

Abstract
Docetaxel is a microtubule inhibitor that has actions in the S and G2–M phase of the cell cycle. The pyrimidine trifluorothymidine (TFT) induces DNA damage and an arrest in the G2–M phase. TFT, as part of TAS‐102, has been clinically evaluated as an oral chemotherapeutic agent in colon and gastric cancer. The aim of the present study was to determine the optimal administration sequence of TFT and docetaxel and to investigate the underlying mechanism of cytotoxicity. Drug interactions were examined by sulforhodamine B assays and subsequent combination index analyses, and for long‐term effects the clonogenic assay was used. A preincubation with docetaxel was synergistic in sulforhodamine B (combination index 0.6–0.8) and clonogenic assays, and was accompanied by a time‐dependent cell death induction (17–36%), the occurrence of polynucleation (22%), and mitotic spindle inhibition as determined by flow cytometry and immunostaining. Interestingly, administration of TFT followed by the combination displayed strong antagonistic activity, and was accompanied by less polynucleation and cell death induction than the synergistic combinations. Western blotting showed that the G2–M‐phase arrest (25–50%) was accompanied by phosphorylation of Chk2 and dephosphorylation of cdc25c in the synergistic combinations. Together, this indicates that synergistic activity requires docetaxel to initiate mitotic failure prior to the activation of TFT damage signaling, whereas antagonism is a result of TFT cell cycle‐arrested cells being less susceptible to docetaxel. Caspase 3 activation was low after docetaxel, suggestive of caspase‐independent mechanisms of cell death. Taken together, our models indicate that combination treatment with docetaxel and TFT displays strong synergy when docetaxel is given first, thus providing clues for possible clinical studies. (Cancer Sci 2008; 99: 2302–2308)
Docetaxel (Taxotere), a taxane, is one of the most important anticancer drugs today. It is part of the standard therapy of various types of solid tumors, such as breast, ovarian, non‐small‐cell lung cancer, androgen‐independent metastatic prostate cancer, and advanced gastric cancer.( 1 , 2 ) Docetaxel promotes tubulin assembly in microtubules and inhibits their depolymerization.( 3 ) In this way, docetaxel interferes with the dynamic changes that occur during the formation of the mitotic spindle. Docetaxel exhibits greater affinity for β‐tubulin, targeting centrosome organization and acting on cells in three phases of the cell cycle (S, G2, and M phases).( 4 ) Treatment with taxanes often leads to side effects as well as drug resistance.( 5 ) Therefore, it is important to develop new strategies that will lead to fewer side effects and improved activity. Various approaches to enhance the efficacy of the taxanes have been investigated( 6 , 7 ) and several taxane combinations are used standardly for various malignancies.( 3 ) The mechanism of action is not always very well understood and may depend on the type of drug or disease. For combinations to be successful a mechanism‐based interaction is preferable.
Trifluorothymidine (TFT) is a cytotoxic agent that might enhance the effect of docetaxel. TFT is part of the novel antitumor drug formulation TAS‐102, consisting of the combination of TFT with a specific inhibitor of thymidine phosphorylase (TPI).( 8 ) Addition of TPI to TFT enhances its bioavailability to tumor cells, as thymidine phosphorylase can break down TFT to an inactive form. TAS‐102 is currently being tested as an oral formulation in phase II clinical studies against both colon and gastric cancer. Murakami et al. previously reported that TFT may be more effective in colorectal cancer cells to overcome (acquired) 5‐fluorouracil (5‐FU) and 5‐fluro‐2'deoxyuride (FdUrd) resistance caused by amplification and subsequent overexpression of thymidylate synthase (TS).( 9 ) One mechanism of action consists of TS inhibition by its monophosphorylated form through a covalent binding to the active site of TS.( 10 , 11 ) TS is one of the major rate‐limiting enzymes in DNA synthesis, and inhibition induces a series of downstream events, eventually leading to cell death.( 12 ) When TFT is further activated to its tri‐phosphorylated form, it can be incorporated into the DNA,( 13 ) which will subsequently result in DNA damage.( 14 ) This may be the major mechanism of action of TFT compared to 5‐FU. This possibly explains why, in contrast to 5‐FU and antifolate‐based TS inhibitors, TFT induces a G2–M‐arrest in cancer cells.( 15 ) Moreover, it is not yet clear whether and how TFT inhibits the various DNA polymerases.
5‐Fluorouracil and the 5‐FU prodrug capecitabine have been combined with docetaxel in various studies and have been reported to be effective in terms of objective response and overall survival.( 6 , 16 , 17 ) In a phase II study where docetaxel was combined with the novel oral fluoropyrimidine formulation S‐1, this combination was shown to be very active and well tolerated.( 18 ) This synergism was probably due to a biochemical modulation of activating and inactivating enzymes of 5‐FU.( 19 ) However, the exact mechanism remains to be determined.
Both docetaxel and TFT show cell cycle‐dependent activity. Therefore, we tested sequential and concurrent combination schedules of the two drugs. Sequential administration of drugs can decrease side effects, which is also an advantage. Moreover, drug combinations with docetaxel and other taxanes were more effective when given sequentially.( 20 )
The aim of the present study was to determine whether TFT can enhance docetaxel sensitivity in colon cancer cells, using various treatment schedules, and to examine the mechanism underlining the synergism to better understand how they can be combined in the clinic.
Materials and Methods
Cell culture and chemicals. Human colon carcinoma cell lines Colo320 and WiDR were obtained from the American Type Culture Collection (ATCC, Teddington, UK) and were cultured as monolayers in Dulbecco's modified Eagle's medium supplemented with 10% heat‐inactivated fetal calf serum and 20 mmol/L HEPES in 25‐cm2 culture flasks (Greiner Bio‐One, Frickenhansen, Germany). Cells were maintained in a humidified 5% CO2 atmosphere at 37°C. TFT was provided by Taiho Pharmaceuticals (Tokushima, Japan) and was dissolved in phosphate‐buffered saline in stock solutions of 20 mmol/L. Docetaxel (Sigma‐Aldrich, Zwijndrecht, the Netherland) was dissolved in dimethylsulfoxide in a stock solution of 20 mmol/L. The stock solutions were stored aliquoted at –20°C.
Drug cytotoxicity assays. Drug cytotoxicity was determined using the sulforhodamine B (SRB) assay.( 21 ) Five thousand cells/well were seeded in 96‐wells plates (Greiner Bio‐One). After 24 h enabling attachment, cells were exposed to increasing concentrations of drugs for 72 h. For the simultaneous combination, cells were exposed to both TFT and docetaxel for 72 h. Sequential combinations consisted of a 24‐h preincubation with either TFT or docetaxel, followed by a 48‐h exposure to the combined drugs. Cells were precipitated with trichloroacetic acid for 1 h at 4°C, colored with SRB, solubilized with Tris, and the optical density (OD) was measured at 495–540 nm. For calculation of the growth‐inhibition curves, OD values were corrected for readings at the day of drug addition. IC50 values (inhibitory concentration of 50%) were subsequently estimated from graphs and are given as mean ± SEM. For simultaneous combinations, fixed ratios based on IC50 values were used to determine the interaction with the multiple drug effect method, in which a combination index (CI) was calculated (Calcusyn Biosoft, Cambridge, UK), based on the method of Chou and Talalay as described previously.( 14 ) A CI < 0.9 indicates synergism and >1.1 indicates antagonism. For each experiment the CI values at fraction‐affected 0.5, 0.75, and 0.9 were averaged and used for calculation of means between experiments. Values below 0.5 are considered to be irrelevant because they represent only a minor growth inhibition.
Clonogenic assay. Cell survival was determined using the clonogenic assay.( 22 ) Single cells were diluted serially to appropriate concentrations (150–2000 cells) and seeded in six‐wells plates in 3 mL medium. After 24 h, cells were exposed to the IC10, IC25, and IC50 values (as determined with the SRB assay) of TFT, docetaxel, and the tested combinations. At 96 h after cell seeding, drug‐containing medium was replaced with fresh medium. After 10 days in drug‐free medium, cells were fixed in ethanol (15 min) and stained with 10% Giemsa (Sigma). Surviving fractions were calculated by dividing the plating efficiency of exposed cells to the plating efficiency of control cells. Theoretical additivity was calculated by multiplication of the surviving fractions of TFT alone with that of docetaxel alone.( 23 )
Western blotting. Colo320 and WiDR cells were seeded at 1.5 × 106 cells per 25‐cm2 culture flask. After 24 h, cells were exposed to IC50 concentrations of TFT alone, docetaxel alone, or the various combination schedules as described for the drug cytotoxicity assays. After treatment, cells were lysed in lysis buffer (10% glycerol, 5 mmol/L ethylenediaminetetraacetic acid, 10 mmol/L Tris‐HCl pH 7.5, 150 mmol/L NaCl, 50 mmol/L β‐glycerophophate, 1% Triton X‐100, 0.04% protease inhibitor cocktail, 1 mmol/L NaVO3) and centrifuged at 13 900 g at 4°C for 10 min. The protein concentration of the supernatant was determined by a Bio‐Rad protein assay (#500‐0006) according to the manufacturer's instructions (Bio‐Rad Laboratories, Veenendaal, the Netherland). For each condition, 30 µg protein was separated on a 10% sodium dodecylsulfate–polyacrylamide (SDS) gel electrophoresis gel and electroblotted onto a polyvinylidene fluoride (PVDF)‐membrane (Millipore, Billerica, MA, USA). Subsequently, the membranes were blocked for 1 h in TBST (10 mmol/L Tris‐HCl, 0.15 mol/L NaCl, 0.05% Tween‐20) with 5% milk powder and subsequently incubated overnight at 4°C with primary antibodies directed against: CDK2 (#2546), phosphorylated CDK2 (Thr160; #2561), Chk1 (#2345), phosphorylated Chk1 (Ser345; #2341), Chk2 (#2662), phosphorylated Chk2 (Thr68; #2661), cdc25c (#4688) and phosphorylated cdc25c (Ser216; #4901) (1:1000; Cell Signaling Technology, Danvers, MA, USA). Following this, membranes were incubated with secondary antibody (1:2000; goat antimouse–horseradish peroxidase or donkey antirabbit–horseradish peroxidase; Dako Cytomation, Glostrup, Denmark) in TBST (2% milk powder) for 1 h at room temperature. β‐Actin served as a loading control for protein amount.
Fluorimetric assay for caspase activity. Caspase 3‐like activity was determined using a spectrofluorimetric assay of proteolytic cleavage of fluorogenic DEVD‐AFC substrates (Clontech Laboratories, Palo Alto, CA, USA).( 24 ) Experiments were carried out according to the manufacturer's instructions. Fluorescence was detected at 400 nm excitation and 505 nm emission (Spectra Fluor Tecan, Salzburg, Austria). Relative caspase activity was calculated in fluorescence units (FU)/h/million cells.
Fluorrescence‐activated cell sorting analysis of cell cycle distribution. Cell cycle analysis and apoptosis measurements were carried out as described previously.( 25 ) In brief, 100 000 cells were seeded in six‐well plates. After treatment, cells were trypsinized, resuspended in medium collected from the matching sample, and centrifuged for 5 min at 322 g. Subsequently, cells were stained with propidium iodide buffer (0.1 mg/mL with 0.1% RNAse A) on ice in the dark. The DNA content of the cells was analyzed by fluorescence‐activated cell sorting (FACS) (FACScan Becton Dickinson, Immunocytometry Systems, San Jose, CA, USA) with an acquisition of 10 000 events. The sub‐G1 peak was used to determine the extent of cell death.
Immunofluorescence staining. Cells were exposed to IC50 concentrations of TFT and docetaxel alone or the various combination schedules. After 72 h exposure, cells were fixed for 30 min in methanol, permeabilized for 10 min with 0.2% Triton X100 in phosphate‐buffered saline (PBS), blocked for 1 h with 3% bovine serum albumin (BSA), and stained for α‐tubulin (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. Subsequently, the secondary antibody (antimouse–fluorescein isothiocyanate; 1:200; Dako Cytomation) was added together with Hoechst 33342 to stain the nuclei (1:400) for 1 h. Subsequently, the coverslip was mounted onto microscope slides using Vectashield (Vector, Burlingame, CA, USA). Fluorescence microscopy was carried out using an inverted Leica DMIRB/E fluorescence microscope (Leica Cambridge, Cambridge, UK). Images were collected using Q500MC Quantimet software V01.01 (Leica Cambridge).
Results
Growth inhibition. The cytotoxic effects of docetaxel and TFT were assessed after 72 h of continuous drug exposure in the two cell lines WiDR and Colo320.( 14 , 15 ) Docetaxel and TFT inhibited the growth of both cell lines in a concentration‐dependent manner (Fig. 1; Table 1). Docetaxel inhibited cell growth at nanomolar concentrations, whereas TFT inhibited cell growth at micromolar concentrations. The IC10, IC25, and IC50 values were determined from the graphs and were used as a basis for the other experiments.
Figure 1.
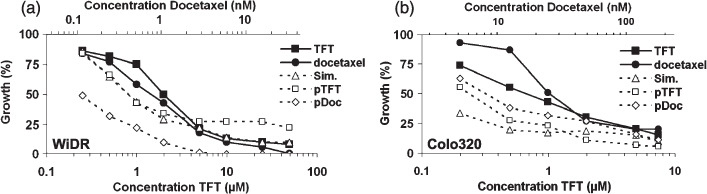
Growth‐inhibition curves of (a) WiDR and (b) Colo320 cells after 72 h exposure to trifluorothymidine (TFT) or docetaxel, or either 72 h exposure simultaneously (Sim) with TFT and docetaxel or 24 h preincubated with TFT (pTFT) or docetaxel (pDoc), followed by a 48 h combination treatment of TFT with docetaxel. All combinations are based on IC50:IC50 ratios. All SEM values were <10%. IC50, inhibitory concentration of 50%.
Table 1.
Growth inhibition of trifluorothymidine (TFT) and docetaxel in colorectal cancer cells: combination analysis of various treatment schedules of TFT combined with docetaxel
| Cell line | Growth inhibition (IC50) | Combination index | |||
|---|---|---|---|---|---|
| TFT (µmol/L) | Docetaxel (nmol/L) | Simultaneous | pTFT | pDoc | |
| WiDR | 3.2 ± 0.3 | 1.7 ± 0.3 | 1.95 ± 0.13 | >10 | 0.62 ± 0.04 |
| Colo320 | 0.6 ± 0.1 | 16 ± 1.4 | 0.76 ± 0.18 | 0.77 ± 0.10 | 0.80 ± 0.20 |
IC50 values were estimated from graphs (Fig. 1) and are given in means of at least three to five independent experiments ± SEM. Average combination index (CI) values were calculated from the fraction affected (FA) from data points with a FA of 0.5, 0.75, and 0.9. CI values < 0.9 indicate synergy; CI = 0.9–1 mean additive; CI > 1.1 means antagonism. Values represent mean CI values ± SEM of three to four independent experiments. pDoc, pre‐exposed to docetaxel; pTFT, pre‐exposed to trifluorothymidine; TFT, trifluorothymidine. IC50, inhibitory concentration of 50%.
Evaluation of combinations. The effect of the combinations was analyzed by determining the CI values from growth‐inhibition curves (Fig. 1). In WiDR cells, the simultaneous combination was antagonistic and strongly antagonistic after pre‐exposure to TFT (pTFT) (Table 1). On the contrary, when WiDR cells were pre‐exposed to docetaxel (pDoc) for 24 h, synergistic activity was detected. In Colo320 cells, all tested combination schedules were synergistic (Table 1).
Cell cycle effects and cell death induction. The sequence in which the cells were exposed to the combinations was important, which may be related to the cell cycle dependency of the drugs in exerting their activity. Therefore, we analyzed the mechanisms underlining the synergistic and antagonistic combinations by determining cell death and cell cycle distribution by FACS analysis on propidium iodide‐stained cells (Fig. 2). In WiDR cells, TFT induced a time‐dependent arrest in G2–M phase and polyploidy. Docetaxel induced an immediate arrest in G2–M phase, with cells progressing to polyploidy and undergoing cell death (Fig. 2a,c). Under antagonistic combinations (simultaneous and pTFT) an increased number of cells accumulated in G2–M phase, and a higher portion of cells died after 72 h, whereas similar levels of polyploidy were detected. For the synergistic pDoc combination, the G2–M arrest was less pronounced, but clearly higher levels of polyploid and dead cells were detected.
Figure 2.
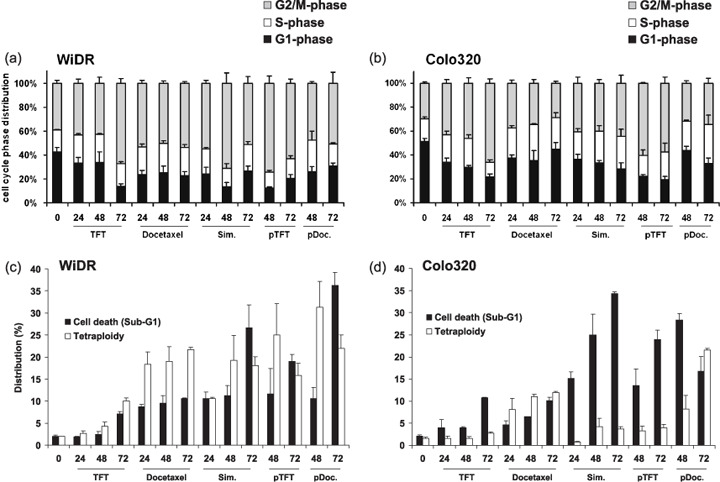
Effect of trifluorothymidine (TFT) IC50 and docetaxel IC50 and the combinations (IC50 ratio) on (a,b) cell cycle distribution and (c,d) cell death and polyploidy in (a,c) WiDR and (b,d) Colo320 cells. Values are means of at least three independent experiments ± SEM. pDoc, pre‐exposed to docetaxel; pTFT, pre‐exposed to trifluorothymidine; Sim, simultaneous exposure.
In Colo320 cells, TFT also induced a time‐dependent G2–M arrest. The G2–M arrest induced by docetaxel was less pronounced (Fig. 2b,d). Combined treatments arrested cells in both the G2–M and S phases of the cell cycle. The pDoc combination resulted in the induction of both cell death and polyploidy, whereas for the other combinations cell death induction alone could account for the synergism. The apparent difference between WiDR and Colo320 cells in polyploidy seems to be related to the extent of cell kill, which was time dependent. The pDoc combination, which was synergistic in both cell lines, worked mostly by the accumulation of polyploidy, after which cell death was induced.
Cell cycle regulation. In order to link the drug‐induced cell cycle effects with molecular mechanisms of cell cycle regulation, the major checkpoints and cell cycle‐dependent kinases Chk1, Chk2, cdc25c, and CDK2 were analyzed by western blotting (Fig. 3).( 26 ) Chk1 and Chk2, when phosphorylated, prevent progression from G1 to S phase and G2 to M phase, respectively. Cdc25c phosphorylation prevents G2 to M progression and upon phosphorylation of CDK2, G1 to S‐phase progression is stimulated.
Figure 3.
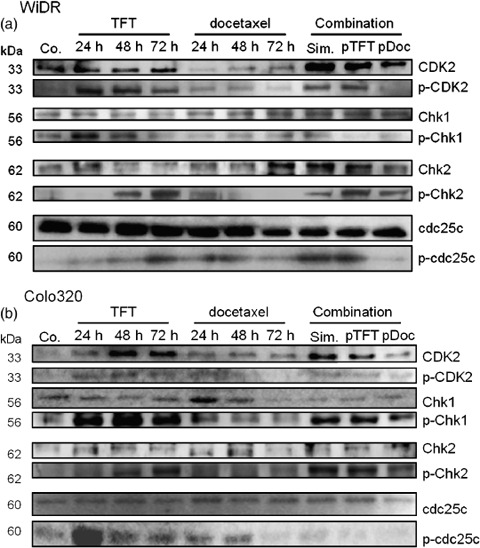
Representative western blot (n = 3) of the expression of cell cycle‐regulating proteins in (a) WiDR and (b) Colo320 cells. Cells were exposed to IC50 concentrations of trifluorothymidine (TFT) and docetaxel alone and to the IC50 ratio‐based combinations for 72 h. Co, control; pDoc, pre‐exposed to docetaxel; p‐, phosphorylated; pTFT, pre‐exposed to trifluorothymidine; Sim, simultaneous exposure.
In WiDR cells, CDK2 was phosphorylated after TFT exposure. In these cells, docetaxel exposure resulted in the downregulation of CDK2, but the phosphorylation levels of CDK2 did not change. The antagonistic combinations (simultaneous and pTFT) resulted in the phosphorylation of CDK2, whereas after the synergistic pDoc combination, CDK2 was not phosphorylated. Chk1 was phosphorylated after 24 h TFT exposure, which reduced in time. Docetaxel did not change Chk1 phosphorylation levels. Chk1 levels hardly changed after the combination, and seemed to decrease after the pTFT combination. Chk2 was phosphorylated in time after TFT exposure, which is in relation to the G2–M arrest found by FACS analysis (Fig. 2). This was in contrast to docetaxel, after which these phosphorylation levels increased after 24 h, but reduced in time to control levels, which explains the less time‐dependent effect of this drug. This may be related to the induction of polyploidy and cell death. After all combination schedules, Chk2 was phosphorylated. Cdc25c was not phosphorylated after the pDoc combination.
In Colo320 cells, TFT exposure resulted in phosphorylation of CDK2, Chk1, Chk2, and cdc25c, which can be related to the time‐dependent induction of G2–M arrest. Docetaxel exposure of Colo320 cells hardly affected CDK2 phosphorylation, activated Chk1, Chk2, and cdc25c after 24 h, which all reduced in time. The combinations resulted in activation of Chk1, Chk2, and dephosphorylated cdc25c. All of the synergistic combinations resulted in dephosphorylation of cdc25c, which may explain why cells arrested to a lower extent in G2–M phase and could progress to polyploidy, resulting in mitotic failure. Probably the synergistic action of the pDoc combination in both cell lines was due to the induction of mitotic failure, after which cells died, whereas after the other combinations the checkpoints activated after cell cycle progression were inhibited in either G2–M phase or polyploidy.
Clonogenic cell survival. For the tested combination schedules there was a time‐dependent induction of cell death and polyploidy. Large polynucleated cells may mask true growth inhibition as determined by the SRB assay. Moreover, other reports about combination studies with taxanes indicate that multiple failure to complete cell division cycles results in induction of polynucleation (mitotic failure), after which cell death is induced.( 27 , 28 ) For determining clonogenic survival, we used the IC10, IC25, and IC50 concentrations as determined in the SRB assay (Fig. 1). At these concentrations, docetaxel had a higher clonogenic survival than TFT in both WiDR and Colo320 cells (Fig. 4). In contrast to the difference in IC50, both WiDR and Colo320 cells had a comparable clonogenic survival after exposure to TFT alone. After exposure to the IC50 concentration of TFT and some of the combinations (IC50:IC50), no colonies were formed. Similar to the SRB assay, the pDoc combination resulted in strong additivity for both cell lines. The other combinations were less active. These long‐term survival data are in agreement with the combined induction of polyploidy and cell death (Fig. 2), which were both strongly induced after the pDoc combination.
Figure 4.
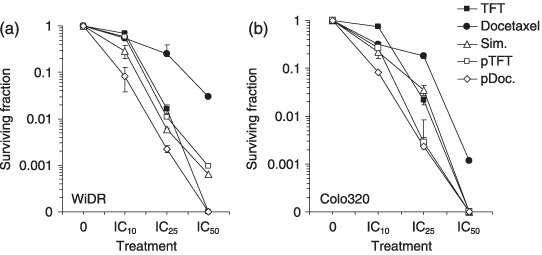
Clonogenic survival of (a) WiDR and (b) Colo320 cells after exposure to trifluorothymidine (TFT), docetaxel, and the combinations. Combination schedules used were IC10 : IC10 and IC25 : IC25 ratios, based on the sulforhodamine B data. Values represent means of at least three independent experiments ± SEM. pDoc, pre‐exposed to docetaxel; pTFT, pre‐exposed to trifluorothymidine; Sim, simultaneous exposure.
Caspase 3 activation. Caspase 3 is an important mediator of the execution phase of the apoptotic process. We determined caspase 3 enzymatic activity and found activation upon TFT exposure in WiDR cells, but only to some extent in Colo320 cells (Fig. 5). Docetaxel activated caspase 3 moderately in both cell lines, although more in Colo320 cells than in WiDR cells. Exposure to any of the combinations did not result in increased activation when compared to exposure to the single agents. Apparently, caspase 3 is not a key factor in triggering apoptosis by these agents, suggesting that other caspases or caspase‐independent mechanisms may be involved.
Figure 5.
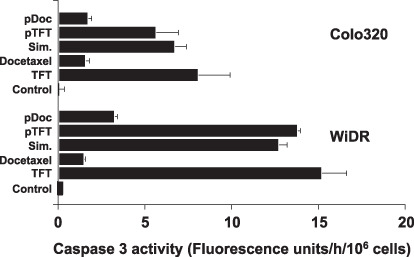
Caspase 3 enzymatic activity after 72 h exposure to trifluorothymidine (TFT) or docetaxel alone or the (sequential) combinations. Values are means ± SEM of two independent experiments. pDoc, pre‐exposed to docetaxel; pTFT, pre‐exposed to trifluorothymidine; Sim, simultaneous exposure.
Effect on microtubules. As both drugs induce G2–M arrest and docetaxel is known to stabilize microtubules, cells were stained immunofluorescently for their cellular tubulin network. Docetaxel exposed cells showed the characteristic aberrant spindle formation, named aster spindles (Fig. 6). TFT treatment resulted in the formation of large multinucleated (polyploidy) cells that seemed to occur prior to the formation of spindles, as mitotic spindles were not (clearly) detected. All combinations of TFT with docetaxel resulted in an increased cell size in both WiDR and Colo320. Multinucleated cells were detected upon treatment with concurrent and pTFT combinations, also displaying increased cell size compared to normal cells. These phenomena were seen more clearly in WiDR cells and are reflected by the increased polyploid cell fraction in the FACS profiles (Fig. 2). Abnormal multipolar aster spindles were seen after all treatment combinations.
Figure 6.
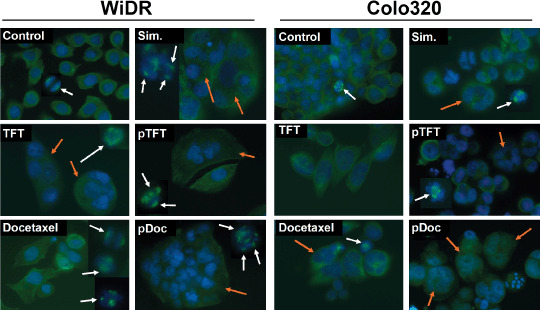
Representative immunofluorescence stainings of α‐tubulin (mitotic spindle; green; white arrows) and Hoechst (DNA, blue; polynucleated cells, orange arrows) (n = 4). Cells were exposed to trifluorothymidine (TFT), docetaxel, simultaneous combination (Sim), preincubation with TFT (pTFT), or a preincubation with docetaxel (pDoc) for 72 h.
Discussion
The present study shows that docetaxel and TFT have synergistic activity, which is most likely related to the combined induction of cell death and mitotic failure. The various combination schedules resulted in different cellular responses. Especially when cells were pretreated with docetaxel before exposure to the combination treatment, WiDR and Colo320 cells both seemed to have similar actions, that is, the induction of both cell death and tetraploidy and inactivation of the cell cycle kinase cdc25c. However, for the other combinations, the response was more cell‐type dependent. Although TFT and docetaxel have different mechanisms of action, both drugs arrested cells in the G2–M phase of the cell cycle,( 15 ) but with different effects on the mitotic spindles. Docetaxel caused the formation of aster spindles, whereas TFT seemed to prevent or induce cell cycle arrest prior to spindle formation. Apparently, these drug‐dependent effects form the basis for synergism.
Clinical data have shown that docetaxel is active against various tumor types.( 3 ) In the present study, docetaxel alone could inhibit cell growth at clinically relevant concentrations. Docetaxel acts by interfering with the formation of the mitotic spindle. This is associated with an arrest in the S‐ and/or G2‐M phase of cell cycle and subsequent cell death induction.( 4 ) Docetaxel is currently being studied in several clinical trials in combination with 5‐FU and oral 5‐FU formulations, such as S‐1, against various cancer types.( 16 , 18 , 19 ) Combining docetaxel with other chemotherapeutic agents has been shown to improve overall survival and responses, but side effects and resistance are recurrent problems. Therefore, TFT (as TAS‐102) could be a new strategy to circumvent resistance and decrease the side effects. TFT has overlapping actions with 5‐FU, and might be a good alternative to 5‐FU. TFT induces cell cycle arrest in the G2–M phase of the cell cycle, whereas 5‐FU has been shown to arrest cells in G1 or S phase.( 29 )
An S‐phase arrest is expected for TS inhibitors, such as nolatrexed and raltitrexed.( 29 ) These drugs clearly showed a strong TS inhibition and arrest in S phase. TFT can also inhibit TS in cancer cells; however, it may be that TFT also has other mechanistic actions apart from TS inhibition. Previously, we showed that TFT can be incorporated into DNA. Moreover, TFT could overcome drug resistance in 5‐FU‐resistant cell lines.( 9 , 30 ) Therefore, TFT may be active against 5‐FU‐resistant colorectal cancer cells. TFT can be activated rapidly as it only needs one activation step, whereas 5‐FU needs two or more activation steps. These differences in response further point out that TS inhibition may not be the main mechanism of action TFT.
Induction of cell cycle arrest is regulated by the activation (phosphorylation) of cell cycle regulators.( 26 ) Interestingly, TFT initially stimulates cells to progress from G1 to S phase (Fig. 2). An explanation for this is that TFT is a nucleoside analog. Exposing cells to nucleosides may stimulate cell cycle progression toward DNA synthesis. The differences in sensitivity between the SRB assay and the clonogenic assay indicate that TFT may need more cycles to cause enough damage to induce cell death. TFT incorporation into the DNA induces DNA strand breaks.( 14 , 15 ) In addition, due to intracellular thymidine depletion after TS inhibition, uracil can be misincorporated into the DNA, thus enhancing the induction of DNA damage.( 31 ) TFT‐induced DNA damage leads to cell cycle arrest in G2 followed by cell death activation in a p53‐independent way, as we have also demonstrated recently.( 32 ) Following activation of the checkpoints Chk1 and Chk2, probably in an ataxia telangiectasia mutated kinase (ATM)‐dependent manner,( 33 ) cdc25c is inactivated, thereby further preventing entry into M phase, inducing arrest in the G2 phase of the cell cycle, after which cell death is induced.
We speculate that the synergistic pDoc combination resulted in the activation of both mitotic catastrophe‐induced cell death and TFT‐induced cell death, whereas the other combinations triggered predominantly either one of the death mechanisms. The pDoc combination demonstrated less activation of the checkpoints and seems to have the highest activity after 48 h, which finally resulted in cell death induction in WiDR and an increase in polyploidy in Colo320 (Fig. 2). The decrease in cell death may be related to the increase in tetraploidy, delaying cell death induction. Interestingly, all synergistic combinations were accompanied by low phosphorylation levels of cdc25c. Recent data suggest that cdc25c plays a complex role during mitosis. In a recent report in which HT29 colon cells were exposed to a cdc25 phosphatase inhibitor, the mitotic spindle was impaired and the segregation of chromosomes became aberrant.( 34 ) The low phosphorylation of cdc25c in the synergistic combinations in our study indicates that this may be important for mitotic catastrophe and subsequent induction of cell death. Apparently, for synergistic activity docetaxel is required to first inflict tubulin damage causing cell cycle arrest and mitotic failure; thereafter, TFT‐induced damage is able to further increase cell death activation. The antagonistic combinations in WiDR were probably due to TFT actions alone, preventing docetaxel from exerting its activity; however, the precise antagonistic mechanism remains unclear. Under these conditions TFT‐induced damage causes activation of the cdc25‐dependent checkpoint, thereby pausing cells and preventing docetaxel from inducing mitotic catastrophe, thus explaining the antagonistic activity. Both the simultaneous and pTFT combination had comparable cell cycle actions, activation of the checkpoints Chk1 and Chk2, strong arrest in G2–M phase, and induction of cell death. These cell cycle‐dependent actions of the drugs may explain why the sequence of the combination is so important when using these types of drugs.
Inhibition of microtubule function by docetaxel leads to the induction of apoptosis.( 4 ) One of the main executioner caspases is caspase 3, which is active in its cleaved form. Caspase 3 was activated upon TFT exposure and only weakly after docetaxel treatment. Furthermore, caspase 3 activation levels did not reflect the levels of cell death induction after combination treatments. Caspase‐independent apoptotic cell death has previously been reported for microtubule inhibitors including paclitaxel, epothilone B, and discodermolide and might involve cathepsin B or another mechanism.( 35 ) Furthermore, in mice with implanted Ly‐TH lymphoma, docetaxel causes tumor lysis rather than apoptosis.( 36 )
In conclusion, these in vitro results provide a rationale for the experimental use of the combination of docetaxel with TFT, in which scheduling is important. Moreover, the in vivo potential of this combination might be enhanced, because TAS‐102 also contains TPI, which besides increasing TFT bioavailability also has antiangiogenic potential. This, combined with reports showing the antiangiogenic potential of taxanes,( 37 ) provides further perspective for using this combination in the clinic.
Acknowledgments
The present study was supported financially by Taiho Pharmaceutical Company, Tokushima, Japan.
References
- 1. Rowinsky EK, Onetto N, Canetta RM, Arbuck SG. Taxol: the first of the taxanes, an important new class of antitumor agents. Semin Oncol 1992; 19: 646–62. [PubMed] [Google Scholar]
- 2. Huizing MT, Misser VH, Pieters RC et al . Taxanes: a new class of antitumor agents. Cancer Invest 1995; 13: 381–404. [DOI] [PubMed] [Google Scholar]
- 3. Montero A, Fossella F, Hortobagyi G, Valero V. Docetaxel for treatment of solid tumours: a systematic review of clinical data. Lancet Oncol 2005; 6: 229–39. [DOI] [PubMed] [Google Scholar]
- 4. Gligorov J, Lotz JP. Preclinical pharmacology of the taxanes: implications of the differences. Oncologist 2004; 9 (Suppl 2): 3–8. [DOI] [PubMed] [Google Scholar]
- 5. Zhao J, Kim JE, Reed E, Li QQ. Molecular mechanism of antitumor activity of taxanes in lung cancer. Int J Oncol 2005; 27: 247–56. [PubMed] [Google Scholar]
- 6. Azzoli CG, Krug LM, Gomez J et al . A phase 1 study of pralatrexate in combination with paclitaxel or docetaxel in patients with advanced solid tumors. Clin Cancer Res 2007; 13: 2692–8. [DOI] [PubMed] [Google Scholar]
- 7. Mrozek E, Ramaswamy B, Young D et al . Phase II study of weekly docetaxel and capecitabine in patients with metastatic breast cancer. Clin Breast Cancer 2006; 7: 141–5. [DOI] [PubMed] [Google Scholar]
- 8. Temmink OH, Emura T, de Bruin M, Fukushima M, Peters GJ. Therapeutic potential of the dual‐targeted TAS‐102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci 2007; 98: 779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murakami Y, Kazuno H, Emura T, Tsujimoto H, Suzuki N, Fukushima M. Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int J Oncol 2000; 17: 277–83. [DOI] [PubMed] [Google Scholar]
- 10. Eckstein JW, Foster PG, Finer‐Moore J, Wataya Y, Santi DV. Mechanism‐based inhibition of thymidylate synthase by 5‐(trifluoromethyl)‐2′‐deoxyuridine 5′‐monophosphate. Biochemistry 1994; 33: 15086–94. [DOI] [PubMed] [Google Scholar]
- 11. Temmink OH, de Bruin M, Comijn EM, Fukushima M, Peters GJ. Determinants of trifluorothymidine sensitivity and metabolism in colon and lung cancer cells. Anticancer Drugs 2005; 16: 285–92. [DOI] [PubMed] [Google Scholar]
- 12. Van Triest B, Pinedo HM, Giaccone G, Peters GJ. Downstream molecular determinants of response to 5‐fluorouracil and antifolate thymidylate synthase inhibitors. Ann Oncol 2000; 11: 385–91. [DOI] [PubMed] [Google Scholar]
- 13. Emura T, Nakagawa F, Fujioka A et al . An optimal dosing schedule for a novel combination antimetabolite, TAS‐102, based on its intracellular metabolism and its incorporation into DNA. Int J Mol Med 2004; 13: 249–55. [PubMed] [Google Scholar]
- 14. Temmink OH, Hoebe EK, van der Born K, Ackland SP, Fukushima M, Peters GJ. Mechanism of trifluorothymidine potentiation of oxaliplatin‐induced cytotoxicity to colorectal cancer cells. Br J Cancer 2007; 96: 231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Temmink OH, Hoebe EK, Fukushima M, Peters GJ. Irinotecan‐induced cytotoxicity to colon cancer cells in vitro is stimulated by pre‐incubation with trifluorothymidine. Eur J Cancer 2007; 43: 175–83. [DOI] [PubMed] [Google Scholar]
- 16. Van Cutsem E, Moiseyenko VM, Tjulandin S et al . Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first‐line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 2006; 24: 4991–7. [DOI] [PubMed] [Google Scholar]
- 17. Di Bartolomeo M, Buzzoni R, Mariani L et al . Feasibility of sequential therapy with FOLFIRI followed by docetaxel/cisplatin in patients with radically resected gastric adenocarcinoma. A randomized phase III trial. Oncology 2006; 71: 341–6. [DOI] [PubMed] [Google Scholar]
- 18. Yoshida K, Ninomiya M, Takakura N et al. Phase II study of docetaxel and S‐1 combination therapy for advanced or recurrent gastric cancer. Clin Cancer Res 2006; 12: 3402–7. [DOI] [PubMed] [Google Scholar]
- 19. Wada Y, Yoshida K, Suzuki T et al . Synergistic effects of docetaxel and S‐1 by modulating the expression of metabolic enzymes of 5‐fluorouracil in human gastric cancer cell lines. Int J Cancer 2006; 119: 783–91. [DOI] [PubMed] [Google Scholar]
- 20. Motwani M, Rizzo C, Sirotnak F, She Y, Schwartz GK. Flavopiridol enhances the effect of docetaxel in vitro and in vivo in human gastric cancer cells. Mol Cancer Ther 2003; 2: 549–55. [PubMed] [Google Scholar]
- 21. Keepers YP, Pizao PE, Peters GJ, van Ark‐Otte J, Winograd B, Pinedo HM. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer 1991; 27: 897–900. [DOI] [PubMed] [Google Scholar]
- 22. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro . Nat Protoc 2006; 1: 2315–19. [DOI] [PubMed] [Google Scholar]
- 23. van Bree C, Castro Kreder N, Loves WJ, Franken NA, Peters GJ, Haveman J. Sensitivity to ionizing radiation and chemotherapeutic agents in gemcitabine‐resistant human tumor cell lines. Int J Radiat Oncol Biol Phys 2002; 54: 237–44. [DOI] [PubMed] [Google Scholar]
- 24. Bröker LE, Huisman C, Ferreira CG, Rodriguez JA, Kruyt FA, Giaccone G. Late activation of apoptotic pathways plays a negligible role in mediating the cytotoxic effects of discodermolide and epothilone B in non‐small cell lung cancer cells. Cancer Res 2002; 62: 4081–8. [PubMed] [Google Scholar]
- 25. Ferreira CG, Tolis C, Span SW et al . Drug‐induced apoptosis in lung cancer cells is not mediated by the Fas/FasL (CD95/APO1) signaling pathway. Clin Cancer Res 2000; 6: 203–12. [PubMed] [Google Scholar]
- 26. Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 2003; 36: 131–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vogel C, Kienitz A, Hofmann I, Müller R, Bastians H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 2004; 23: 6845–53. [DOI] [PubMed] [Google Scholar]
- 28. Blagosklonny MV. Prolonged mitosis versus tetraploid checkpoint: how p53 measures the duration of mitosis. Cell Cycle 2006; 5: 971–5. [DOI] [PubMed] [Google Scholar]
- 29. Backus HH, Pinedo HM, Wouters D et al . Differences in the induction of DNA damage, cell cycle arrest, and cell death by 5‐fluorouracil and antifolates. Oncol Res 2000; 12: 231–9. [DOI] [PubMed] [Google Scholar]
- 30. Emura T, Murakami Y, Nakagawa F, Fukushima M, Kitazato K. A novel antimetabolite, TAS‐102 retains its effect on FU‐related resistant cancer cells. Int J Mol Med 2004; 13: 545–9. [PubMed] [Google Scholar]
- 31. Webley SD, Welsh SJ, Jackman AL, Aherne GW. The ability to accumulate deoxyuridine triphosphate and cellular response to thymidylate synthase (TS) inhibition. Br J Cancer 2001; 85: 446–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bijnsdorp IV, Kruyt FA, Fukushima M, Peters GJ. Trifluorothymidine induces cell death independently of p53. Nucleosides Nucleotides Nucleic Acids 2008; 27: 699–703. [DOI] [PubMed] [Google Scholar]
- 33. Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26: 7773–9. [DOI] [PubMed] [Google Scholar]
- 34. Cazales M, Boutros R, Brezak MC, Chaumeron S, Prevost G, Ducommun B. Pharmacologic inhibition of CDC25 phosphatases impairs interphase microtubule dynamics and mitotic spindle assembly. Mol Cancer Ther 2007; 6: 318–25. [DOI] [PubMed] [Google Scholar]
- 35. Bröker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G. Cathepsin B mediates caspase‐independent cell death induced by microtubule stabilizing agents in non‐small cell lung cancer cells. Cancer Res 2004; 64: 27–30. [DOI] [PubMed] [Google Scholar]
- 36. Schimming R, Mason KA, Hunter N, Weil M, Kishi K, Milas L. Lack of correlation between mitotic arrest or apoptosis and antitumor effect of docetaxel. Cancer Chemother Pharmacol 1999; 43: 165–72. [DOI] [PubMed] [Google Scholar]
- 37. Bijman MN, van Nieuw Amerongen GP, Laurens N, van Hinsbergh VW, Boven E. Microtubule‐targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther 2006; 5: 2348–57. [DOI] [PubMed] [Google Scholar]