scholar]
- 30. Tornkvist A, Parpal S, Gustavsson J, Stralfors P. Inhibition of Raf‐1 kinase expression abolishes insulin stimulation of DNA synthesis in H4IIE hepatoma cells. J Biol Chem 1994; 269: 13919–21. [PubMed] [Google Scholar]
- 31. Nishitani S, Takehana K. Pharmacological activities of branched‐chain amino acids: augmentation of albumin synthesis in liver and improvement of glucose metabolism in skeletal muscle. Hepatol Res 2004; 30S: 19–24. [DOI] [PubMed] [Google Scholar]
- 32. Nishitani S, Takehana K, Fujitani S, Sonaka I. Branched‐chain amino acids improve glucose metabolism in rats with liver cirrhosis. Am J Physiol Gastrointest Liver Physiol 2005; 288: G1292–300. [DOI] [PubMed] [Google Scholar]
- 33. Doi M, Yamaoka I, Nakayama M, Sugahara K, Yoshizawa F. Hypoglycemic effect of isoleucine involves increased muscle glucose uptake and whole body glucose oxidation and decreased hepatic gluconeogenesis. Am J Physiol Endocrinol Metab 2007; 292: E1683–93. [DOI] [PubMed] [Google Scholar]
- 34. Urata Y, Okita K, Korenaga K, Uchida K, Yamasaki T, Sakaida I. The effect of supplementation with branched‐chain amino acids in patients with liver cirrhosis. Hepatol Res 2007; 37: 510–6. [DOI] [PubMed] [Google Scholar]
- 35. Marchesini G, Brizi M, Bianchi G et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 2001; 50: 1844–50. [DOI] [PubMed] [Google Scholar]
- 36. Powell EE, Jonsson JR, Clouston AD. Steatosis: co‐factor in other liver diseases. Hepatology 2005; 42: 5–13. [DOI] [PubMed] [Google Scholar]
- 37. Moriya K, Fujie H, Shintani Y et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 1998; 4: 1065–7. [DOI] [PubMed] [Google Scholar]
- 38. Moriya K, Yotsuyanagi H, Shintani Y et al. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol 1997; 78: 1527–31. [DOI] [PubMed] [Google Scholar]
- 39. Shintani Y, Fujie H, Miyoshi H et al. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology 2004; 126: 840–8. [DOI] [PubMed] [Google Scholar]
- 40. Ohata K, Hamasaki K, Toriyama K et al. Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer 2003; 97: 3036–43. [DOI] [PubMed] [Google Scholar]
- 41. Ikejima K, Takei Y, Honda H et al. Leptin receptor‐mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 2002; 122: 1399–410. [DOI] [PubMed] [Google Scholar]
- 42. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol 2002; 37: 206–13. [DOI] [PubMed] [Google Scholar]
- 43. Friedman S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008; 134: 1655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamaguchi K, Yang L, McCall S et al. Diacylglycerol acyltranferase 1 anti‐sense oligonucleotides reduce hepatic fibrosis in mice with nonalcoholic steatohepatitis. Hepatology 2008; 47: 625–35. [DOI] [PubMed] [Google Scholar]
- 45. Giovannucci E, Michaud D. The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology 2007; 132: 2208–25. [DOI] [PubMed] [Google Scholar]
- 46. Yoshiji H, Noguchi R, Kitade M et al. Branched‐chain amino acids suppress insulin‐resistance‐based hepatocarcinogenesis in obese diabetic rats. J Gastroenterol 2009; 44: 483–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Incidence and multiplicity of hepatic neoplasms and FCA and serum levels of ALT in db/db, +/+ and B6 mice.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item