

Abstract
The regulatory mechanism of endometrial carcinoma and the signal transduction pathways involved in hormone action are poorly defined. It has become apparent that the G protein‐coupled receptor (GPR) 30 mediates the non‐genomic signaling of 17β‐estradiol (E2). Here we show that GPR30 is highly expressed in endometrial cancer tissues and cancer cell lines and positively regulates cell proliferation and invasion. GPR30 expression was detected in 50 human endometrial carcinomas. The transcription level of GPR30 was significantly higher in the tissue of endometrial carcinoma than in normal endometrium (P < 0.05). Immunohistochemical assays revealed that the positive expression rate of GPR30 protein in endometrial carcinoma tissue (35/50, 70%) was statistically higher than in normal endometrium tissue (8/30, 26.67%) (χ2 = 14.16, P = 0.0002). GPR30 overexpression was correlated with high‐grade endometrial carcinoma. GPR30 expression was also found in two human endometrial cancer cell lines: RL95‐2 (estrogen receptor positive) and KLE (estrogen receptor negative). The roles of GPR30 in proliferative and invasive responses to E2 and G1, a non‐steroidal GPR30‐specific agonist, in RL95‐2 and KLE cell lines were then explored. We showed that E2 and G1 could initiate the MAPK/ERK mitogen‐activated protein kinase pathway in both cell lines. What's more, E2 and G1 promoted KLE and RL95‐2 proliferation and stimulated matrix metalloproteinase production and activity via the GPR30‐mediated MEK/ERK mitogen‐activated protein kinase pathway, as well as increased interleukin‐6 secretion. These findings suggest that GPR30‐mediated non‐genomic signaling could play an important role in endometrial cancer. (Cancer Sci 2009; 100: 1051–1061)
Abbreviations:
- BSA
bovine serum albumin
- CV
coefficient of variation
- DMEM
Dulbecco's modified Eagle's medium
- E2
17β‐estradiol
- EGFR
epidermal growth factor receptor
- ELISA
enzyme‐linked immunosorbent assay
- ER
estrogen receptor
- ERK
extracellular regulated kinase
- FBS
fetal bovine serum
- FigO
International Federation of Obstetrics and Gynecology
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GFP
green fluorescent protein
- GPR
G protein‐coupled receptor
- ICC
immunocytochemistry
- IL
interleukin
- JNK
c‐jun N‐terminal kinase
- MAPK
mitogen‐activated protein kinase
- MEK
MAPK/ERK kinase
- MMP
matrix metalloproteinase
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PI3K
phosphatidylinositol 3‐kinase
- PTX
pertussis toxin
- RT
reverse transcription
- SAPK
Stress‐activated protein kinase
- SDS
sodium dodecylsulfate
- shRNA
short hairpin RNA
Estrogens regulate a remarkable array of physiological processes, ranging from regulation of the menstrual cycle and reproduction to the modulation of bone density, brain function, and behavior.( 1 ) The mechanism responsible for these diverse effects involves estrogen binding to the ER. In the ‘classical’ or ‘genomic’ mechanism, estrogen molecules diffuse into the cell and bind to the ER, which are members of the nuclear hormone receptors and interact with the estrogen response element located in the regulatory region of target genes. The resulting fluctuations in mRNA and the proteins they encode underlie an overall physiological response that takes place within hours following estrogen exposure.( 2 , 3 , 4 )
Many physiological estrogen responses occur in this time frame. However, there are rapid biochemical and physiological responses to estrogen occurring more rapidly (within seconds to minutes) than gene transcription events attributed to the ER (over the course of several hours) that cannot be accounted for by changes in gene expression mediated by nuclear ER. Recently, GPR30, a member of the seven‐transmembrane GPCR family, has been suggested to be a transmembrane ER that can mediate rapid and transcription‐independent estrogen signaling in different cell types.( 5 )
It was demonstrated that GPR30 is responsible for activation of MAPK, adenylyl cyclase, and PI3K in cells lacking classical nuclear ER.( 5 , 6 , 7 ) GPR30 responding to estrogen is involved in a broad spectrum of physiological functions, for example, oocyte maturation, pubertal adrenal development, and liver injury attenuation.( 8 , 9 , 10 ) Expression of GPR30 protein has been observed in a number of cancer cell lines.( 7 , 11 , 12 , 13 , 14 ) In addition, GPR30 has been demonstrated to mediate the proliferative effects of both estrogen and tamoxifen in endometrial cancer cells and thyroid cancer cells.( 11 , 14 ) Taken together, these observations suggest that GPR30 may play a role in the regulation of estrogen actions.
Given that estrogen is now considered the classic etiological factor for endometrial tumorgenesis,( 2 ) and GPR30 is considered a transmembrane receptor of estrogen, we undertook a study of the association between GPR30 and endometrial cancer, with the hope that such associations might provide insights into the causal mechanism of endometrial cancer cell proliferation and invasion. In this present study, we detected GPR30 expression in 30 human endometrial carcinomas and in two human endometrial cancer cell lines. We found that transcriptional and translational levels of GPR30 were significantly higher in the tissue of endometrial carcinoma than in normal endometrium. We then examined the roles of GPR30 in proliferative and invasive responses to E2 and G1, a non‐steroidal GPR30‐specific agonist in both ER‐negative and ER‐positive endometrial carcinoma cell lines, which showed an important regulatory role for GPR30 via the MEK/ERK MAPK pathway in endometrial carcinoma.
Materials and Methods
Tissue collection. Fifty cases of uterine endometrial carcinoma were obtained from patients who underwent initial hysterectomy at the Obstetrics and Gynecology Department, Shanghai First People's Hospital, Shanghai Jiao Tong University from April 2004 to March 2006. The stages and histological grades of these tumors were established according to the criteria of FIGO.( 15 ) The features of the population‐based endometrial carcinoma tissue samples are shown in Table 1. All of the tissues were sectioned and reviewed by an independent pathologist to verify the diagnosis and histology. Thirty normal endometrial samples were also obtained from patients who underwent hysterectomy to treat other types of diseases such as uterine myoma and adenomyosis (16 cases of proliferative phase, 14 cases of secretory phase). Half of the tissue samples were examined histologically and by immunochemical assay, and the remaining portions of the samples were frozen at –80°C until RNA or protein extraction. The project was approved by the Human Investigation Ethical Committee of Shanghai Jiao Tong University Affiliated First People's Hospital, and informed consent for the experimental use of surgical samples was obtained from all patients.
Table 1.
Clinicopathological variables in the patients entered into this study
| Characteristic | No. patients (%) | |
|---|---|---|
| n | % | |
| Total | 50 | 100 |
| Age (years) | ||
| ≤50 | 17 | 34 |
| >50 | 33 | 66 |
| FIGO stage | ||
| Stage I | 24 | 48 |
| Stage II | 10 | 20 |
| Stage III | 16 | 32 |
| Grade | ||
| G1 | 10 | 20 |
| G2 | 26 | 52 |
| G3 | 14 | 28 |
| Histological type | ||
| Endometrioid | 42 | 84 |
| Squamous | 4 | 8 |
| Undifferentiated | 4 | 8 |
| Myometrial invasion | ||
| ≤1/2 | 36 | 72 |
| >1/2 | 14 | 28 |
| Peritoneal cytology | ||
| Negative | 42 | 84 |
| Positive | 8 | 16 |
Cell culture and transfections. Two human endometrial cell lines, KLE (ER negative) and RL95‐2 (ER positive), were grown in DMEM/F12 Media (11030; Gibco, Auckland, NZ) supplemented with 10% FBS (S1810; Biowest, Nuaillé, France), 100 units/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere of 5% CO2, 95% air at 37°C. Cells were transfected with shRNA constructs against GPR30 in the pGFP‐V‐RS (TG316565; OriGene Technologies, Rockville, MD) and HuSH 29‐mer non‐effective against enhanced GFP vector (pRS.TR30003; OriGene Technologies) using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. All transfections are carried out in six‐well plates.
Reverse transcription‐PCR for GPR30 transcription. Total RNA was extracted from human endometrial carcinoma tissue, normal endometrial tissue, KLE, and RL95‐2 cells with Tri‐reagent (TR118; Molecular Research Center, Cincinnati, OH, USA). The cDNA was generated with oligo(dT)18 primers using the Revert Aid First Strand cDNA Synthesis Kit (K1622; Fermentas Life Science, Opelstrasse, St. Leon‐Rot, Germany). The 50‐µL PCR amplification of single‐strand cDNA was carried out with 35 cycles of denaturation (94°C) for 60 s, annealing (55°C) for 30 s and elongation (72°C) for 30 s using PerfectShot Ex Taq (loading dye mix) (DRR05TA; Takara, Dalian, China). The primer sequences are shown in Table 2. The amplified DNA was fractionated by 2% agarose gel electrophoresis, and ethidium bromide‐stained bands were photographed using Scion Image 4.02Beta software (Scion Corporation, Frederick, MD, USA).
Table 2.
Primer sequences of G protein‐coupled receptor (GPR) 30 and β‐actin for reverse transcription (RT)–polymerase chain reaction (PCR) and real‐time PCR
| PCR | mRNA | Size (bp) | Primer sequence | Position |
|---|---|---|---|---|
| RT‐PCR | GPR30 NM_001039966 | 240 | Sense: 5′AGTCGGATGTGAGGTTCAG3′ | 1981–1999 |
| Antisense: 5′TCTGTGTGAGGAGTGCAAG3′ | 2220–2202 | |||
| β‐Actin NM_001101 | 586 | Sense: 5′CATGATGGAGTTGAAGGTAG3′ | 337–355 | |
| Antisense: 5′ACACCTTCTACAATGAGCTG3′ | 922–903 | |||
| Real‐time PCR | GPR30 NM_001039966 | 217 | Sense: 5′‐GCCTGCCGCTGCAGGAAACATTTC‐3′ | 2323–2346 |
| Antisense: 5′‐CTCGGCGGTGCGCTAGCTAGCTCAG‐3′ | 2539–2519 | |||
| β‐Actin NM_001101 | 251 | Sense: 5′CAGCCATGTACGTTGCTATCCAGG‐3′ | 462–485 | |
| Antisense: 5′AGGTCCAGACGCAGGATGGCATG‐3′ | 612–590 |
Immunostaining. For immunohistochemistry, paraffin sections (5 µm) of endometrial carcinoma tissue and normal endometrial tissue were dehydrated in PBS and underwent antigen retrieval with sodium citrate buffer (10 mM, pH 6.0). For immunocytochemical staining, KLE and RL95‐2 growing on coverslips were cultured for 48 h. The coverslips were fixed in 4% paraformaldehyde for 20 min at room temperature, washed in PBS, and permeabilized for 10 min with 0.25% Triton X‐100 in PBS. Then the tissue sections and cell coverslips were incubated with 0.3% H2O2 in PBS for 15 min and 1% BSA in Phosphate Buffered Saline with Tween‐20 (PBST) for 30 min to block non‐specific binding of the antibodies. The samples were then incubated with rabbit polyclonal anti‐human GPR30 antibody (10 mg/mL; ab12563; Abcam, Cambridge Science Park, Cambridge, UK), mouse anti‐human ERα antibody (ready‐to‐use GT201702; Gene Tech Company, Shanghai, China), or rabbit or mouse IgG overnight at 4°C in a humid chamber. After washing with PBS, the sections and coverslips were overlaid with peroxidase‐labeled goat anti‐rabbit IgG (H + L) antibody (1:200; 074‐1506; KPL, Gaitherburg, MD, USA) or peroxidase‐labeled rabbit anti‐mouse IgG (H + L) antibody (ready‐to‐use GK507705; Gene Tech Company) and the reaction was developed with 3,3‐diaminobenzidine. Next, sections and coverslips were counterstained with hematoxylin. The immunohistochemical and immunocytochemical results were evaluated by a pathologist.
Proliferation assay. KLE and RL95‐2 cells were seeded in 96‐well plates at 2 × 105 cells/mL and cultured for 24 h. Cells, including transfected cells, were starved for 24 h by replacing the media with phenol red‐free and serum‐free media (DMEM/F12, 11039; Gibco) followed by the addition of G1, a non‐steroidal GPR30‐specific agonist (10−11 M–10−7 M; 371705; Calbiochem, Darmstadt, Germany), E2 (17β‐estrial, 10−11 M–10−7 M; E2758‐250MG; Sigma Aldrich, St. Louis, MO, USA), or G1 and E2 in the presence or absence of the MAPK inhibitor U0126 (30 µM; 9903; Cell Signaling Technology, Beverly, MA, USA) or PTX (200 ng/mL; 516560; Calbiochem). The cells were further cultured for 48 h with phenol red‐free DMEM/F12 containing 5% dextran‐coated charcoal‐treated FBS (S181F; Biowest). Cell proliferation was evaluated using Cell Counting Kit‐8 (CK04‐11; Dojindo Molecular Technologies, Gaithersburg, MD, USA). The controls were treated with vehicle (0.1% dimethyl sulfoxide/phenol red‐free DMEM/F12).
Sandwich ELISA assay for MAPK in the KLE and RL95‐2 cell lines. KLE and RL95‐2 cells were lysed with Cell Lysis Buffer (9803; Cell Signaling Technology), then the levels of p38, p‐MEK1/2, p‐ERK1/2, and p‐SAPK/JNK in these cell lysates were determined using the PathScan MAPK Multi‐Target Sandwich ELISA Kit (7274; Cell Signaling Technology) according to the manufacturer's instructions.
Sodium dodecylsulfate gel electrophoresis and western blotting. For MAPK detection, protein was extracted from the cells by suspension in RIPA buffer (1 × PBS, 1% Nonidet NP‐40, 0.1% SDS) containing a cocktail of protease inhibitors (11257200; Roche Diagnostics, Mannheim, Germany) at a concentration of 107 cells/mL. For detection of GPR30 and ERα expression, protein was extracted using Mem‐PER Eukaryotic Membrane Protein Extraction Reagent (89826; Pierce, Rockford, IL, USA) containing Complete Mini Cocktail and NE‐PER Nuclear and Cytoplasmic Extraction Reagents (78833; Pierce) from endometrial carcinoma tissue and normal endometrial tissue, respectively. All extracts were lysed according to the protocols. Total protein content was determined by the Bradford protein method using the BCA protein assay kit (23227; Pierce). Protein (15 µg) was loaded onto precast 4% stacking, 10% Tris‐glycine gels, separated by gel electrophoresis followed by electrophoretic blotting onto a PVDF membrane. After washing with Tris‐buffered saline, membranes were blocked with 5% BSA/PBS for 1 h. Membranes were incubated with rabbit anti‐human MEK1/2, rabbit anti‐human phosphorylated‐MEK1/2, rabbit anti‐human ERK1/2 or rabbit anti‐human phosphorylated‐ERK1/2 (1:1000 for all antibodies; 9121S, 9122S, 4695, and 4370S all from Cell Signaling Technology, respectively), rabbit anti‐human ERα and anti‐human ERα antibodies (1:250; sc‐542, Santa Cruz, CA, USA and ab39724, Abcam, respectively) at 4°C overnight. The membranes were then washed three times with Tris‐buffered saline with Tween‐20 (TBST), incubated with peroxidase‐labeled goat anti‐rabbit IgG (H + L) antibody for 60 min and washed three times with TBST. Equivalent protein loading was controlled by monitoring GAPDH expression using a horseradish peroxidase‐conjugated monoclonal mouse anti‐GAPDH (KC‐5G5; Kangchen, Shanghai, China). Immunoblots were visualized by enhanced chemiluminescence (34095 Supersignal; Pierce) according to the manufacturer's instructions.
Gelatin zymography. KLE and RL95‐2 cells were seeded in 24‐well plates at 1 × 106 cells/mL. After starvation for 24 h, cells were treated with G1 (10−8 M) or 17β‐estrial (10−8 M), plus U0126 or PTX in phenol‐red free and serum‐free medium for 48 h. Cultured medium was collected and concentrated using the BCA protein assay kit. The proteolytic activity of both MMP‐9 and MMP‐2 was measured by the gelatin zymography described by Yoshizaki et al., with some modifications.( 16 ) Briefly, the cultured supernatants collected for the invasion assay, containing 10 µg of total protein, were mixed with SDS loading buffer, applied to each lane, and separated on 10% acrylamide gels containing 0.2% gelatin. After electrophoresis, the gels were incubated in 2.5% Triton X‐100 solution for 60 min at room temperature with gentle agitation to remove SDS and then incubated in reaction buffer (50 mM Tris buffer l [pH 7.5], 200 mM NaCl, and 10 mM CaCl2) at 37°C for 24 h. After reaction, the gels were stained for 30 min with staining solution (0.1% Coomassie Brilliant Blue, 30% methanol, and 10% acetic acid). Then the gels were rinsed in the same solution, but without Coomassie Brilliant Blue. The gels were photographed and assayed using Scion Image 4.02Beta software. The gelatinolytic activity was visualized as a clear white band against a dark background of stained gelatin. The proteolytic activity of MMP was equal to the intensity of MMP‐9 or MMP‐2 of the treatment group over that of the control.
Enzyme‐linked immunosorbent assay for determination of IL‐6 production, MMP‐2, and MMP‐9 in KLE and RL95‐2 cells. KLE and RL95‐2 cells were seeded in 24‐well plates at 1 × 106 cells/mL, cultured, and starved as previously described. Cells were incubated with G1 (10−8 M), 17β‐estrial (10−8 M), or in the presence or absence of U0126 or PTX. The cells were further cultured for 48 h with phenol red‐free DMEM/F12 containing 5% dextran‐coated charcoal‐treated FBS and the supernatants were harvested. Each supernatant was centrifuged at 200g and stored at –80°C until detection. The concentrations of human IL‐6, MMP‐2, and MMP‐9 were determined by ELISA kits (IL‐6, BMS213/2; MMP‐9, BMS2016/2; both from Bender MedSystems, Campus Vienna Biocenter, Vienna, Austria; MMP‐2, SMP200; R&D Systems, Minneapolis, MN, USA) according to the manufacturers’ instructions. The IL‐6 assay sensitivity was 0.92 pg/mL, and the intra‐assay and inter‐assay CV were 3.4 and 5.2%, respectively. The MMP‐9 assay sensitivity was 0.05 ng/mL, with intra‐assay and inter‐assay CV of 7.3 and 10.2%, respectively. For MMP‐2, the assay sensitivity was 0.16 ng/mL, and the intra‐assay and inter‐assay CV were 4.8 and 7.6%, respectively.
Statistics. Categorical data were analyzed by χ2 ‐test, χ2 continuity correction test, binary logistic regression test or Spearman test. Measurement data were treated using t‐test or one‐way anova analysis of variance, following least significant difference (equal variances assumed), or Tamhane's test (equal variances not assumed) for multiple comparisons. All tests were carried out with Statistical Package for the Social Sciences software version 15.0 (Chicago, IL, USA). Differences were considered statistically significant if P < 0.05.
Results
Overexpression of GPR30 in endometrial carcinoma. We investigated the transcription of GPR30 in endometrial carcinomas and normal endometrium using RT‐PCR. GPR30 expression was highly increased in endometrial carcinoma compared with normal endometrium (Fig. 1a,b), the ratio of the band intensities of GPR30 to that of β‐actin was 0.95 ± 0.07 to 0.40 ± 0.04, respectively (Fig. 1c). The transcription of GPR30 in endometrial carcinomas was defined as 13.24‐fold compared to normal endometrium by real‐time quantitative PCR (Fig. 1d). Next, to verify whether the GPR30 protein was translated in endometrial carcinoma, we examined the levels of GPR30 protein in carcinomas and normal endometrium through immunochemistry and western blotting. Compared with normal endometrium, the levels of GPR30 protein were found to be markedly increased in endometrial carcinoma, which was in accordance with the results of RT‐PCR (Fig. 1e–g).
Figure 1.
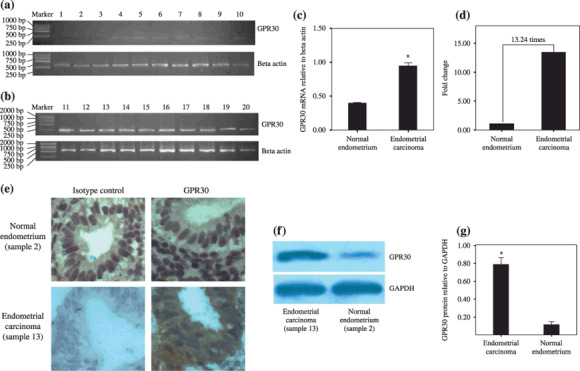
G protein‐coupled receptor (GPR) 30 transcription and overexpression in endometrial carcinoma. (a) Normal endometrium tissue: samples 1–6, endometrium of proliferative phase; samples 7–10, endometrium of secretory phase. (b) Endometrial carcinoma: samples 11–20, 10 carcinoma samples. Note: the marker contains the fragments (from top to foot of gel) 2000, 1000, 750, 500, 250, and 100 bp. (c) The y‐axis shows the ratio of optical density of GPR30 to β‐actin (mean ± SD) from (b). *P < 0.05. (d) The relative mRNA levels of GPR30 in normal endometrium and endometrial cancer by real‐time polymerase chain reaction. Ten normal endometrial tissues and 10 endometrial carcinoma tissues were used. (e) Immunochemistry for GPR30 in normal endometrium tissue and endometrial carcinoma. Normal endometrium, endometrial carcinoma, isotype control detected by rabbit IgG, and GPR30 expression detected by rabbit anti‐human GPR30 antibody are shown. Magnification, ×400. (f) Western blotting analysis for GPR30 protein levels in normal endometrium tissue and endometrial carcinoma. (g) showed the densitometric analysis. *P < 0.05. Fifty endometrial carcinoma samples and 17 normal endometrium samples were used in the reverse transcription–polymerase chain reaction, immunochemistry, and western blotting. The results were highly reproducible and these pictures are representative. GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
After the statistical analyses, the remarkably significant difference of GPR30 expression between normal endometrium and endometrial carcinoma was presented. Seventy percent (35/50) of endometrial carcinomas expressed GPR30, whereas only 26.67% of normal endometrium (8/30) were GPR30 positive (χ2 = 14.16, P = 0.0002) (Table 3).
Table 3.
Statistical difference between G protein‐coupled receptor (GPR) 30 expression in normal endometrium and endometrial carcinoma
| GPR30 expression | χ2 | P‐value* | ||
|---|---|---|---|---|
| Positive | Negative | |||
| Normal endometrium | 8 | 22 | 14.16 | 0.0002 |
| Endometrial carcinoma | 35 | 15 | ||
χ2 test.
To investigate whether the translation of GPR30 in endometrial carcinoma was relevant to the available clinical data, we analyzed the FigO stage, histological grade, myometrial invasion, and peritoneal cytology in these endometrial samples. Translation of GPR30 didn't correlate positively with FigO stage, myometrial invasion, or peritoneal cytology (Table 4), but it did correlate with the pathological grade of endometrial carcinoma (P = 0.029, Binary logistic regression test).
Table 4.
Association between G protein‐coupled receptor (GPR) 30 expression and clinicopathological variables
| Clinicopathological data | No. patients | GPR30 expression | P‐value | ||
|---|---|---|---|---|---|
| n | % | Positive | Negative | ||
| FIGO stage | |||||
| Stage I | 24 | 48 | 18 | 6 | 0.119* |
| Stage II | 10 | 20 | 7 | 3 | |
| Stage III | 16 | 32 | 10 | 6 | |
| Grade | |||||
| G1 | 10 | 20 | 5 | 5 | 0.029* |
| G2 | 26 | 52 | 18 | 8 | |
| G3 | 14 | 28 | 12 | 2 | |
| Myometrial invasion | |||||
| ≤1/2 | 36 | 72 | 23 | 13 | 0.863 † |
| >1/2 | 14 | 28 | 10 | 4 | |
| Peritoneal cytology | |||||
| Negative | 42 | 84 | 23 | 19 | 0.987 † |
| Positive | 8 | 16 | 5 | 3 | |
Binary logistic regression test.
χ2 continuity correction test.
Estrogen receptor‐negative endometrial carcinoma expressed GPR30. Endometrial carcinomas are classified into two classic types on the basis of an estrogenic state, type 1 (ER positive) and type 2 (ER negative).( 17 ) Due to GPR30 overexpression in endometrial carcinoma, it is necessary to determine whether GPR30 overexpression correlated with ER. We measured ER expression in these endometrial carcinomas. GPR30 was expressed in ER‐negative carcinoma (Fig. 2; Table 1), but GPR30 had no correlation with ER expression (r = –0.027, P = 0.853; Table 5).
Figure 2.
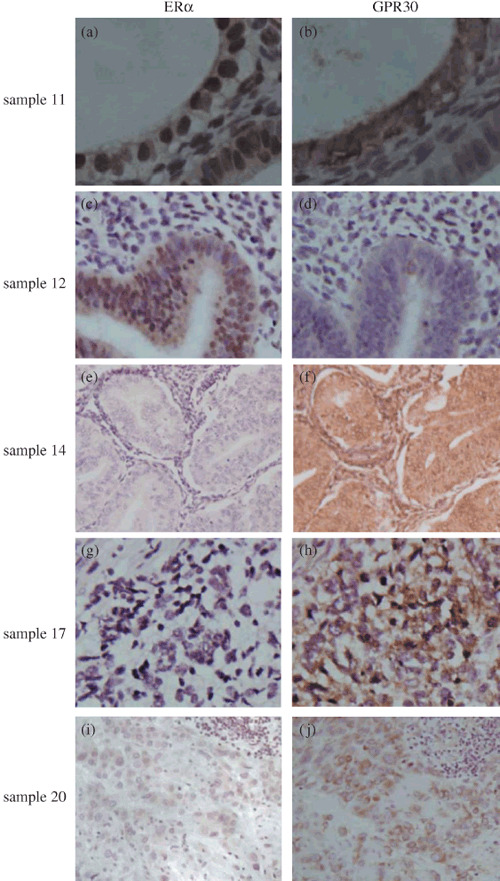
G protein‐coupled receptor (GPR) 30 expressed in estrogen receptor (ER)‐negative endometrial carcinoma. a and b, c and d, e and f, g and h, I and j are from the same endometrial carcinoma, respectively. (a–f) Endometrioid carcinoma, (g,h) adenosquamous endometrial carcinoma, (i,j) undifferentiated endometrial carcinoma. (a,c) ER protein expression was observed in the nuclei of tumor cells. (b,f,h,j) Carcinoma cells showed GPR30 staining in the cytoplasm and membrane. (d) GPR30 expression was not found in this carcinoma. (e,g) No of ERα expression. (i) Weak staining for ERα in the nuclei of tumor cells.
Table 5.
Correlation of GPR 30 and estrogen receptor (ER) expression in endometrial cancer
| ER | GPR30 | r | P‐value* | |
|---|---|---|---|---|
| Positive | Negative | |||
| Positive | 20 | 9 | –0.027 | 0.853 |
| Negative | 15 | 6 | ||
Spearman test.
Estrogen receptor‐positive and ‐negative endometrial cancer cell lines expressed GPR30. To determine whether GPR30 functioned in endometrial cancer, we first measured GPR30 expression in endometrial cancer cell lines.
As expected, both KLE and RL95‐2 cells were stained with GPR30 in the cytoplasm and membrane (Fig. 3e,f). ER protein expression was only observed in the nucleus of RL95‐2 (Fig. 3c), which implied a potentially important role for GPR30 in endometrial cancer. Therefore we further studied the effects of GPR30 on the endometrial cancer cell lines and the signaling pathway involved.
Figure 3.
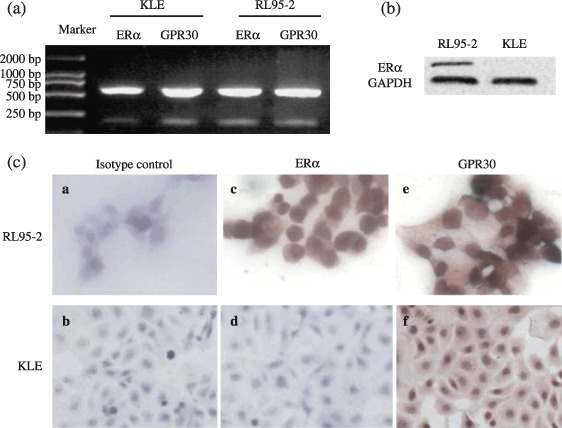
G protein‐coupled receptor (GPR) 30 expression in endometrial cancer cell lines. RL95‐2 cells were estrogen receptor (ER) positive whereas KLE cells were ER negative. (a–c) ERα mRNA and protein were highly expressed in RL95‐2 but hardly expressed in KLE. (c) ERα protein expression was observed in the nuclei of RL95‐2, but GPR30 immunostaining was found in the cytoplasm and membranes of RL95‐2 and KLE. Rabbit IgG and mouse IgG were used as isotype controls; only one isotype control is displayed here. (a,c,e) Magnification, ×400; (b,d,f) ×200.
G protein‐coupled receptor 30 signaling initiated the MEK/ERK MAPK pathway in endometrial cancer cell lines. Our prior reports indicated that several genes related to the MAPK signaling pathway are upregulated in type II endometrial carcinoma.( 18 ) Moreover, estrogen triggers signaling cascades linked to the MAPK pathway.( 19 , 20 , 21 ) We examined activation of the MAPK pathway stimulated by E2 and G1, a specific agonist for GPR30, using the PathScan MAP Kinase Multi‐Target Sandwich ELISA Kit.
As shown in Figure 4, E2 and G1 triggered p‐ERK1/2 and p‐MEK1/2 in KLE and RL95‐2 cells compared to vehicle (P < 0.05; Fig. 4a,b). However, G1 had more power to activate p‐ERK1/2 and p‐MEK1/2 than E2 in ER‐negative KLE cells (P < 0.05; Fig. 4a,c). Although both E2 and G1 initiated p‐ERK1/2 and p‐MEK1/2 in ER‐positive RL95‐2 cells, the activation difference between E1 and G1 was not statistically significant (P > 0.05; Fig. 4b,e). In addition, p38 could also be more phosphorylated by E2 than G1 in KLE cells (P < 0.05; Fig. 4a), which didn't occur in RL95‐2 cells. We also found that the SAPK/JNK MAPK signaling pathway was not involved in signaling activation of estrogen through classic receptors or a new transmembrane receptor (Fig. 4a,b,d,f).
Figure 4.
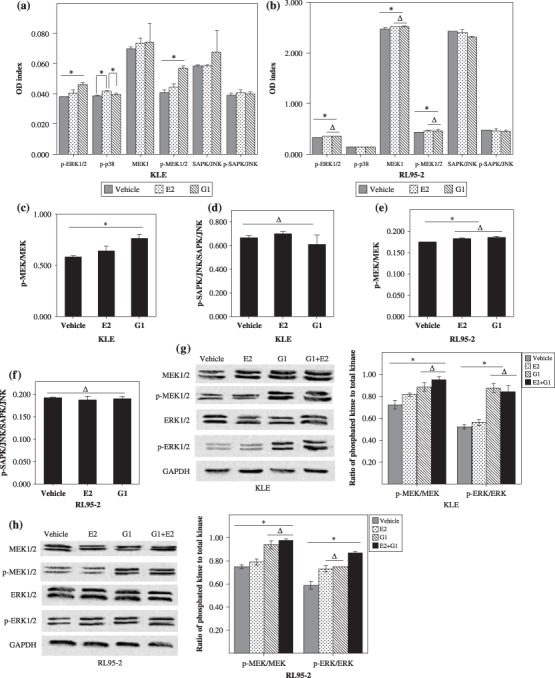
G protein‐coupled receptor (GPR) 30 signaling initiated the mitogen‐activated protein kinase (MAPK) pathway in endometrial cancer cell lines. (a–f) Enzyme‐linked immunosorbent assays (ELISA) for MAPK status in KLE and RL95‐2 cell lines. RL95‐2 and KLE were seeded in 10‐cm dishes at 1 × 106/mL, cultured, and starved. Then the cells were incubated with 10−8 M G1 or 10−8 M 17β‐estradiol (E2) for 30 min. Next the cells were lysed with cell lysis buffer (400 µL per 10‐cm dish). The supernatants were collected after centrifugation at 10 000g at 4°C for 10 min, and were used for the sandwich ELISA, reading absorbance at 450 nm according to the manufacturers’ protocols. Status of the MAPK pathway activated by G1 and E2 in (a) KLE and (b) RL95‐2 cells. The ratios of p‐mitogen‐activated protein kinase (MEK)/MEK in (c) KLE and (e) RL95‐2 cells, and the ratios of p‐SAPK/JNK/SAPK/JNK in (d) KLE and (f) RL95‐2. (g–h) Western blotting for MAPK status in KLE and RL95‐2 cell lines. The bar graphs show the ratio histograms for the western blotting densitometry. *P < 0.05, ΔP > 0.05. GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; JNK, c‐Jun NH2‐terminal kinase; SPAK, Stress‐activated protein kinase.
G protein‐coupled receptor 30 signaling via the MEK/ERK MAPK pathway stimulated cell proliferation of endometrial cancer cell lines. We carried out a proliferation assay to test whether GPR30 signaling could have effects on endometrial cancer cell proliferation. As shown in Figure 5, all doses of E2 and low concentrations of G1 enhanced the proliferation of cell lines including KLE and RL95‐2 after 48 h culture. The high concentration of 10−7 M G1 showed downregulation of proliferation in both KLE and RL95‐2 cells (P < 0.05; Fig. 5a,c). There was no statistical difference in the promotion of KLE proliferation with either 10−11 M–10−8 M G1 or 10−10 M–10−8 M E2 (P > 0.05; Fig. 5a,b). However, the proliferation‐promoting activity induction peaked at a concentration of 10−8 M E2 and 10−8 M G1 in RL95‐2 cells (P < 0.05; Fig. 5c,d). Both 10−8 M G1 and 10−8 M E2 showed the same proliferation‐stimulating activity in ER‐negative KLE cells (P > 0.05; Fig. 5e). In contrast, E2 had a greater capacity to stimulate proliferation in ER‐positive RL95‐2 cells than G1 (P < 0.05, Fig. 5f). Nevertheless, the ability of G1 and E2 to promote proliferation was not concentration‐dependent in either KLE or RL95‐2 cells. Thus, a complicated estrogen–ER regulation mechanism may be involved in ER‐positive and ER‐negative endometrial cancer cells.
Figure 5.
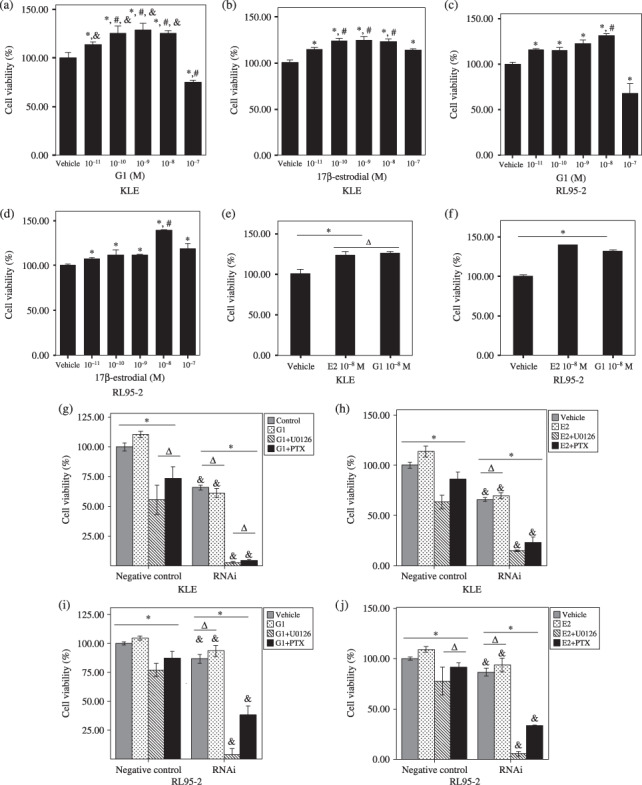
G protein‐coupled receptor (GPR) 30 via the MEK/ERK mitogen‐activated protein kinase (MAPK) pathway stimulates cell proliferation of endometrial cancer cell lines. KLE and RL95‐2 cells were seeded in 24‐well plates at 1 × 106 cells/mL and cultured for 24 h with Dulbecco's modified Eagle's medium (DMEM)/F12 containing 10% fetal bovine serum. The cells were then starved for another 24 h by replacing the media with phenol red‐free and serum‐free DMEM/F12, followed by addition of different doses of 17β‐estradiol (E2) (10−11 M–10−7 M) or G1 (10−11 M–10−7 M), 10−8 M E2 or 10−8 M G1 in the presence of pertussis toxin (PTX) (200 ng/mL) or U0126 (30 µM). Cells were further cultured for 48 h before evaluation of cell proliferation. Vehicle: 0.1% dimethyl sulfoxide/phenol red‐free and serum‐free DMEM/F12. (a) *P < 0.05 versus vehicle; #P < 0.05 versus 10−11 M G1; &P < 0.05 versus 10−7 M G1. (b) *P < 0.05 versus vehicle; #P < 0.05 versus 10−11 M and 10−7 M E2. (c,d) *P < 0.05 versus vehicle; #P < 0.05 versus the others. (g–j) Negative control: cells transfected with HuSH 29‐mer non‐effective against enhanced green fluorescent protein (pRS). RNAi: cells transfected with HuSH 29‐mer short hairpin RNA against GPR30 in pGFP‐V‐RS vector. *P < 0.05; ΔP > 0.05; &P < 0.05 versus cells with the same treament in the negative control.
We also showed that the stimulation of proliferation by E2 and G1 could be disrupted by PTX, an inhibitor of G‐protein signaling,( 22 ) and U0126, an inhibitor of MEK/ERK MAPK, in ER‐negative KLE cells and ER‐positive RL95‐2 cells (P < 0.05; Fig. 5g,h). Moreover, after transfection with shRNA against GPR30, the proliferation of KLE and RL95‐2 did decline even with G1 or E2 treatment, compared with the negative control group (P < 0.05; Fig. 5h–j). These data suggest that G1 and E2 stimulated proliferation via GPR30 signaling in both ER‐negative and ER‐positive endometrial carcinoma cells through the GPR30‐mediated MEK/ERK MAPK pathway.
G protein‐coupled receptor 30 signaling via the MEK/ERK MAPK pathway increased MMP production and activity in endometrial cancer cells. Because G1 and E2 promoted KLE and RL95‐2 cell proliferation via GPR30 signaling, we speculated that the non‐genomic effects invoked by G1 and E2 via GPR30 signaling may mediate the biology of endometrial cancer cells. Invasion is a major prognostic marker and causes cancer malignancy. We detected whether G1 and E2 had effects on MMP production and activity via GPR30 signaling. As shown in Figure 6(a–d), MMP‐9 and MMP‐2 production increased when KLE and RL95‐2 cells were treated with G1 and E2. Moreover, the increase in secretion of MMP‐9 and MMP‐2 caused by G1 and E2 could be blocked by the presence of U0126 and PTX (P < 0.05). MMP activity could be increased by G1 and E2 compared with the vehicle group as well, with U0126 and PTX blocking this promotion (P < 0.05, Fig. 6e,f). These data implied that GPR30 signaling via the MEK/ERK MAPK pathway increased MMP production and activity in endometrial cancer cells.
Figure 6.
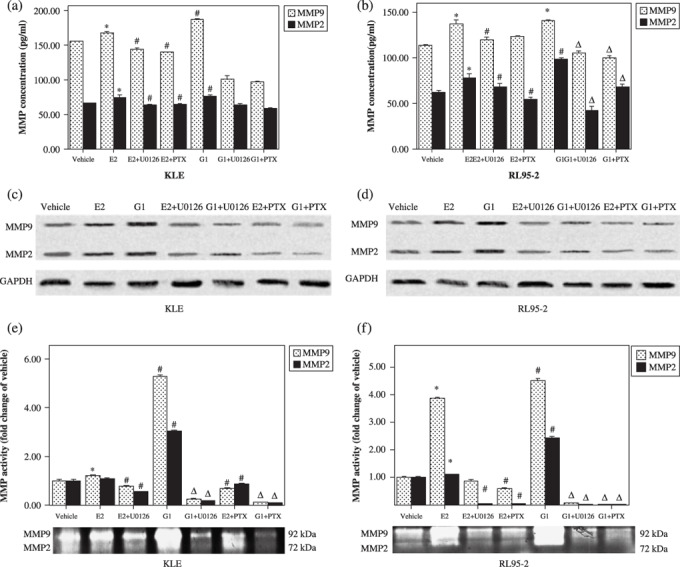
G protein‐coupled receptor (GPR) 30 signaling via the MEK/ERK mitogen‐activated protein kinase (MAPK) pathway increased matrix metalloproteinase (MMP) production and activity in endometrial cancer cells. (a,b) Enzyme‐linked immunosorbent assay (ELISA) data for MMP‐2 and MMP‐9. Data are expressed as mean ± SD. *P < 0.05 compared with vehicle; #P < 0.05 compared with vehicle and 17β‐estradiol (E2); ΔP < 0.05 compared with vehicle and G1. (c,d) Western blotting showing MMM‐9 and MMP‐2 protein expression. (e,f) Gelatin zymography showing MMP‐9 and MMP‐2 activity. The y‐axis displays normalization of MMP‐9 and MMP‐2 band intensity to the vehicle group. Data are expressed as mean ± SD. *P < 0.05 compared with vehicle; #P < 0.05 compared with vehicle and E2; ΔP < 0.05 compared with vehicle and G1.
G protein‐coupled receptor 30 signaling via the MEK/ERK MAPK pathway increased IL‐6 production in endometrial cancer cells. IL‐6 has been previously shown to regulate cell growth of a variety of human cancers, and high serum levels of this cytokine have been correlated with shorter survival in patients harboring several carcinomas.( 23 , 24 ) We therefore used ELISA to test IL‐6 secretion in KLE and RL95‐2 cells treated with G1 or E2, with PTX or U0126. IL‐6 production increased when KLE and RL95‐2 cells were treated with G1 and E2 (Fig. 7). Moreover, the increase in secretion caused by G1 and E2 could be blocked by the presence of U0126 and PTX.
Figure 7.
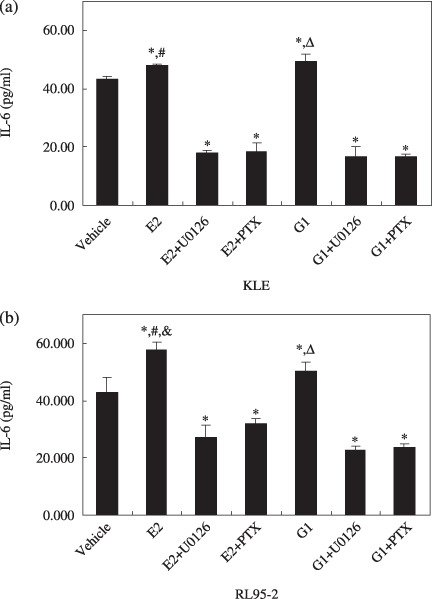
G protein‐coupled receptor (GPR) 30 signaling increased interleukin (IL)‐6 production by endometrial cancer cells. RL95‐2 and KLE were seeded in 24‐well plates at 1 × 106 cells/mL, cultured, and starved. Then the cells were incubated with 10−8 M G1 or 10−8 M 17β‐estradiol (E2) plus U0126 or pertussis toxin (PTX) for 48 h. The supernatants were harvested, centrifuged at 10 000g for 5 min, and the sediments discarded. The supernatants were stored at –80°C until enzyme‐linked immunosorbent assay. (a) IL‐6 secretion in KLE cells. *P < 0.05 versus vehicle; #P < 0.05 versus E2 + U0126 and E2 + PTX; ΔP < 0.05 versus G1 + U0126 and G1 + PTX. (b) IL‐6 secretion in KLE cells. *P < 0.05 versus vehicle; &P < 0.05 versus G1; P < 0.05 versus E2 + U0126 and E2 + PTX; ΔP < 0.05 versus G1 + U0126 and G1 + PTX.
Discussion
Although estrogen is now considered the classic etiological factor for endometrial tumorgenesis,( 2 ) the modulation mechanisms of estrogen that are involved in endometrial cancer are still poorly understood and need to be elucidated urgently.
Here we suggested an important role for GPR30, an ER that is a member of the seven‐transmembrane GPCR family, in endometrial cancer. First, GPR30 was expressed in the plasma membranes of both endometrial carcinoma cells and endometrial carcinoma cells. Until now, there remains controversy over the site of GPR30 expression. GPR30 expression has previously been detected in an intracellular tubuloreticular network, which is a compartment of the endoplasmic reticulum.( 4 ) GPR30 expression has also been observed in the plasma membrane.( 25 , 26 ) Interestingly, it has been shown that G protein γ subunits can translocate as βγ complexes from the plasma to the Golgi complex or the endoplasmic reticulum, which makes it possible for GPCR on the cell surface to direct an active component of a G protein to intracellular membranes.( 27 ) From that point, it is certainly possible that under the appropriate conditions, intracellular GPR30 could exist or translocate to the cell surface or vice versa.
Second, in contrast with normal endometrium, the translation and transcription levels of GPR30 increased in endometrial carcinoma (Fig. 1; Table 3). Furthermore, GPR30 was overexpressed in ER‐negative endometrial carcinoma and more frequently in endometrial carcinoma with high pathological grade (Fig. 2; Table 4). Expression of GPR30 protein has been observed in a number of cancer cell lines.( 7 , 11 , 12 , 13 , 14 ) Studies using transformed cell lines suggest that GPR30 expression is likely more widespread in malignancies.( 28 ) In breast carcinomas, overexpression of GPR30 is significantly associated with tumor size (>2 cm), the presence of distant metastases, and human epidermal growth factor receptor (HER)‐2/neu overexpression, which indicates that GPR30 overexpression may be a predictor of biologically aggressive disease in breast cancer.( 29 ) In endometrial carcinomas, GPR30 overexpression occurs more frequently in endometrial carcinomas exhibiting deep myometrial invasion, high‐grade biologically aggressive histological subtypes, advanced stage, and lower survival rate.( 28 ) These evidence reinforced our speculation that GPR30 plays an important role in endometrial cancer. In our study, we found that 8 of 22 normal endometrial tissues expressed GPR30 protein (Table 3). Most interestingly, there were six tissues in secretory phase among these samples (data not shown), which meant that GPR30 may be developmentally regulated in the estrous cycle. As shown in hamster ovary, GPR30 expression in ductal epithelia of gland, granulosa, and thecal cell layers of ovary exhibits extrous cycle‐dependent changes.( 30 ) Further study needs to be done to test estrous cycle modulation of GPR30 in the human menstrual cycle and whether this modulation affects endometrial cancer. Although it was reported that expression of GPR30 may represent a mechanism to overcome ER loss in hormonally regulated cancers such as breast, endometrial, and ovarian cancers,( 31 ) we demonstrated that GPR30 overexpression was also detected in ER‐negative endometrial carcinoma, but the overexpression was not associated with ER expression. This data suggested the presence of a complicated correlation between GPR30 and ER. Interaction and modulation of E2 and GPR30 are expected.
Third, two endometrial cancer cell lines, KLE and RL95‐2, were stained with GPR30 in the cytoplasm and membrane (Fig. 3). E2 and G1 triggered the MEK/ERK MAPK pathway in ER‐negative KLE cells and ER‐positive RL95‐2 cells. GPR30‐dependent activation of MAPK–ERK1/2 via EGFR transactivation was first described in 2000.( 6 ) Subsequently, estrogen‐mediated adenylyl cyclase activation by GPR30 has also been demonstrated. Moreover, GPR30 is the sole receptor responsible for mediating PI3K activation in response to estrogen via EGFR transactivation in ER‐negative breast cancer cells.( 31 ) Our studies reported previously that genes related to the MAPK signaling pathway are upregulated in ER‐negative endometrial carcinoma.( 18 ) As our results show, although ER was not expressed in KLE, it still activated the MEK/ERK MAPK pathway, which supported a powerful interpretation for our studies. Taken together, the GPR30‐mediated MEK/ERK MAPK pathway is involved in endometrial carcinoma, including in ER‐negative endometrial carcinoma.
Proliferation, invasion, and metastasis are major prognostic markers of carcinoma. Here, we report that E2‐mediated or G1‐mediated GPR30 signaling via the MEK/ERK MAPK pathway could stimulate KLE and RL95‐2 cell proliferation, and promote the production and activity of MMP‐2 and MMP‐9. MMP are a family of structurally related zinc‐ and calcium‐dependent endopeptidases that can degrade extracellular matrix components. MMP‐mediated proteolysis is involved in various physiological and pathological processes, such as cancer invasion and metastasis. MMP‐2 and MMP‐9 contribute to tumor invasion. Tumor invasion and angiogenesis are impaired by the combined deficiency in both metalloproteinases.( 32 ) Our results provide evidence that the production and activity of MMP‐2 and MMP‐9 were upregulated after G1 and E2 treatment in both KLE and RL95‐2 cells. Notably, KLE lacked classic ER but expressed GPR30. This implied that tumors deficient in classic ER did remain estrogen responsive through GPR30 activation. The activation of GPR30 signaling is critical and might be responsible for the aggressiveness of both ER‐negative and ER‐positive endometrial carcinoma by modifying tumor proliferation and invasion through its action on MMP‐2 and MMP‐9. PTX is an exotoxin produced by Bordetella pertussis that modifies GTP‐binding regulatory proteins (G proteins), thus interfering with G protein signal transduction of GPCR.( 22 ) Blocking GPR30 by the addition of PTX would interrupt the signaling cascades. We revealed that the proliferation stimulation and invasion promotion by E2 and G1 could be blocked by PTX and MEK/ERK MAPK inhibitor in both ER‐negative and ER‐positive endometrial carcinoma cells, which did imply the involvement of estrogenic GPR30‐mediated non‐genomic signaling.
Finally, we tested IL‐6 secretion in KLE and RL95‐2 cells treated with G1 or E2 and with PTX or U0126. IL‐6 is a pleiotropic cytokine endowed with a variety of effects on hematopoiesis, the immune system, and acute‐phase responses. Studies regarding cancers of the prostate, ovary, and head and neck have further demonstrated that the activation of IL‐6 is associated with tumor growth.( 33 , 34 , 35 ) High serum levels of this cytokine have been correlated with shorter survival in patients harboring renal carcinoma, prostate cancer, and ovarian carcinoma.( 23 ) Furthermore, IL‐6 is highly expressed in uterine serous papillary carcinoma, a type‐2 endometrial cancer, and is released to high concentrations in the serum of uterine serous papillary carcinoma patients.( 24 ) We found that E2 and G1 could increase IL‐6 production through the GPR30‐mediated MEK/ERK MAPK signaling pathway, which suggests that IL‐6 is involved in the regulation of endometrial cancer by estrogenic GPR30‐mediated non‐genomic signaling.
In summary, we demonstrated that GPR30 overexpression was detected in endometrial carcinomas, including in ER‐negative carcinomas, and its overexpression was closely associated with the pathological grade of endometrial carcinoma. GPR30 expressed in ER‐negative and ER‐positive endometrial cancer cells and GPR30 signaling could promote cell proliferation, IL‐6 secretion, and the invasion potential of ER‐negative endometrial cancer cells via the GPR30‐mediated MEK/ERK MAPK signaling pathway, which suggested that GPR30 signaling plays an important role in endometrial carcinoma, including ER‐negative endometrial carcinoma. Surprisingly, classical ER antagonists such as tamoxifen and ICI182, 780 are agonists of GPR30.( 36 ) Tamoxifen can result in ERK1/2 and PI3K activation via GPR30 in GPR30‐positive cells but does not stimulate PI3K in ERα‐expressing cells, demonstrating that complicated differential effects and an intricate reciprocal action of ERα and GPR30 exist. Further research is warranted to elucidate the functions and significance of GPR30 and the regulation of classic ER and transmembrane GPR30, which will lead to a deeper understanding of tumorgenesis and provide improved treatment options for endometrial cancer.
Acknowledgments
The present study was supported by National Natural Science Foundation of China (no. 30672236 and no. 30872760, X.‐P.W.), the Key Project of Shanghai Municipal Health Bureau (no. 2005ZD002, X.‐P.W.), and China Postdoctoral Science Foundation (no. 20080440627, Y.‐Y.H.).
References
- 1. Hewitt SC, Deroo BJ, Korach KS. A new mediator for an old hormone? Science 2005; 307: 1572–3. [DOI] [PubMed] [Google Scholar]
- 2. Shang Y. Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nature Rev Cancer 2006; 6: 360–8. [DOI] [PubMed] [Google Scholar]
- 3. Pietras RJ, Levin ER, Szego CM. Estrogen receptors and cell signaling. Science 2005; 310: 51–3. [DOI] [PubMed] [Google Scholar]
- 4. Thornton JW, Need E, Crews D. Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science 2003; 301: 1714–17. [DOI] [PubMed] [Google Scholar]
- 5. Revankar CM, Cimino DF, Sklar LA et al . A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005; 307: 1625–30. [DOI] [PubMed] [Google Scholar]
- 6. Filardo EJ, Quinn JA, Bland KI et al . Estrogen‐induced activation of Erk‐1 and Erk‐2 requires the G protein‐coupled receptor homolog, GPR30, and occurs via trans‐activation of the epidermal growth factor receptor through release of HB‐EGF. Mol Endocrinol 2000; 14: 1649–60. [DOI] [PubMed] [Google Scholar]
- 7. Filardo EJ, Quinn JA, Frackelton Jr et al . Estrogen action via the G protein‐coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP‐mediated attenuation of the epidermal growth factor receptor‐to‐MAPK signaling axis. Mol Endocrinol 2002; 16: 70–84. [DOI] [PubMed] [Google Scholar]
- 8. Wang C, Prossnitz ER, Roy SK. Expression of G protein‐coupled receptor 30 in the hamster ovary: differential regulation by gonadotropins and steroid hormones. Endocrinology 2007; 148: 4853–64. [DOI] [PubMed] [Google Scholar]
- 9. Baquedano MS, Saraco N, Berensztein E et al . Identification and developmental changes of aromatase and estrogen receptor expression in prepubertal and pubertal human adrenal tissues. J Clin Endocrinol Metab 2007; 92: 2215–22. [DOI] [PubMed] [Google Scholar]
- 10. Hsieh YC, Yu HP, Frink M et al . G protein‐coupled receptor 30‐dependent protein kinase A pathway is critical in nongenomic effects of estrogen in attenuating liver injury after trauma‐hemorrhage. Am J Pathol 2007; 170: 1210–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carmeci C, Thompson DA, Ring HZ et al . Identification of a gene (GPR30) with homology to the G‐protein‐coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997; 45: 607–17. [DOI] [PubMed] [Google Scholar]
- 12. Vivacqua A, Bonofiglio D, Recchia AG et al . The G protein coupled receptor GPR30 mediates the proliferative effects induced by 17β‐estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol 2006; 20: 631–46. [DOI] [PubMed] [Google Scholar]
- 13. Albanito L, Madeo A, Lappano R et al . G protein coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β‐estradiol and selective GPR30 ligand G‐1 in ovarian cancer cells. Cancer Res 2007; 67: 1859–66. [DOI] [PubMed] [Google Scholar]
- 14. Vivacqua A, Bonofiglio D, Albanito L et al . 17β‐Estradiol, genistein and 4‐hydroxytamoxifen induce the proliferation of thyroid cancer cells through the G protein‐coupled receptor GPR30. Mol Pharmacol 2006; 70: 1414–23. [DOI] [PubMed] [Google Scholar]
- 15. Benedet JL, Bender H, Jones H 3rd et al . FIGO staging classifications and clinical practice guidelines in the management of gynecologic cancers. FIGO Committee on Gynecologic Oncology. Int J Gynaecol Obstet 2000; 70: 209–62. [PubMed] [Google Scholar]
- 16. Yoshizaki T, Sato H, Maruyama Y et al . Increased expression of membrane type 1‐matrix metalloproteinase in head and neck carcinoma. Cancer 1997; 79: 139–44. [DOI] [PubMed] [Google Scholar]
- 17. Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol 2007; 2: 57–85. [DOI] [PubMed] [Google Scholar]
- 18. Cai B, Liu L, Wan XP et al . Comparison of the molecular classification with FIGO stage and histological grade on endometrial cancer. Eur J Gynaecol Oncol 2007; 28: 451–60. [PubMed] [Google Scholar]
- 19. Migliaccio A, Di Domenico M, Castoria G et al . Tyrosine kinase/p21ras/MAPkinase pathway activation by estradiol‐receptor complex in MCF‐7 cells. EMBO J 1996; 15: 1292–300. [PMC free article] [PubMed] [Google Scholar]
- 20. Martin MB, Franke TF, Stoica GE et al . A role for AKT in mediating the estrogenic functions of epidermal growth factor and insulin‐like growth factor I. Endocrinology 2000; 141: 4503–11. [DOI] [PubMed] [Google Scholar]
- 21. Filardo EJ. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G‐protein‐coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol 2002; 80: 231–8. [DOI] [PubMed] [Google Scholar]
- 22. Burnette WN. Perspectives in recombinant pertussis toxoid development. In: Koff WC, Six HR, eds. Vaccine Research and Development. New York: Marcel Dekker, 1992: 143–96. [Google Scholar]
- 23. Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin‐6 in immunity, inflammation and cancer. Trends Mol Med 2008; 14: 109–19. [DOI] [PubMed] [Google Scholar]
- 24. Bellone S, Watts K, Cane’ S et al . High serum levels of interleukin‐6 in endometrial carcinoma are associated with uterine serous papillary histology, a highly aggressive and chemotherapy‐resistant variant of endometrial cancer. Gynecol Oncol 2005; 98: 92–8. [DOI] [PubMed] [Google Scholar]
- 25. Thomas P, Pang Y, Filardo EJ et al . Identity of an estrogen membrane receptor coupled to a G‐protein in human breast cancer cells. Endocrinology 2005; 146: 624–32. [DOI] [PubMed] [Google Scholar]
- 26. Funakoshi T, Yanai A, Shinoda K et al . G protein‐coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun 2006; 346: 904–10. [DOI] [PubMed] [Google Scholar]
- 27. Saini DK, Kalyanaraman V, Chisari M et al . A family of G protein βγ subunits translocate reversibly from the plasma membrane to endomembranes on receptor activation. J Biol Chem 2007; 282: 24 099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith HO, Leslie KK, Singh M et al . GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol 2007; 196: 386–9. [DOI] [PubMed] [Google Scholar]
- 29. Filardo EJ, Graeber CT, Quinn JA et al . Distribution of GPR30, a seven membrane‐spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res 2006; 12: 6359–66. [DOI] [PubMed] [Google Scholar]
- 30. Wang C, Prossnitz ER, Roy SK. Expression of GPR30 in the hamster ovary: differential regulation by gonadotropins and steroid hormones. Endocrinology 2007; 554: 4853–64. [DOI] [PubMed] [Google Scholar]
- 31. Prossnitz ER, Arterburn JB, Sklar LA. GPR30: a G protein‐coupled receptor for estrogen. Mol Cell Endocrinol 2007; 265–266: 138–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002; 2: 161–74. [DOI] [PubMed] [Google Scholar]
- 33. Syed V, Ulinski G, Mok SC et al . Reproductive hormone‐induced, STAT3‐mediated interleukin 6 action in normal and malignant human ovarian surface epithelial cells. J Natl Cancer Inst 2002; 94: 617–29. [DOI] [PubMed] [Google Scholar]
- 34. Sriuranpong V, Park JI, Amornphimoltham P et al . Epidermal growth factor receptor‐independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res 2003; 63: 2948–56. [PubMed] [Google Scholar]
- 35. Giri D, Ozen M, Ittmann M. Interleukin‐6 is an autocrine growth factor in human prostate cancer. Am J Pathol 2001; 159: 2159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ariazi EA, Ariazi JL, Cordera F et al . Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem 2006; 6: 181–202. [PubMed] [Google Scholar]