

Abstract
Despite current risk‐directed therapy, approximately 15–20% of pediatric patients with acute lymphoblastic leukemia (ALL) have relapses. Recent genome‐wide analyses have identified that an alteration of IKZF1 is associated with very poor outcomes in B‐cell progenitor ALL. In this study, we determined the prognostic significance of IKZF1 deletions in patients with childhood ALL. This study analyzed 242 pediatric B‐cell progenitor ALL patients in Taiwan. We developed a simple yet sensitive multiplex quantitative PCR coupled with capillary electrophoresis to accurately determine the allele dose of IKZF1, and high resolution melting was used for mutation screening for all coding exons of IKZF1. Twenty‐six (10.7%) pediatric B‐cell progenitor ALL patients were found to harbor these deletions. Most of the deletions were broader deletions that encompassed exon 3 to exon 6, consistent with previous reports. Genomic sequencing of IKZF1 was carried out in all cases and no point mutations were identified. Patients with IKZF1 deletions had inferior event‐free survival (P < 0.001), and overall survival (P = 0.0016). The association between IKZF1 deletions and event‐free survival was independent of age, leukocyte count at presentation, and cytogenetic subtype by multivariate Cox analysis (P = 0.003, hazard ratio = 2.45). This study indicates that detection of IKZF1 deletions upon diagnosis of B‐cell progenitor ALL may help to identify patients at risk of treatment failure. IKZF1 deletions could be incorporated as a new high‐risk prognostic factor in future treatment protocols. To the best of our knowledge, this is the first study to examine the poor prognosis of IKZF1 deletions in an Asian population. (Cancer Sci 2011; 102: 1874–1881)
The cure rate of childhood acute lymphoblastic leukemia (ALL) is >80% in most advanced countries, and is expected to reach 90% in the near future.( 1 , 2 ) Treatment success depends on the progressive intensification of standard chemotherapy and the development of detailed risk classification schemes that tailor the intensity of therapy to the predicted risk of relapse. Risk stratification in B‐cell progenitor ALL is based on a number of recurring chromosomal abnormalities, including hyperdiploidy, hypodiploidy, translocations t(12;21)(ETV6‐RUNX1), t(9;22)(BCR–ABL1), and t(1;19)(TCF3‐PBX1), rearrangement of the mixed‐lineage leukemia (MLL) gene, and an early response to chemotherapy (minimal residual disease).( 1 ) Nonetheless, 10–20% of patients will experience a relapse. Relapsed ALL ranks as the fourth most common childhood malignancy, with an overall survival rate of only 30%.( 3 )
The heterogeneity of childhood ALL likely depends on the activation of different leukemogenic pathways defining susceptibility to current treatment protocols. Childhood ALL can be grouped by genome‐wide expression profiling, and the gene expression profiles also have close relationships with recurrent chromosomal abnormalities, immuno‐phenotypes, treatment outcomes, drug resistance, and minimal residual disease. Gene expression signatures have been constructed and proposed as new prognostic markers in treatment protocols.( 4 , 5 , 6 , 7 , 8 , 9 , 10 ) Even though RNA‐based risk markers may be useful in the future, quantification of mRNA in clinical samples is technically demanding and is strongly dependent on pre‐analytic handling of the bone marrow. In principle, DNA‐based markers are more robust and may be more applicable in practice.
The recent application of high‐resolution genomic profiling has extended the understanding of genetic lesions underlying childhood ALL. Using a single‐nucleotide polymorphism (SNP) array, Mullighan et al. identified an average of six copy number alterations per patient of childhood ALL. Mutations of genes encoding transcriptional regulators of B lymphoid development, including PAX5, EBF1, and IKZF1, occur in more than 40% of patients with B‐cell progenitor ALL.( 11 , 12 , 13 , 14 ) Deletions of IKZF1, which encodes lymphoid transcription factor IKAROS, are very frequent in BCR–ABL1 positive ALL and in the progression of CML to lymphoid blast crisis.( 12 ) A comprehensive analysis of high‐risk precursor B cell ALL carried out by Mullighan et al. ( 15 ) revealed that genetic alterations of IKZF1 were associated with a high risk of relapse. This strong association between IKZF1 and adverse outcomes was confirmed in a Dutch DCOG‐ALL9 cohort. Martinelli et al. ( 16 ) also showed that IKZF1 deletions were likely to be genomic alterations that significantly affect the prognosis of BCR–ABL1 positive ALL in adults. These studies suggest that deletions of IKZF1 may be a new prognostic marker to identify high‐risk ALL patients who will fail initial chemotherapy.
Multiplex quantitative PCR is a new approach to detect gene deletions, duplications, and rearrangements. In recent years, several new techniques have been developed for the quantitative assay of PCR products. Capillary electrophoresis is a simple, high‐performance, reliable, high‐resolution, time‐saving, and low labor‐intensive technique that has promise as a sensitive and specific tool for separating biomolecules and detecting variations in DNA.( 17 ) High resolution melting (HRM) is a method for large‐scale mutation analysis.( 18 ) This study applied these methods to carry out a comprehensive analysis to identify the genetic alterations of IKZF1 in childhood ALL, and investigate the prognostic impact of IKZF1 deletions in Taiwan.
Patients and Methods
Patients and protocols. Viable diagnostic bone marrow (BM) or peripheral blood was obtained from 242 children with B‐precursor ALL from May 1995 to August 2009 at the National Taiwan University Hospital, China Medical University Hospital, Chang Gung Memorial Hospital–Kaohsiung, and National Cheng Kung University Hospital (Taiwan). Seventy‐seven patients were treated with TPOG‐ALL‐93 protocols, 27 with TPOG‐97‐VHR, and 138 with TPOG‐ALL‐2002 protocols. The diagnosis of ALL was based on morphologic findings of BM aspirates and immuno‐phenotype analyses of leukemic cells by flow cytometry. Conventional cytogenetic analyses were done as part of the routine work‐up.
The treatment protocols were previously described.( 19 , 20 , 21 ) Patients were considered to have standard‐risk (SR, low risk in other studies) ALL if they were between 1 and 9 years of age presenting with a leukocyte count of <10 × 109 cells/L, or were between 2 and 7 years of age presenting with a leukocyte count between 10 × 109 and 50 × 109 cells/L. Patients were considered to have high‐risk (HR, intermediate risk in other studies) ALL if they were between 1 and 9 years of age presenting with a leukocyte count between 50 × 109 and 100 × 109 cells/L, or between 1 and 2 or 7 and 10 years of age presenting with a leukocyte count between 10 × 109 and 50 × 109 cells/L. In addition, those with central nervous system leukemia or cranial nerve palsy at diagnosis, or expression of myeloid antigens for TPOG‐ALL‐93 and those with central nervous system leukemia (cerebrospinal fluid white blood cells with blasts), cranial nerve palsy, testicular leukemia, or pre‐B ALL with TCF‐PBX1 fusion for TPOG‐ALL‐2002 were also considered to be at high risk. Patients with at least one of the following were assigned to the very high‐risk (VHR, high risk in other studies) group: age below 1 year, initial leukocyte count >100 × 109 cells/L, lymphoblastic lymphoma with more than 25% lymphoblasts in the bone marrow, hypodiploidy, HR patients with poor treatment response, and the presence of BCR–ABL1, MLL–AF4, or other MLL rearrangements in pre‐B ALL.
The patients were prospectively assigned to one of three risk groups (standard, high, and very high) based on their presenting clinical features and the biological features of their leukemic cells. The risk‐directed Taiwan Pediatric Oncology Group (TPOG) protocols consist of multiple chemotherapeutic agents of different intensities. The treatment protocol was upgraded if complete remission was not achieved after initial induction therapy. Events were defined as any relapse, death, or secondary malignancy. The Institutional Review Board of National Taiwan University Hospital approved the study and all of the participants provided written informed consent in accordance with the Declaration of Helsinki. Details of the protocols and risk group assignment have been published elsewhere.( 19 , 20 , 21 )
Determination of IKZF1 deletions by multiplex PCR with capillary electrophoresis. As almost all IKZF1 deletions involve exon 1 to exon 6,( 12 , 15 ) two multiplex quantitative PCR reactions were designed to amplify these exons. FGFR2 and FBN1 were used as internal controls to determine the relative allele dosage of the gene tested. Two multiplex quantitative PCR amplifications (multiplex I included the FGFR2 and FBN1 genes, and exons 1, 3, and 5 of the IKZF1 gene; multiplex II included the FGFR2 and FBN1 genes, and exons 2, 4, and 6 of the IKZF1 gene) were designed in a final volume of 25 μL containing 100 ng genomic DNA, 0.28 μM each of FGFR2 and FBN1 primers, 0.12–0.8 μM each primer of the exon of interest, 200 μM dNTPs, 2 mM MgCl2, and 0.5 units of AmpliTaq Gold enzyme (Applied Biosystems, Foster City, CA, USA) in 1× buffer II (10 mM Tris‐HCl, pH 8.3, 50 mM KCl) as provided by the manufacturer. The sequencing of these designed primers is listed in Table S1
Amplification was carried out using an MBS thermocycler (ThermoHybaid, Ashford, UK) with an initial denaturation step at 95°C for 10 min, followed by 25 cycles of denaturation at 94°C for 30 s, annealing at 62°C for 45 s, extension at 72°C for 45 s, and a final extension step at 72°C for 10 min.
For rapid DNA separation and detection, a high‐performance DNA analysis capillary electrophoresis system with a CK‐5000 disposable cartridge (eGene, Irvine, CA, USA) was used according to the manufacturer’s instructions. The gel‐matrix in the gel cartridge consisted of a proprietary linear polymer with ethidium bromide dye. Briefly, 2 μL unpurified multiplex quantitative PCR products was directly diluted 10‐fold with 18 μL deionized H2O. The samples were placed in the instrument sample tray, automatically injected into the capillary channel, and subjected to electrophoresis by selecting the OM500.mtd method from the BioCalculator software (eGene). The sample injection voltage was 5 kV with a sample injection time of 20 s followed by separation voltage of 5 kV and separation time of 500 s. Quantification of DNA fragments was based on the integrated peak area automatically determined by the BioCalculator software.
Determination of IKZF1 mutations by HRM for mutation analysis. We designed several PCR reactions for point mutation analysis in all coding regions of IKZF1. Samples with abnormal peaks (Fig. S1, exon 5 as an example) were selected for sequencing. Details of the primers and PCR conditions for HRM analysis are listed in Table S2.
Statistical analysis. Fisher’s exact test was used to compare baseline clinical variables across groups for categorical data, and the non‐parametric Mann–Whitney U‐test was applied for continuous variables. A P‐value <0.05 (two‐sided) was considered significant.
The overall survival (OS) was calculated using the Kaplan–Meier method and the log–rank test was used to compare differences between survival curves. The OS was measured from the protocol commencement date until the date of death regardless of cause, excluding patients who were alive at the last follow‐up. Event‐free survival (EFS) was defined only for patients who achieved complete remission, and was measured from the date of attaining complete remission until the date of relapse. Patients with no reports of relapse by the end of follow‐up were censored on the date of last follow‐up.
Cox proportional hazard models were constructed for EFS and OS, with covariates that included sex, white blood cell (WBC) count (<10 000/μL, 10 000–50 000/μL, 50 000–100 000/μL, and >100 000/μL), age (<1 year, between 1 and 10 years, and >10 years), the status of IKZF1 deletions, and genetic subtypes. Stepwise backward selection was carried out. All calculations were done using the SAS software package, version 9 (SAS Institute, Cary, NC, USA).
Results
Multiplex PCR with capillary electrophoresis detected recurrent deletions. Of the 242 patients analyzed, 26 (10.7%) harbored IKZF1 deletions (Fig. 1). The incidence of IKZF1 deletions was consistent with a previous report on unselected childhood B‐progenitor ALL patients,( 11 ) but was lower than that reported for high‐risk patients.( 15 ) All of the 26 deletions were hemizygous, and exons 3–6 were most involved (Table 1), similar to previous studies.( 12 , 15 ) High resolution melting was used to screen the point mutations in all of the coding exons of IKZF1, however no patient was detected in this cohort.
Figure 1.
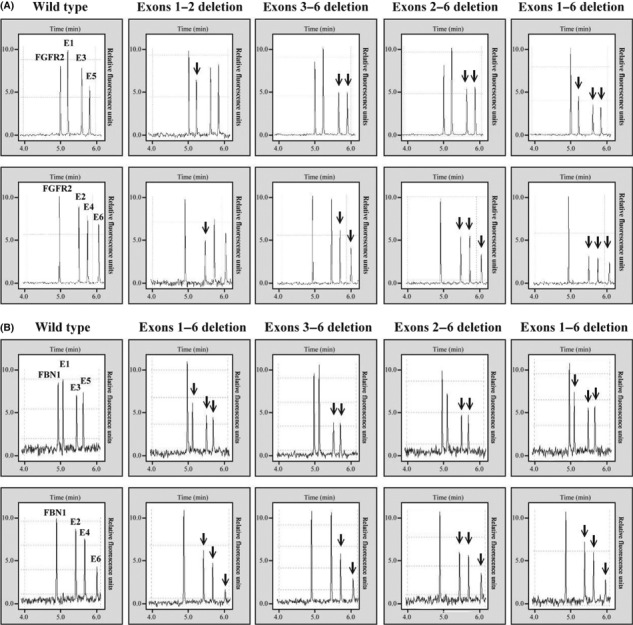
Separation of amplified DNA fragments by capillary electrophoresis on the high‐performance DNA analysis (HDA) system. (A) Multiplex PCR I with FGFR2, exons 1, 3, and 5 of IKZF1 gene. (B) multiplex PCR II with FGFR2, exons 2, 4, and 6 of IKZF1 gene. Arrows indicate the deletion of the target exon.
Table 1.
Deletion patterns in patients with IKZF1 deletions (n = 26)
| Patient | Exons involved | BCR/ABL1 status | Hemizygous or homozygous deletions |
|---|---|---|---|
| 1 | 1–3 | Negative | Hemizygous |
| 2 | 3–6 | Positive | Hemizygous |
| 3 | 2–6 | Positive | Hemizygous |
| 4 | 3–6 | Positive | Hemizygous |
| 5 | 1–2 | Negative | Hemizygous |
| 6 | 2–6 | Negative | Hemizygous |
| 7 | 3–6 | Negative | Hemizygous |
| 8 | 3–5 | Negative | Hemizygous |
| 9 | 3–6 | Negative | Hemizygous |
| 10 | 2–6 | Positive | Hemizygous |
| 11 | 2 | Negative | Hemizygous |
| 12 | 3–6 | Negative | Hemizygous |
| 13 | 3–6 | Negative | Hemizygous |
| 14 | 2–6 | Negative | Hemizygous |
| 15 | 2–6 | Negative | Hemizygous |
| 16 | 2–6 | Negative | Hemizygous |
| 17 | 2–6 | Negative | Hemizygous |
| 18 | 3–5 | Positive | Hemizygous |
| 19 | 2–5 | Negative | Hemizygous |
| 20 | 3–6 | Negative | Hemizygous |
| 21 | 3–6 | Negative | Hemizygous |
| 22 | 1–2 | Negative | Hemizygous |
| 23 | 1–6 | Negative | Hemizygous |
| 24 | 1–6 | Negative | Hemizygous |
| 25 | 3–6 | Positive | Hemizygous |
| 26 | 3–6 | Positive | Hemizygous |
Patient characteristics. The clinical characteristics of the patients at the time of diagnosis are shown in Table 2. There were 126 boys and 116 girls with a median age of 4.99 years (range, 0–18 years). Eighty‐six patients were at SR, 95 at HR, and 61 at VHR. The TPOG revised the chemotherapy from TPOG‐ALL‐93 (for SR and HR patients) and TPOG‐97‐VHR (for VHR patients) to TPOG‐ALL‐2002 in 2002. The TPOG‐ALL‐2002 protocol is still in use for treatment of childhood ALL in Taiwan. The clinical features of these patients according to the different protocol periods are detailed in Table 2.
Table 2.
Clinical characteristics of patients with childhood acute lymphoblastic leukemia who participated in this study (n = 242)
| Variable | TPOG‐ALL‐93 + TPOG‐97‐VHR (n = 104) | TPOG‐ALL‐2002 (n = 138) | All patients (n = 242) |
|---|---|---|---|
| Gender | |||
| Male | 57 (54.8) | 69 (50.0) | 126 (52.1) |
| Female | 47 (45.2) | 69 (50.0) | 116 (47.9) |
| Age | |||
| Mean ± SD | 5.79 ± 3.84 | 6.58 ± 4.47 | 6.24 ± 4.22 |
| <1 | 7 (6.7) | 4 (2.9) | 11 (4.6) |
| 1–9 | 80 (76.9) | 105 (76.1) | 185 (76.5) |
| ≥10 | 17 (16.4) | 29 (21.0) | 46 (19.0) |
| WBC | |||
| <10 | 46 (44.2) | 54 (39.1) | 100 (41.3) |
| 10–49 | 26 (25.0) | 48 (34.8) | 74 (30.6) |
| 50–99 | 11 (10.6) | 11 (8.0) | 22 (9.1) |
| >100 | 21 (20.2) | 25 (18.1) | 46 (19.0) |
| Risk group | |||
| SR | 34 (32.7) | 52 (37.7) | 86 (35.5) |
| HR | 43 (41.4) | 52 (37.7) | 95 (39.3) |
| VHR | 27 (26.0) | 34 (24.6) | 61 (25.2) |
| IKZF1 | |||
| Deletion | 10 (9.6) | 16 (11.6) | 26 (10.7) |
| Non | 94 (90.4) | 122 (88.4) | 216 (89.3) |
Results are shown as n (%). HR, high risk; Non, wild‐type; SR, standard risk; VHR, very high risk; WBC, white blood cell.
IKZF1 deletions associated with very poor clinical outcomes. The EFS and OS were analyzed for patients with and without IKZF1 deletions. Patients with IKZF1 deletions had a trend of high initial WBC count and received the VHR protocols. Patients with IKZF1 deletions had inferior 5‐ and 10‐year EFS than those without (15.42 ± 8.0%vs 77.89 ± 3.0% and 15.42 ± 8.0%vs 76.35 ± 3.2%, respectively, P < 0.0001), and inferior 5‐ and 10‐year OS (47.58 ± 11.1%vs 78.21 ± 3.0% and 38.07 ± 12.3%vs 77.47 ± 3.1%, respectively, P = 0.0016) (Fig. 2).
Figure 2.
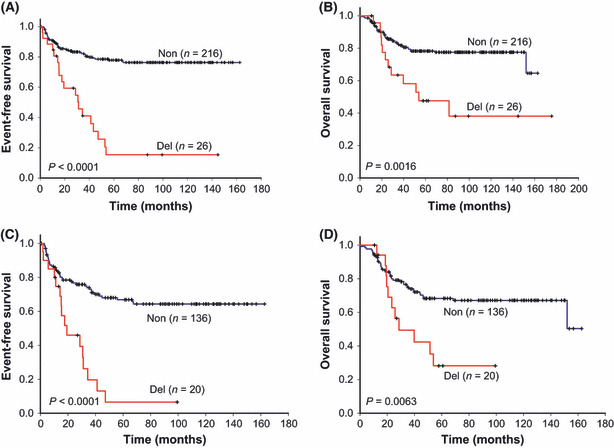
Survival curves of childhood acute lymphoblastic leukemia patients with and without IKZF1 deletions. (A,B) All patients with acute lymphoblastic leukemia who participated in this study (n = 242). (C,D) Patients from this study who were grouped as high‐risk and very high‐risk. Non, wild‐type IKZF1; Del, deletions of IKZF1.
Excluding SR patients, the EFS and OS were analyzed for HR and VHR patients. Those with IKZF1 deletions still had inferior 5‐year EFS and OS than those without (6.56 ± 6.3%vs 66.88 ± 44% and 28.24 ± 11.8%vs 68.32 ± 4.3%; P < 0.0001 and 0.0063, respectively) (Fig. 2).
There was no difference in 5‐year EFS and OS for patients treated with TPOG‐ALL‐2002 or TPOG‐ALL‐93 and TPOG‐97‐VHR in this study cohort (Fig. 3A,B). For patients treated before 2002, patients with IKZF1 deletions had inferior 5‐ and 10‐year EFS than those without (20.00 ± 12.7%vs 74.37 ± 4.6% and 20.00 ± 12.7%vs 72.05 ± 4.8%, respectively; P = 0.0009), and inferior 5‐ and 10‐year OS (40.00 ± 15.5%vs 70.21 ± 4.8% and 30.00 ± 14.5%vs 69.15 ± 4.8%, respectively; P = 0.0355) (Fig. 3C,D). For patients treated after 2002, patients with IKZF1 deletions had inferior 5‐year EFS and OS than those without (11.11 ± 10.1%vs 80.26 ± 4.1%, P < 0.0001, and 57.14 ± 14.6%vs 85.21 ± 3.7%, P = 0.004, respectively) (Fig. 3E,F).
Figure 3.
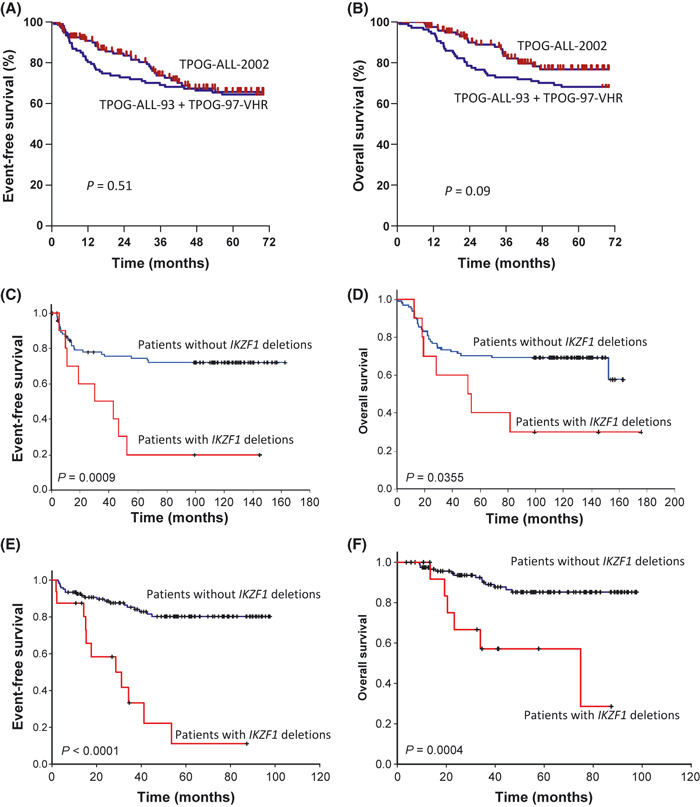
Survival curves of childhood acute lymphoblastic leukemia patients in this study. (A,B) Comparison of 5‐year event‐free survival (EFS) and overall survival (OS) for patients treated with TPOG‐ALL‐2002 or TPOG‐ALL‐93 + TPOG‐97‐VHR protocols. (C,D) Comparison of EFS and OS for patients with or without IKZF1 deletions treated with TPOG‐ALL‐93 and TPOG‐97‐VHR. (E,F) Comparison of EFS and OS for patients with or without IKZF1 deletions treated with TPOG‐ALL‐2002.
Multivariate analysis. In multivariate analysis, considering age, presenting leukocyte count, sex, cytogenetic changes, IKZF1 deletions, and step‐wise variable selection (P < 0.5 for model entry and <0.25 to remain in the mode), IKZF1 deletions remained significantly associated with EFS at a P threshold of 0.05 (hazard ratio (HR) 2.45, 95% confidence interval (CI) 1.36–4.44, P = 0.0001). BCR–ABL1 (HR 4.20, CI 1.92–9.22, P = 0.0003), MLL rearrangements (HR 4.55, CI 1.51–13.67, P = 0.007), and high WBC counts (HR 1.65, CI 1.31–2.08, P < 0.0001) were also significantly associated with the event in this model (Table 3).
Table 3.
Multivariable Cox regression analysis of event‐free survival for patients with childhood acute lymphoblastic leukemia who participated in this study
| Variable | Hazard ratio | 95% HR | CI | P‐value |
|---|---|---|---|---|
| All patients (n = 242) | ||||
| IKZF1 | 2.45 | 1.36 | 4.44 | 0.0030 |
| WBC | 1.65 | 1.31 | 2.08 | <0.0001 |
| t (9;22) | 4.20 | 1.92 | 9.22 | 0.0003 |
| MLL | 4.55 | 1.51 | 13.67 | 0.0070 |
| Patients with high and very high risk (n = 156) | ||||
| IKZF1 | 2.07 | 1.07 | 3.98 | 0.0300 |
| WBC | 1.49 | 1.16 | 1.91 | 0.0020 |
| t (9;22) | 4.06 | 1.83 | 8.99 | 0.0010 |
| MLL | 3.28 | 1.11 | 9.69 | 0.0320 |
| Patients treated with TPOG‐ALL‐2002 (n = 138) | ||||
| IKZF1 | 2.92 | 1.27 | 6.72 | 0.0120 |
| WBC | 1.77 | 1.22 | 2.58 | 0.0030 |
| t (9;22) | 4.33 | 1.50 | 12.51 | 0.0070 |
CI, confidence interval; HR, hazard ratio; WBC, white blood cell.
If the analysis was restricted to the HR and VHR patients, similar results were obtained and IKZF1 deletions still had a poor prognostic impact (HR 2.07, CI 1.07–3.98, P = 0.030) (Table 3). In addition, for patients treated with the current protocol (TPOG‐ALL‐2002), the IKZF1 deletions remained poor prognostic factors (HR 2.92, CI 1.27–6.72, P = 0.012) (Table 3).
Discussion
In this comprehensive analysis of the IKZF1 gene, IKZF1 deletions were identified as an independent poor prognostic marker in B‐cell progenitor childhood ALL in Taiwan. To the best of our knowledge, this is the first report of IKZF1 deletions and the poor prognostic value in an Asian population. Although the SNP array is the most comprehensive method for detecting deletions or other genetic changes, a capillary system coupled with multiplex PCR to type the IKZF1 deletions is also feasible in routine clinical practice.
Precise assessment of the relapse hazard for individual patients with childhood ALL followed by adjustment of treatment intensity is central to the successful management of childhood ALL. In most protocols, risk stratification is based primarily on the patient’s clinical features such as age, WBC counts, and genetic backgrounds of the leukemic cells.( 22 ) For many years, cytogenetics had a major prognostic value in childhood ALL. Patients with BCR–ABL1 and MLL gene rearrangements have poor outcomes, but these genetic changes cannot account for all relapsed patients.( 22 ) Genome‐wide, gene‐expression profiling offers a powerful new approach to the study of leukemia cell biology, and potentially provides a new molecular classification of leukemia. Searching for genetic changes related to treatment response across different regimens may provide clues about the general mechanisms that regulate drug sensitivity in leukemic cells and refine the current risk‐classification system. IKZF1 alteration was identified in 10% of childhood ALL in an unselected cohort in the first, large genome‐wide study of childhood ALL.( 11 ) However, IKZF1 was deleted in 83.7% of BCR–ABL1 patients.( 12 ) BCR–ABL1 is notorious for its poor treatment response. The same group also identified the poor prognostic implications of IKZF1 deletions in children with B‐cell progenitor ALL. The IKZF1 deletions correlated with increased incidence of relapse and resistance to chemotherapy. The poor prognostic impact of IKZF1 alterations was independent of BCR–ABL1 translocations and other known risk factors. This strong association between IKZF1 and adverse outcomes was confirmed by Kuiper et al. in a Dutch DCOG‐ALL9 cohort.( 23 ) In an adult study with subgroups of Philadelphia chromosome patients, IKZF1 was associated with a high relapse rate.( 16 ) These findings highlight the prognostic significance of IKZF1 deletions in ALL. In the current study, 19 of 26 patients with IKZF1 deletions had subsequent relapses. Six patients with BCR–ABL1 also had these deletions and all suffered relapses. Deletions of IKZF1 were associated with poor EFS and OS in our cohort of Taiwanese patients with childhood ALL. Even selecting only HR and VHR patients for analysis, the EFS and OS of patients with deletions remained poor. In addition, most patients with IKZF1 deletions under the TPOG‐ALL‐2002 suffered from relapses and only had 11.11 ± 10.1% of 5‐year EFS in this study. The result hinted that patients with IKZF1 deletions cannot be cured under the current protocol in Taiwan. New therapeutic strategies, such as hematopoietic stem cell transplantation, may be considered for patients with IKZF1 deletions in future clinical trials in Taiwan. One limitation of this study is the unknown status of MRD of these patients, so we could not analyze the relationship of MRD and IKZF1 deletions.
Although patients with the Philadelphia chromosome have a high incidence of IKZF1 deletions, IKZF1 deletions are not restricted to this subtype of patients. In the current study, only six of 26 patients with this deletion had BCR–ABL1. Several recent reports have also indicated that IKZF1 deletions are not restricted to the subtype of ALL with BCR–ABL1, and such deletions appear in normal cytogenetic patients.( 15 , 24 , 25 ) There is a HR subgroup of BCR–ABL1‐negative ALL that is characterized by IKZF1 deletions, and it has a genetic profile similar to cases with BCR–ABL1 fusion. These findings have important clinical implications. Currently, few tests can rapidly identify patients who lack known chromosomal alterations and have a high risk of relapse. The data here validates that detection of IKZF1 alterations should be considered upon diagnosis of childhood ALL, particularly in those with clinical HR features (e.g. high leukocyte count). However, not all patients with IKZF1 deletions succumb to relapse. Wannders et al. used MRD and IKZF1 alterations to predict a 79% relapse rate in childhood ALL in the DCOG‐ALL9 protocol( 26 ). Furthermore, Harvey et al. carried out gene expression profiles and copy‐number profiles in HR patients, and the HR patients could be separated into eight groups. IKZF1 deletions were found in several cluster groups. Patients with IKZF1 deletions had extremely good outcomes in cluster R6, in contrast with poor outcomes in cluster R8.( 27 ) Gene expression cluster, MRD, and IKZF1 alterations may be able to refine the current classification system of B‐cell ALL.
The incidence of IKZF1 alterations in childhood ALL is different to the above reports. In unselected risk patients, IKZF1 has been noted in approximately 9% of B‐ALL.( 11 ) In one subsequent smaller series (n = 40), IKZF1 deletions were detected in one case.( 14 ) In a Japanese study analyzing 399 pediatric B‐ALL patients using 250 K or 50 K arrays, IKZF1 deletions were detected in <2% of total cases.( 13 ) In a report by Den Boer et al.,( 28 ) IKZF1 deletions developed in 73% of patients with Philadelphia chromosome‐positive ALL, and 39% of BCR/ABL1‐like ALL. In the most recent report of Kuiper et al.,( 27 ) IKZF1 deletions were predominant in relapsed samples in paired analysis. A Japanese study showed low frequencies of IKZF1 deletions in a Japanese population.( 13 ) They later hypothesized that the IKZF1 deletions may have been missed by the 50 K and 250 K SNP arrays that were used in their study.( 29 ) In our study, the incidence was approximately 10% in an unselected patient risk group, comparable with previous reports.
The SNP array is the most comprehensive method for detecting IKZF1 deletions. In this study, we used multiplex PCR coupled with a capillary system to screen for these deletions, which can assay several exons at the same time. Two PCR reactions, including exons 1, 3, and 5, and exons 2, 4, and 6, respectively, were designed to check the deletion status of IKZF1, and 26 patients were identified. All suffered from hemizygous deletions, which is consistent with a previous report, however, no patient had point mutations of coding exons. Nonetheless, the most important advantages of our system are the relatively low cost and simple comparison with SNP array findings.
Recently, in addition to IKZF1 deletions, other studies have also identified a group of HR childhood ALL patients harboring a BCR–ABL1‐like gene expression. This group of patients suffered from similar poor outcomes as patients with BCR–ABL1. Mullighan et al. ( 30 ) identified JAK mutations in HR childhood ALL, and the presence of JAK mutations was also significantly associated with alteration of IKZF1. Yoda et al. and Harvey et al. used different methods of approach to identify whether CRLF2 is overexpressed in HR pediatric B‐ALL patients that lack common chromosomal translocations.( 24 , 27 , 31 , 32 ) CRLF2 alterations were significantly associated with JAK mutations and clinical outcomes.( 31 ) Recently, the prognostic impact of CRLF2 alterations has been validated in the Berlin‐Frankfurt‐Münster (BFM) protocol. However, many patients with JAK mutations and CRLF2 expression, similar to patients with IKZF1 deletions, lack other identified known chromosomal alterations. This implies that these newly identified markers may be incorporated into future protocols as poor prognostic factors, and could be new therapeutic targets in the future.
In conclusion, multiplex PCR can be used as a cost‐effective and rapid method to identify the deletion status of IKZF1. IKZF1 abnormalities were associated with poor outcomes in the Taiwanese population in this study. To the best of our knowledge, this is the first report to validate the poor prognostic value of IKZF1 deletions. Including alterations of IKZF1 as a HR factor in ALL and new therapeutic strategies for this subgroup of patients should be considered.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. High resolution melting plots for exon5 of IKZF1. Samples with abnormal peaks were picked for sequencings. All the mutations were in the intron but not the exon.
Table S1. Polymerase chain reaction primers and conditions used for multiplex PCR in the capillary electrophoresis assay.
Table S2. Primers used for PCR–high resolution melting analysis of all coding exons in the IKZF1 gene.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
The authors wish to thank all of the patients who participated in this study and their parents. The authors also thank Ching‐Tzu Yen, Jing‐Fang Lin, Ying‐Hui Su, Ya‐Ping Li, Yu‐Ju Chen, Chien‐Yu Lin, and Mei‐Ya Fang for their technical assistance. The authors also acknowledge TPOG and the Childhood Cancer Foundation in Taiwan for their assistance in data collection and management. This work was supported by the Excellent Research Projects of National Taiwan University (Grant No. BM01‐05) and 98R0452 by the National Taiwan University, from the National Taiwan University Hospital (NTUH.99‐M1412) and by Mr. Chang Han‐Sen’s research fund for pediatric blood disease.
References
- 1. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet 2008; 371: 1030–43. [DOI] [PubMed] [Google Scholar]
- 2. Pui CH, Campana D, Pei D et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 2009; 360: 2730–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Einsiedel HG, von Stackelberg A, Hartmann R et al. Long‐term outcome in children with relapsed ALL by risk‐stratified salvage therapy: results of trial acute lymphoblastic leukemia‐relapse study of the Berlin‐Frankfurt‐Munster Group 87. J Clin Oncol 2005; 23: 7942–50. [DOI] [PubMed] [Google Scholar]
- 4. Ferrando AA, Neuberg DS, Staunton J et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002; 1: 75–87. [DOI] [PubMed] [Google Scholar]
- 5. Yeoh EJ, Ross ME, Shurtleff SA et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002; 1: 133–43. [DOI] [PubMed] [Google Scholar]
- 6. Holleman A, Cheok MH, den Boer ML et al. Gene‐expression patterns in drug‐resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med 2004; 351: 533–42. [DOI] [PubMed] [Google Scholar]
- 7. Lugthart S, Cheok MH, den Boer ML et al. Identification of genes associated with chemotherapy crossresistance and treatment response in childhood acute lymphoblastic leukemia. Cancer Cell 2005; 7: 375–86. [DOI] [PubMed] [Google Scholar]
- 8. Flotho C, Coustan‐Smith E, Pei D et al. A set of genes that regulate cell proliferation predicts treatment outcome in childhood acute lymphoblastic leukemia. Blood 2007; 110: 1271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kang H, Chen IM, Wilson CS et al. Gene expression classifiers for relapse‐free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B‐precursor acute lymphoblastic leukemia. Blood 2010; 115: 1394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cario G, Stanulla M, Fine BM et al. Distinct gene expression profiles determine molecular treatment response in childhood acute lymphoblastic leukemia. Blood 2005; 105: 821–6. [DOI] [PubMed] [Google Scholar]
- 11. Mullighan CG, Goorha S, Radtke I et al. Genome‐wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446: 758–64. [DOI] [PubMed] [Google Scholar]
- 12. Mullighan CG, Miller CB, Radtke I et al. BCR‐ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008; 453: 110–4. [DOI] [PubMed] [Google Scholar]
- 13. Kawamata N, Ogawa S, Zimmermann M et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high‐resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood 2008; 111: 776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuiper RP, Schoenmakers EFPM, van Reijmersdal SV et al. High‐resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 2007; 21: 1258–66. [DOI] [PubMed] [Google Scholar]
- 15. Mullighan CG, Su X, Zhang J et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360: 470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martinelli G, Iacobucci I, Storlazzi CT et al. IKZF1 (Ikaros) deletions in BCR‐ABL1‐positive acute lymphoblastic leukemia are associated with short disease‐free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP Report. J Clin Oncol 2009; 27: 5202–7. [DOI] [PubMed] [Google Scholar]
- 17. Hung CC, Chen CP, Lin SP et al. Quantitative assay of deletion or duplication genotype by capillary electrophoresis system: application in Prader‐Willi syndrome and Duchenne muscular dystrophy. Clin Chem 2006; 52: 2203–10. [DOI] [PubMed] [Google Scholar]
- 18. Vossen RH, Aten E, Roos A, den Dunnen JT. High‐resolution melting analysis (HRMA): more than just sequence variant screening. Hum Mutat 2009; 30: 860–6. [DOI] [PubMed] [Google Scholar]
- 19. Liang DC, Hung IJ, Yang CP et al. Unexpected mortality from the use of E. coli L‐asparaginase during remission induction therapy for childhood acute lymphoblastic leukemia: a report from the Taiwan Pediatric Oncology Group. Leukemia 1999; 13: 155–60. [DOI] [PubMed] [Google Scholar]
- 20. Lin WY, Liu HC, Yeh TC, Wang LY, Liang DC. Triple intrathecal therapy without cranial irradiation for central nervous system preventive therapy in childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 2008; 50: 523–7. [DOI] [PubMed] [Google Scholar]
- 21. Liang DC, Yang CP, Lin DT et al. Long‐term results of Taiwan Pediatric Oncology Group studies 1997 and 2002 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24: 397–405. [DOI] [PubMed] [Google Scholar]
- 22. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006; 354: 166–78. [DOI] [PubMed] [Google Scholar]
- 23. Kuiper RP, Waanders E, van der Velden VH et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B‐ALL. Leukemia 2010; 24: 1258–64. [DOI] [PubMed] [Google Scholar]
- 24. Harvey RC, Mullighan CG, Chen IM et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B‐progenitor acute lymphoblastic leukemia. Blood 2010; 115: 5312–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuiper RP, Waanders E, van der Velden VH et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B‐ALL. Leukemia 2010; 24: 1258–64. [DOI] [PubMed] [Google Scholar]
- 26. Waanders E, van der Velden VHJ, van der Schoot CE et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia 2011; 25: 254–8. [DOI] [PubMed] [Google Scholar]
- 27. Harvey RC, Mullighan CG, Wang X et al. Identification of novel cluster groups in pediatric high‐risk B‐precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome‐wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010; 116: 4874–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Den Boer ML, van Slegtenhorst M, De Menezes RX et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome‐wide classification study. Lancet Oncol 2009; 10: 125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okamoto R, Ogawa S, Nowak D et al. Genomic profiling of adult acute lymphoblastic leukemia by single nucleotide polymorphism oligonucleotide microarray and comparison to pediatric acute lymphoblastic leukemia. Haematologica 2010; 95: 1481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mullighan CG, Zhang J, Harvey RC et al. JAK mutations in high‐risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA 2009; 106: 9414–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoda A, Yoda Y, Chiaretti S et al. Functional screening identifies CRLF2 in precursor B‐cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA 2010; 107: 252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cario G, Zimmermann M, Romey R et al. Presence of the P2RY8‐CRLF2 rearrangement is associated with a poor prognosis in non‐high‐risk precursor B‐cell acute lymphoblastic leukemia in children treated according to the ALL‐BFM 2000 protocol. Blood 2010; 115: 5393–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. High resolution melting plots for exon5 of IKZF1. Samples with abnormal peaks were picked for sequencings. All the mutations were in the intron but not the exon.
Table S1. Polymerase chain reaction primers and conditions used for multiplex PCR in the capillary electrophoresis assay.
Table S2. Primers used for PCR–high resolution melting analysis of all coding exons in the IKZF1 gene.
Supporting info item
Supporting info item
Supporting info item