

Abstract
Epidermal growth factor receptor (EGFR) gene alterations have been found in human lung cancers. However, there is no information on the factors inducing EGFR mutations. In rodents, K‐ras mutations are frequently found in many lung carcinogenesis models, but hitherto, Egfr mutations have not been reported. Their presence was therefore investigated in representative lung carcinogenesis models with 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone (NNK), N‐nitrosobis(2‐hydroxypropyl)amine (BHP), 2‐amino‐3,8‐dimethylimidazo[4,5‐f]quinoxaline (MeIQx) and ethyl carbamate (urethane), as well as X‐ray irradiation. With the chemical carcinogenesis models, no mutations were detected in Egfr, which is in clear contrast to the high rates observed in either codon 12 or 61 of K‐ras (21/23 of the lung tumors induced with NNK, 4/5 with MeIQx, 1/4 with urethane and 7/18 with BHP). However, in the X‐ray‐induced lung tumors, Egfr mutations with amino acid substitution were observed in exons 18 and 21 (4/12, 33%), but no activating mutation of K‐ras was detected. In addition, one and four silent mutations were identified in K‐ras (exon 1) and Egfr (exons 18, 20 and 21), respectively. Most mutations in both Egfr and K‐ras were G/C→A/T transitions (7/8, 88% and 31/34, 91%, respectively). Although, the mutational patterns in equivalent human lesions were not completely coincident, this first report of Egfr mutations in an experimental lung tumor model suggests that X‐rays or other factors producing oxygen radicals could cause EGFR mutations in some proportion of lung cancers in humans. (Cancer Sci 2008; 99: 241–245)
Lung cancer is the major cause of death in both sexes in Japan and many parts of the world( 1 , 2 ) so analysis of causative factors and development of preventive methods is important, in addition to advances in diagnostic and therapeutic methods. Genetic alterations (KRAS, TP53 etc.) in lung cancers have been studied in this context.( 3 )
Epidemiologic studies of lung cancers have pointed to many risk factors including tobacco smoking, air pollution, occupational environments, and ionizing radiations including radon.( 4 ) Interestingly, dietary habits have also been demonstrated to influence the risk of lung cancer, with well‐cooked red meat consumption demonstrated as a risk factor.( 5 , 6 ) Tobacco smoking appears to be correlated with KRAS mutations.( 3 )
Recently, somatic mutations of the epidermal growth factor receptor (EGFR) gene, a tyrosine kinase of the ErbB family, have been reported to be frequent in human lung adenomas and adenocarcinomas, especially in Asians, women, and non‐smokers.( 7 , 8 , 9 , 10 ) However, factors inducing EGFR mutations are quite unclear. To prevent the presently increasing rates of lung adenocarcinomas, this question demands our urgent attention.
Experimental animal models of lung carcinogenesis have been established to elucidate mechanisms and to allow screening for enhancing and suppressing factors. Representative carcinogens inducing high incidences of lung cancers include: 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone (NNK), found in tobacco smoke; N‐nitrosobis(2‐hydroxypropyl)amine (BHP), a synthesized carcinogen not existing in nature; 2‐amino‐3,8‐dimethylimidazo[4,5‐f]quinoxaline (MeIQx), a heterocyclic amine having mutagenicity and carcinogenicity, which exists in cooked meat and fish;( 5 , 6 , 11 ) ethyl carbamate (urethane) and X‐rays. Mutations of K‐ras have been reported in mouse and rat tumors, including NNK‐ and BHP‐induced lung adenomas and adenocarcinomas.( 12 , 13 ) However, alterations of Egfr have hitherto not been identified.
In the present study, to assess possible mutational factors impacting on Egfr, we investigated genetic alterations in Egfr exons 18–21, frequently found in human lung cancer( 7 , 8 , 9 , 10 ) in a series of animal lung neoplasms induced by NNK, BHP, MeIQx, urethane and X‐rays. For comparison, K‐ras exons 1 and 2 were also analyzed.
Materials and Methods
Chemicals. NNK and urethane were purchased from Sigma (St Louis, MO, USA), MeIQx from Nard Institute (Nishinomiya, Japan), and BHP from Nakarai Tesuque (Kyoto, Japan).
Animal treatments. Experimental animals were purchased from Japan SLC, Inc. (Shizuoka, Japan) and each experimental treatment started after adaptation for a week.
To obtain animal lung tumor samples, animal experiments with five lung carcinogenesis models were carried out as follows.
For NNK‐induced lung tumors, 7‐week‐old female A/J mice were given a single dose of NNK (2 mg/0.1 mL saline/mouse, i.p.), and then maintained without additional treatment until sacrificed at week 52. This experiment was conducted by M. Yokohira and K. Imaida.
For MeIQx‐induced lung tumors, 7‐week‐old female A/J mice were given 600 p.p.m. MeIQx in a basal diet for 12 weeks, and were then maintained on the basal diet without MeIQx until sacrificed at week 32. This experiment was also conducted by M. Yokohira and K. Imaida.
For the urethane‐induced lung tumors, 8‐week‐old female A/J mice were given a single dose of urethane (250 mg/kg, i.p.), and then maintained without additional treatment until sacrifice at week 50. This experiment was conducted by N. Takasuka.
The protocol for BHP‐induced lung tumors was based on previous reports( 14 ) and the experiment was conducted by M. Tsutsumi.
The protocol for X‐ray‐induced lung tumors was conducted by Y. Yamada and Y. Oghiso.( 15 ) Briefly, for local thoracic X‐irradiation, female Wistar (W/M) strain rats at the ages of 100–120 days were exposed to 3.0 Gy of X‐rays and then maintained without additional treatment until sacrifice when moribund, dead, or at 24 months.
All the studies were conducted according to the Guidelines for Animal Experiments in the respective facilities.
Tissue preparation and DNA extraction. Upon sacrifice, the lungs were immediately excised and portions were fixed in neutrally buffered 10% formalin and embedded in paraffin. Two serial thin sections were made, one of 3 µm thickness to be stained with hematoxylin and eosin for histological examination, and the other of 8 µm thickness for DNA extraction.
For analysis of K‐ras and Egfr mutations, paraffin‐embedded lung neoplastic lesions (alveolar hyperplasia [AH], adenoma [Ad] and adenocarcinoma [AC]) from each animal model were used. Neoplastic lesions were scraped off from paraffin sections using needles and DNA was extracted using DEXPAT (TaKaRa Shuzo, Shiga, Japan).
Polymerase chain reaction (PCR). K‐ras and Egfr gene fragments were amplified by PCR from lung DNA samples. PCR primers were synthesized at Operon Biotechnologies Inc. (Tokyo, Japan) with oligonucleotide purification cartridge grade. The sequences and target codons are listed in Table 1. Different primer sets were used according to each sample quality. In some cases, one exon was analyzed with two short PCR products overlapping partially.
Table 1.
Oligonucleotide primers for PCR amplification
| Gene | Animal | Treatment | Exon | Primer sequence (5’‐3’) | Target codons † | Annealing temperature | |
|---|---|---|---|---|---|---|---|
| K‐ras | Mouse | MeIQx, NNK | 1 | F; ACTGAGTATAAACTTGTGGT | R; CCTCTATCGTAGGGTCGTAC | 9–30 | 53°C |
| 2 | F; AAGTAGTAATTGATGGAGAA | R; TTATGGCAAATACACAAAGA | 50–77 | 53°C | |||
| Urethane | 1 | F; AGGCCTGCTGAAAATGACTG | R; CCTCTATCGTAGGGTCGTAC | 4–30 | 55°C | ||
| 2 | F; AAGTAGTAATTGATGGAGAA | R; TGGTGAATATCTTCAAATGATTTAGT | 50–86 | 53°C | |||
| Rat | BHP | 1 | F; AGGCCTGCTGAAAATGACTG | R; GCAGCATTTACCTCTATCGT | 4–33 | 53°C | |
| 2 | F; AAGTAGTAATTGATGGAGAA | R; TGGTGAATATCTTCAAATGATTTAGT | 50–86 | 53°C | |||
| X‐ray | 1 | F; TGACTGAGTATAAACTTGTGGTAGTTG | R; TCGTAGGATCATATCATTCCACAAAG | 11–26 | 57°C | ||
| 2 | F; AAGTAGTAATTGATGGAGAA | R; GGCAAATACACAAAGAAAGC | 50–76 | 53°C | |||
| Egfr | Mouse | MeIQx, NNK | 18 | F; CTCCCTTCTTCACAGCTCG | R; TCTCCAGGATGTTACCTTATAC | 692–727 | 55°C |
| 19 | F; TTCTTAATCTCAGGGTCTCT | R; GAAAACTCACGTCAAGGAT | 734–760 | 55°C | |||
| 20 | F; GTCCTTACCTTGTAGGAAGC | R; TCCCAACGTGCTTACCTTTG | 766–823 | 55°C | |||
| 21 | F; GGGCATGAACTACCTGGAAG | R; AGGACTTACTTTGCCCCCCTC | 833–873 | 55°C | |||
| Urethane | 18 | F; CTCGTGGAACCTCTCACACC | R; ATGTTACCTTATACACTGTGCCAAATG | 697–723 | 55°C | ||
| 19 | F; CAAGTTAATGTCAGCCCTCTTC | R; TAAAAGAAAACTCACGTCAAGGATTTC | 731–759 | 55°C | |||
| 20 | F; AGGAAGCCTATGTGATGGCTA | R; GACGTAGTCCAGGAGGCAAC | 771–797 | 60°C | |||
| 21 | F; CCTCTGTATTTCAGGGCATG | R; ACTCCCAGGACTTACTTTGC | 828–875 | 55°C | |||
| Rat | BHP | 18 | F; TGGTGCTAGCATCTCTGGTC | R; AGTCCAGACCTGTCTCCAGG | 689–729 | 55°C | |
| 19 | F; CAGGTTAATGTCAGCCCTCTTC | R; GGAAACCGTGGTTAGCAAGA | 730–762 | 55°C | |||
| 20 | F; CCCATCAGCCAAGAAACAAT | R; GTACTCCAGGGGGCAGACCT | 763–824 | 55°C | |||
| 21 | F; GGGCATGAACTACCTGGAAG | R; AGGACTTACTTTGCCCCCCTC | 832–872 | 55°C | |||
| X‐ray | 18 | F1; TGGTGCTAGCATCTCTGGTC | R1; CTCCTGAACCCAGAACTTTGA | 689–715 | 55°C | ||
| F2; GGAGAAGCTCCGAACCAAG | R2; AGTCCAGACCTGTCTCCAGG | 704–729 | 55°C | ||||
| 19 | F; CAGCCCTCTTCTTAATCTCAGG | R; GCAAGACATAAAAGGAAACTCACA | 731–762 | 55°C | |||
| 20 | F1; CACATGTGTTGTCCTTACCTTG | R1; AACCATAGGGCATGAGTTGTG | 763–790 | 55°C | |||
| F2; ACCTCCACTGTCCAGCTCAT | R2; GCAGACCTTCCAATGTGCTTA | 791–824 | 55°C | ||||
| 21 | F1; TGAAGCGTCTTCTGTGTTTCA | R1; TTGGCCAGTCCAAAATCTGT | 825–854 | 55°C | |||
| F2; TACTGGTAAAGACACCACAGCA | R2; GCTTCCTGACTTATTCTCAGGACT | 852–876 | 55°C | ||||
Corresponding to mouse and rat codons. BHP, N‐nitrosobis(2‐hydroxypropyl)amine; Egrf, epidermal growth factor receptor; MeIQx, 2‐amino‐3,8‐dimethylimidazo[4,5‐ƒ]quinoxaline; NNK, 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone; PCR, polymerase chain reaction.
PCR for analysis of the gene alterations was performed in 50 µL of reaction mixture consisting of 0.5 µM of each primer, 10 × PCR buffer (Applied Biosystems, Foster City, CA, USA), 200 µM each dNTP, 2.5 U AmpliTaq Gold (Applied Biosystems) and 0.5–5 µL of template DNA. The mixture was heated at 94°C for 9 min and subjected to 50 cycles of denaturation (94°C, 30 s), annealing (at each temperature in Table 2, 30 s) and extension (72°C, 1 min) using a thermal cycler, DNA Engine PTC‐200 (Bio‐Rad Laboratories Inc. Hercules, CA, USA).
Table 2.
Incidences of mutations with amino acid substitution and silent mutations of the K‐ras and Egfr genes in animal lung neoplasms
| Treatment | Animal | K‐ras | Egfr | ||||
|---|---|---|---|---|---|---|---|
| Exon 1 | Exon 2 | Exon 18 | Exon 19 | Exon 20 | Exon 21 | ||
| Mutations with amino acid substitution | |||||||
| NNK | Mice | 21/23 (91%) | 0/22 | 0/23 | 0/22 | 0/15 | 0/14 |
| MeIQx | Mice | 4/5 (80%) | 0/5 | 0/5 | 0/4 | 0/4 | 0/5 |
| Urethane | Mice | 0/8 | 1/4 (25%) | 0/4 | 0/4 | 0/5 | 0/3 |
| BHP | Rats | 7/18 (39%) | 0/14 | 0/18 | 0/18 | 0/17 | 0/18 |
| X‐ray | Rats | 0/12 | 0/11 | 3/12 (25%) | 0/11 | 0/12 | 1/11 (9%) |
| Silent mutations | |||||||
| X‐ray | Rats | 1/12 (8%) | 0/11 | 1/12 (8%) | 0/11 | 2/12 (17%) | 1/11 (9%) |
BHP, N‐nitrosobis(2‐hydroxypropyl)amine; Egrf, epidermal growth factor receptor; MeIQx, 2‐amino‐3,8‐dimethylimidazo[4,5‐ƒ]quinoxaline; NNK, 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone.
Single strand conformation polymorphism (SSCP) analysis. SSCP analysis was conducted by the method of Orita et al. with modifications.( 16 ) PCR products were treated using ExoSAP‐IT (USB Corp., Cleveland, OH, USA) before application to SSCP analysis. Four and a half µL of 95% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol were added to 0.5 µL PCR products treated by ExoSAP‐IT, heated at 90°C for 3 min, cooled at 4°C for 1 min and then applied to 5–20% gradient polyacrylamide gel (e‐PAGEL, ATTO corporation, Tokyo, Japan).
Electrophoresis was carried out at 300 V for 1.5 h at 4°C and the gels were soaked in 10% trichloroacetate and in 50% methanol for 10 min each. DNA bands were detected by silver staining using 2D Silver Staining Solution II (Daiichi Pure Chemicals Co. Ltd, Tokyo, Japan). Detected shifted bands were homogenated, heated and centrifuged with DEXPAT to extract DNA and again applied to PCR and direct sequencing for verification of the mutation.
Direct DNA sequencing. With 2 µL of the ExoSAP‐IT‐treated PCR products and 5′ or 3′ of each PCR primer (Table 1), cycle sequencing reactions were carried out using a DYEnamic ET terminator cycle sequencing kit (GE Healthcare UK Ltd, Amersham Place, Little Chalfont, Buckinghamshire, England) and the sequences were determined with an ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
Results
Histological findings. Lung neoplastic lesions induced by each treatment mostly originated from alveolar type II cells or bronchiolar Clara cells. Almost all histopathological types of lung neoplasms with each treatment were epithelial types, being classified into AH, Ad, AC, adenosquamous carcinoma and squamous cell carcinoma categories. A total of 66 lesions, classified into 4 AHs, 7 Ads, and 12 ACs from NNK, 2 AHs and 3 Ads from MeIQx, 8 Ads from urethane, 15 Ads and 3 ACs from BHP, 4 Ads and 8 ACs from X‐ray‐treated animal lungs, respectively, were used in the present mutational analyses.
K‐ras alterations in lung neoplastic lesions. Activating mutations of the K‐ras gene at codons 12 and 61 were detected in neoplastic lesions induced by NNK (21/23; 91%), MeIQx (4/5; 80%), urethane (1/4; 25%) and BHP (7/18; 39%), respectively, but not in X‐ray‐induced tumors (Table 2). Histological classifications in neoplasms with K‐ras mutations were 3 AHs, 7 Ads and 11 ACs from NNK, 1 AH and 3 Ads from MeIQx, 1 Ad from urethane, 5 Ads and 2 ACs from BHP‐induced neoplasms. Only one silent mutation was detected in a X‐ray‐induced Ad (1/12; 8% in Table 2). Mutations were mostly G/C→A/T transitions. In one neoplasm, each in the MeIQx, urethane and X‐ray models, G/C→C/G (AH), A/T→T/A (Ad) and A/T→T/A (Ad) transversions, respectively, were observed (Table 3). Particular histological differences were not observed with each mutation pattern.
Table 3.
Mutation patterns for the K‐ras and Egfr genes in lung neoplasms
| Treatment | Animal | Gene | Exon | Nucleotide alteration | Amino acid substitution † | Frequency |
|---|---|---|---|---|---|---|
| NNK | Mice | K‐ras | 1 | GGT→GAT | G12D | 21/21 |
| MeIQx | Mice | K‐ras | 1 | GGT→GAT | G12D | 3/4 |
| GGT→GCT | G12A | 1/4 | ||||
| Urethane | Mice | K‐ras | 2 | CAA→CTA | Q61L | 1/1 |
| BHP | Rats | K‐ras | 1 | GGT→GAT | G12D | 7/7 |
| X‐ray | Rats | K‐ras | 1 | GGA→GGT | G10 | 1/1 |
| Egfr | 18 | CCC→CTC | P695L | 1/8 | ||
| GGA→GAA | G697E | 1/8 | ||||
| GTT→GTC | V718 | 1/8 | ||||
| GGT→GAT | G720D | 1/8 | ||||
| 20 | ACC→ACT | T784 | 1/8 | |||
| CAG→CAA | Q788 | 1/8 | ||||
| 21 | CAC→CAT | H836 | 1/8 | |||
| GGT→GAT | G864D | 1/8 |
Corresponding to mouse and rat codons. BHP, N‐nitrosobis(2‐hydroxypropyl)amine; Egrf, epidermal growth factor receptor; MeIQx, 2‐amino‐3,8‐dimethylimidazo[4,5‐ƒ]quinoxaline; NNK, 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone.
Egfr gene alterations in lung neoplastic lesions. Lung neoplastic lesions induced by NNK, MeIQx, urethane and BHP were found to harbor activating K‐ras mutations, but not Egfr mutations. On the other hand, Egfr mutations with amino acid substitution were detected in X‐ray‐induced tumors (4/12; 33% in Table 2). Representative mutation charts are shown in Fig. 1. All the Egfr mutations were reconfirmed by sequencing of independent PCR products derived from the original template DNA. Mutation sites were mostly found in exon 18 (3/4) and the remainder in exon 21 (1/4). In addition to the mutations with amino acid substitution, silent mutations were also detected (4/12, 33%) in exons 18 (1/4), 20 (2/4) and 21 (1/4), respectively, two of the four silent mutations overlapping with mutations causing amino acid substitution. None of the tumors with Egfr mutations had K‐ras mutations. Egfr alteration sites were not located in specific codons, but distributed over exons 18–21 (except 19) (Table 3). Tumors harboring Egfr mutations with amino acid substitution were histologically classified as 1 Ad and 3 ACs all with G/C→A/T patterns. Histological classifications of tumors with silent mutations of Egfr were 1 Ad with G/C→A/T, 3 ACs with 2 G/C→A/T and T/A→C/G patterns, respectively. Among these tumors, two ACs exhibited both missense and silent mutations, 1 AC with G/C→ A/T at codon 697 in exon 18 and G/C→A/T at codon 784 in exon 20, and 1 AC with G/C→A/T at codon 720 in exon 18 and T/A→C/G at codon 718 in exon 18, respectively. In addition, we further classified histological subtypes of the tumors with or without the Egfr mutations. Typical histological subtypes of X‐ray‐induced tumors used in the present study are shown in Fig. 2. Out of six tumors with the Egfr mutations, three were papillary type and the other three were solid type, while one papillary, one solid, three acinar and one bronchiolo‐alveolar types were included in six tumors without the Egfr mutation. The papillary and solid types were frequent in the tumors with the Egfr mutations compared to the tumors without the Egfr mutation in the X‐ray model.
Figure 1.
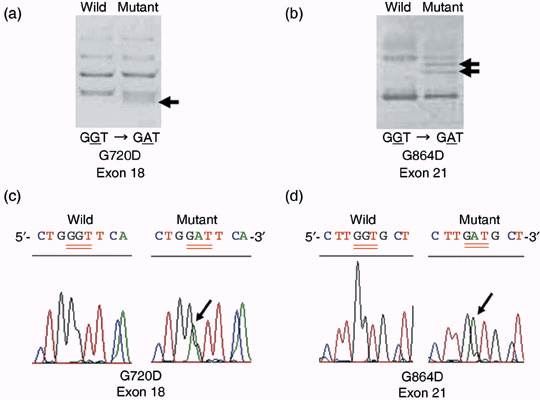
Representative examples of single strand conformation polymorphism analysis (a, b) and charts of Egfr mutations confirmed by direct sequencing (c, d). Arrows in panels (a) and (b) indicate shifted bands associated with mutations. Panels a, c and b, d show G/C→A/T mutations found at codon 720 in exon 18 and at codon 864 in exon 21, respectively. Both arrows in panel (b) indicate the same mutation pattern of GGT to GAT at codon 864.
Figure 2.
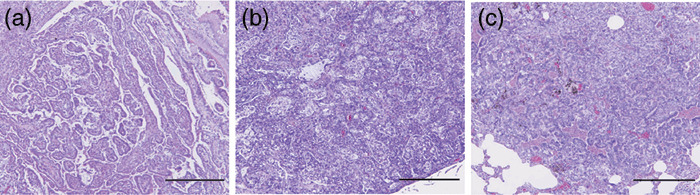
Typical histological subtypes of X‐ray‐induced rat lung tumors with and without the Egfr mutations. (a) Papillary type adenocarcinoma. (b) Solid type adenocaricnoma. (c) Acinar type adenocarcinoma. The tumors of (a) and (b) exhibit the Egfr mutations and (c) is without the Egfr mutation. Histological classifications refer to our previous study.( 15 , 20 ) Scale bars, 500 µm.
Discussion
In the present study of mutation profiles of K‐ras and Egfr in chemical and X‐ray‐induced lung carcinogenesis animal models, Egfr alterations were detected in 4 of 12 (33%) X‐ray‐induced tumors. To our knowledge, this is the first demonstration of mutations in Egfr in an animal lung carcinogenesis model. These mutations detected in this study are thought to be somatic, because experimental animals used for X‐ray lung carcinogenesis were inbred and no mutations were detected in non‐neoplastic parts of lung sections used for extracting DNA samples. The Egfr mutation sites and patterns found in X‐ray‐induced tumors have no codon specificity and were scattered over exons 18–21 (except 19). An amino acid substitution at codon 720 is reported as a mutation site in human lung cancer (corresponding to codon 719 in human EGFR).( 10 ) This similarity suggests that the rat lung carcinogenesis model induced by X‐rays may reflect, in part, human lung carcinogenesis with EGFR mutation. However, the other mutations differed from the most frequent mutations in human cases.
Yuan et al. reported that T/A→G/C substitutions, a change frequently detected in EGFR of human lung cancer, were induced by a 5.0 Gy dose of X‐ray irradiation in mouse cells.( 17 ) They speculated that this mismatch substitution resulted from repair activity of polymerase‐β. However, in the present study, T/A→G/C substitutions were not detected in rat lung tumors induced in vivo by 3.0 Gy of X‐ray irradiation. The differences between this study's data and Yuan's data could be due to differences in irradiation doses and species dependence regarding metabolism and repair systems.
In X‐ray‐induced tumors, silent mutations in Egfr and K‐ras were also observed (Table 3). Generally, these have been believed to not affect events of protein levels, but a recent study demonstrated that silent mutations may also alter the conformation and activity of a protein.( 18 ) Although half of the detected silent mutations (2/4) in Egfr overlapped with mutations generating amino acid substitution, some role in lung carcinogenesis could not be ruled out, at least in the other two cases.
In the histological evaluation, Egfr mutations were detected in both Ad and AC induced by X‐rays in the lung, suggesting a possible involvement in an early stage of the neoplastic process, as with activating K‐ras mutations. In the present study, the papillary type was frequently observed in the tumors with the Egfr mutations compared to the tumors without the Egfr mutations in the X‐ray model. In humans, the papillary type was also predominant in lung cancers with EGFR mutations.( 19 ) There are some similarities of the histological subtype between X‐ray‐induced lung tumors and human lung cancers with EGFR mutations.
In our previous study, immunohistochemical staining for surfactant apoprotein A and Clara cell 10 kDa protein have revealed that most of the lung tumors induced by X‐rays originated from either type II alveolar or Clara cells.( 20 ) On the bases of our previous data, we compared the expression of SP‐A and CC‐10 between the tumors with and without the EGFR mutations in the present study. However, no specificity of cell differentiation phenotypes was observed between tumors with and without the Egfr mutations.
In the present study, we conducted immunostaining for the Egfr downstream molecules of phospholyrated ERK (pERK) and Akt (pAkt). However, pERK and pAkt were mostly negative in both X‐ray‐induced tumors with and without the Egfr mutations (data not shown). No specificity of the expression patterns of pERK and pAkt were observed between tumors with and without the Egfr mutations. In the human lung cancer, Ikeda et al. reported that pAkt expression was significantly associated with the codon 858 mutation in the exon 21, but not in the exon 19 deletions, while pERK did not have any correlation.( 21 ) The Egfr mutation patterns and sites detected in the present study were different from the above two mutations of human lung cancer. Moreover, to investigate the influence of the Egfr mutations detected in the present study on cell proliferation activity, we also conducted immunostaining for proliferating cell nuclear antigen (PCNA). The ratio of PCNA positive cells tended to be higher in X‐ray‐induced lung tumors with the Egfr mutations (22.1 ± 6.9%[mean ± standard deviation]) than those without the Egfr mutation (13.9 ± 3.7%). Further analysis such as transfection study in cell culture system is warranted to clarify the biological effect of the Egfr mutations, found in the present study.
The nitroso compounds, NNK and BHP, are well known to frequently induce K‐ras mutations with G/C→A/T transitions in animal models( 12 , 13 ) as confirmed in our present study. Lung tumors induced by MeIQx and urethane, non‐smoking factors, were also found to harbor K‐ras, but none of them featured any Egfr mutations. These data suggest that mutation hotspots might differ between chemical and X‐ray mutagenesis.
Most chemical carcinogens modify DNA bases by forming adducts, whereas X‐rays are known to cause genomic DNA damage, mostly having indirect effects, by producing oxygen radicals derived mainly from O2 and H2O molecules in vivo. These different mechanisms of DNA damages may contribute to different gene targets. Indeed, the coexistence of both K‐ras and Egfr mutations was not observed in the present study as observed in human cases. In addition to X‐rays, microparticles, a factor of air pollutions derived from various industrial activities, might therefore also be likely to induce EGFR mutations because of their ability to produce oxygen radicals.( 22 )
Recently, the proportion of lung adenocarcinomas is increasing in our country. The prevalence of lowtar filter cigarettes makes smokers inhale more deeply and this is suggested to be a cause for recent increases. However, the reason for the increases of lung adenocarcinomas, especially with the EGFR mutations among non‐smokers in Asia, is unclear. Ashakumary et al. reported that administration of a high‐fat diet increases the concentration of lipid peroxides in rat lung tissue.( 23 ) Recent changes of lifestyle may contribute to the increases of lung adenocarcinomas. The fact that endogenous oxygen radicals are produced by chronic inflammation is of interest in this context.( 24 ) Clearly, the causative factors for EGFR mutations warrant further attention.
As an animal model for human lung adenocarcinomas, transgenic mice expressing mutant EGFR of human patterns in alveolar epithelium have been established.( 25 ) That model is considered to be useful for developing therapeutic methods for human lung adenocarcinomas bearing EGFR mutations. The present model of X‐ray‐induced rat lung adenocarcinoma with the Egfr mutations may also be useful for studying lung carcinogenesis processes and developing therapeutic methods.
In conclusion, though the mechanisms of X‐ray lung carcinogenesis have yet to be fully elucidated at the molecular level, in the present study we predominantly detected Egfr mutations in X‐ray‐induced lung tumors of rats. X‐ray irradiation or endogenous factors producing oxygen radicals may thus cause EGFR mutations in some proportion of human lung cancers.
Acknowledgments
Grant support: This work was supported by Grants‐in‐Aid for Cancer Research and for the Third Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan.
The authors would like to express their appreciation to Dr Tatsuhiro Shibata and Dr Koji Tsuta (National Cancer Center Research Institute, Tokyo, Japan) for their helpful suggestions, and also to Ms. Sachiko Nakai and Ms. Megumi Yamaguchi (Saiseikai Chuwa Hospital, Nara, Japan) for their technical assistance.
T. Kitahashi is an Awardee of the Research Resident Fellowship from the Foundation for Promotion of Cancer Research (Japan) for the Third Term Comprehensive 10‐Year Strategy for Cancer Control during the performance of this study.
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer 2001; 94: 153–6. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Siegel R, Ward E et al . Cancer statistics, 2006. CA Cancer J Clin 2006; 56: 106–30. [DOI] [PubMed] [Google Scholar]
- 3. Le Calvez F, Mukeria A, Hunt JD et al . TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res 2005; 65: 5076–83. [DOI] [PubMed] [Google Scholar]
- 4. Subramanian J, Govindan R. Lung cancer in never smokers. J Clin Oncol 2007; 25: 561–70. [DOI] [PubMed] [Google Scholar]
- 5. Sinha R, Kulldorff M, Swanson CA et al . Dietary heterocyclic amines and the risk of lung cancer among Missouri women. Cancer Res 2000; 60: 3753–6. [PubMed] [Google Scholar]
- 6. Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci 2004; 95: 290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paez JG, Janne PA, Lee JC et al . EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 8. Pao W, Miller V, Zakowski M et al . EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shigematsu H, Lin L, Takahashi T et al . Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97: 339–46. [DOI] [PubMed] [Google Scholar]
- 10. Tam IY, Chung LP, Suen WS et al . Distinct epidermal growth factor receptor and KRAS mutation patterns in non‐small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res 2006; 12: 1647–53. [DOI] [PubMed] [Google Scholar]
- 11. Kato T, Ohgaki H, Hasegawa H et al . Carcinogenicity in rats of a mutagenic compound, 2‐amino‐3,8‐dimethylimidazo[4,5‐f]quinoxaline. Carcinogenesis 1988; 9: 71–3. [DOI] [PubMed] [Google Scholar]
- 12. Kawano R, Takeshima Y, Inai K. Effects of K‐ras gene mutations in the development of lung lesions induced by 4‐(N‐methyl‐n‐nitrosamino)‐1‐(3‐pyridyl)‐1‐butanone in A/J mice. Jpn J Cancer Res 1996; 87: 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kitada H, Tsutsumi M, Tsujiuchi T et al . Frequent mutations of Ki‐ras but no mutations of Ha‐ras and p53 in lung lesions induced by N‐nitrosobis(2‐hydroxypropyl)amine in rats. Mol Carcinog 1996; 15: 276–83. [DOI] [PubMed] [Google Scholar]
- 14. Konishi Y, Kondo H, Denda A, Takahashi S, Inui S. Lung carcinomas induced by oral administration of N‐bis(2‐hydroxypropyl)nitrosamine in rats. In: Severi L, ed. Tumors of Early Life in Man and Animals. Perugia, Italy: Perugia University Press, 1978; 637–49. [Google Scholar]
- 15. Oghiso Y, Yamada Y. Comparisons of pulmonary carcinogenesis in rats following inhalation exposure to plutonium dioxide or X‐ray irradiation. J Radiat Res (Tokyo) 2003; 44: 261–70. [DOI] [PubMed] [Google Scholar]
- 16. Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single‐strand conformation polymorphisms. Proc Natl Acad Sci USA 1989; 86: 2766–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuan J, Yeasky TM, Rhee MC, Glazer PM. Frequent T:A→G:C transversions in X‐irradiated mouse cells. Carcinogenesis 1995; 16: 83–8. [DOI] [PubMed] [Google Scholar]
- 18. Kimchi‐Sarfaty C, Oh JM, Kim IW et al . A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science 2007; 315: 525–8. [DOI] [PubMed] [Google Scholar]
- 19. Takano T, Ohe Y, Sakamoto H, Tsuta K et al . Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non‐small‐cell lung cancer. J Clin Oncol 2005; 23: 6829–37. [DOI] [PubMed] [Google Scholar]
- 20. Oghiso Y, Yamada Y. Immunohistochemical study on cellular origins of rat lung tumors induced by inhalation exposures to plutonium dioxide aerosols as compared to those by X‐ray irradiation. J Radiat Res (Tokyo) 2002; 43: 301–11. [DOI] [PubMed] [Google Scholar]
- 21. Ikeda S, Takabe K, Inagaki M, Funakoshi N, Suzuki K, Shibata T. Correlation between EGFR gene mutation pattern and Akt phosphorylation in pulmonary adenocarcinomas. Pathol Int 2007; 57: 268–75. [DOI] [PubMed] [Google Scholar]
- 22. Nikula KJ, Snipes MB, Barr EB et al . Comparative pulmonary toxicities and carcinogenicities of chronically inhaled diesel exhaust and carbon black in F344 rats. Fundam Appl Toxicol 1995; 25: 80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ashakumary L, Vijayammal PL. Effect of nicotine on antioxidant defence mechanisms in rats fed a high‐fat diet. Pharmacology 1996; 52: 153–8. [DOI] [PubMed] [Google Scholar]
- 24. Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg 2006; 391: 499–510. [DOI] [PubMed] [Google Scholar]
- 25. Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down‐regulation of the receptors. Genes Dev 2006; 20: 1496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]