

Abstract
Mitochondrial DNA (mtDNA) repair systems are thought to be associated with the susceptibility of cancer cells to anticancer agents. The present study investigated the relationship between the susceptibility to γ‐rays and the mtDNA repair ability of oral squamous cell carcinoma (OSC) cell lines. The levels of 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG) and mtDNA common deletion in both nuclear and mitochondrial DNA of OSC‐2, OSC‐3 and OSC‐6 cells (radio‐sensitive cell lines) after γ‐ray‐irradiation were higher than those of OSC‐1, OSC‐4 and OSC‐5 cells (radio‐resistant cell lines). Compared with OSC‐2, OSC‐3 and OSC‐6 cells, OSC‐1, OSC‐4 and OSC‐5 cells had higher levels of activity of phosphoinositide‐3 kinase (PI‐3K)/Akt and more strongly expressed 8‐hydroxyguanine DNA glycosylase (OGG1), DNA polymerase γ (POLG) and mitochondrial transcription factor A (Tfam). Down‐regulation of these mtDNA‐repair‐associated molecules by the RNA interference technique enhanced the susceptibility of OSC‐2 and OSC‐5 cells to γ‐rays, and the expression of Tfam and POLG was down‐regulated by inhibitors of PI‐3K/Akt signaling. These results indicate that the inhibition of mtDNA repair capacity by PI‐3K/Akt signal inhibitors and OGG1 down‐regulator in cancer cells may be a useful strategy for cancer treatment when combined with ionizing irradiation and chemotherapeutic drugs. (Cancer Sci 2008; 99: 2230–2237)
There are multiple patterns of cell death, including necrosis, apoptosis, senescence, autophagy and mitotic catastrophe.( 1 ) Cells are always exposed to danger of impairment induced by extrinsic factors, such as ultraviolet and chemicals, and intrinsic factors, such as reactive oxygen species (ROS) and endogenous cytokines. Of the patterns of cell death, apoptosis has been most aggressively studied and it has been revealed recently that apoptosis is closely associated not only with organ development and senescence but also with many diseases and their pathogenesis.( 2 ) Research on apoptosis has clarified the details of apoptosis‐inducing signaling systems and their regulatory systems, suggesting the possibility of new therapeutic approaches to some intractable human diseases.( 3 , 4 , 5 , 6 )
Chemotherapeutic drugs and ionizing irradiation induce ROS generation in cancer cells, and the generated ROS impair proteins, lipids and nucleic acids by their strong oxidative activities.( 7 , 8 ) Human cells, especially cancer cells, possess integrated protection systems against ROS‐inducing impairment.( 7 , 9 , 10 ) The DNA‐repairing system of cancer cells is therefore disadvantageous to cancer treatment. Our accumulated studies have indicated a variety of ROS roles in γ‐ray‐ and anticancer‐drug‐induced apoptosis of oral squamous cell carcinoma (OSC) cells as follows. (1) Generated ROS increase the expression of pro‐apoptotic Bcl‐2 family proteins and decrease the expression of antiapoptotic Bcl‐2 family proteins.( 11 ) (2) Intracellular ROS levels are closely correlated with apoptosis induction.( 7 ) Conversely, the activity of manganese‐superoxide dismutase (Mn‐SOD) is negatively correlated with apoptosis.( 7 ) (3) Apoptosis is more strongly induced in manganese superoxide dismutase (Mn‐SOD) antisense‐transfected OSC cells than in mock‐transfected OSC cells.( 7 , 12 ) However, we have not examined the damage of mitochondrial DNA by ROS.
Human mitochondrial DNA (mtDNA) consists of 16 569 bp. MtDNA possesses a double‐strand loop structure and encodes 13 respiration‐associated polypeptides and oxidative‐phosphorylation‐associated polypeptides, 2 ribosomal RNAs and 22 transfer RNAs.( 13 ) MtDNA is more easily damaged than nuclear DNA because mtDNA lacks histone proteins, and mitochondria are the main site of ROS generation.( 14 , 15 , 16 ) To counteract the oxidative damage of mtDNA, cells are equipped with several defense mechanisms. The first step of DNA damage by ROS is oxidation of the component bases. The damaged bases are mainly repaired by the base excision repair system, which is initiated by the excision of the damaged bases by specific DNA glycosylases such as 8‐hydroxyguanine DNA glycosylase (OGG1), and the DNA synthesis system, of which DNA polymerase γ (POLG) is a component.( 17 , 18 , 19 ) It was recently demonstrated that mitochondrial transcription factor A (Tfam), which was firstly identified as a mitochondrial transcription factor, is involved in not only the transcription of mtDNA but also the replication of mtDNA, recognition of mtDNA damage, stabilization of mtDNA and, indirectly, in repair of mtDNA.( 20 ) It was also reported that the expression of Tfam was controlled by the phosphoinositide‐3 kinase (PI‐3K)/Akt‐mediated signal(s) through activation of its transcriptional factor, nuclear respiratory factor‐1 (Nrf1).( 21 ) These mitochondrial repair systems are therefore thought to be associated with the susceptibility of cancer cells to anticancer agents. Supporting this notion, the susceptibility of cancer cells to ionizing irradiation is inversely correlated with the activity of OGG1.( 22 , 23 ) However, there have been few studies on the relationship between these repair systems in mitochondria and the susceptibility to anticancer agents of head and neck squamous cell carcinoma cells.( 24 )
Here, we studied the relationships among the activities of PI‐3K/Akt, the expression of OGG1, POLG and Tfam, and the susceptibility of OSC cells to γ‐rays, and we obtained the following results: γ‐ray‐resistant OSC cells had a high level of PI‐3K/Akt activity and they strongly expressed OGG1, POLG and Tfam. In addition, we found that down‐regulation of these mtDNA‐repair‐associated molecules by inhibition of the PI‐3K/Akt signal enhanced the susceptibility of OSC cells to γ‐rays.
Materials and Methods
Cell lines. OSC cell lines established in our laboratory were used. OSC‐2, OSC‐3 and OSC‐6 (γ‐ray sensitive) and OSC‐1, OSC‐4 and OSC‐5 (γ‐ray resistant) cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Nissui Pharmaceutical, Tokyo, Japan) supplemented with 10% (v/v) heat‐inactivated fetal bovine serum, 2 mM l‐glutamine and penicillin–streptomycin at 37°C in humidified air with 5% CO2.
MTT assay. The susceptibility of OSC cell lines to ionizing radiation was determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐2H‐tetrazolium bromide (MTT, Sigma‐Aldrich, St. Louis, MO, USA) colorimetric assay. OSC‐1 to OSC‐6 cells (5 × 103 cells/well) were cultured in 96‐well microplates for 12 h and then irradiated at a dose of 30 Gy using a 137Cs source. After 48 h, the cells in each well were washed with 200 µL of phosphate‐buffered saline (PBS) and incubated with 100 µL of 2% (w/v) MTT in a solution of 0.05 M Tris‐HCl (pH 7.6), 0.5 mM MgCl2, 2.5 mM CoCl2 and 0.25 M disodium succinate at 37°C for 30 min. The cells were fixed by the addition of 100 µL of 4% (v/v) formaldehyde in 0.2 M Tris‐HCl (pH 7.6), and after a 5 min incubation at room temperature, the liquid was removed and the wells were allowed to dry. Each well was rinsed with 200 µL of water and the cells were solubilized by the addition of 100 µL dimethyl sulfoxide (DMSO). The colored formazan product was measured using a Thermo MAX microplate reader (Molecular Devices, Sunnyvale, CA, USA) at a wavelength of 562 nm.
Apoptosis assay. After the indicated treatment, cells were trypsinized, washed once with complete medium and stained with propidium iodide and FITC‐conjugated annexin V (Sigma‐Aldrich) according to the manufacturer's instructions. The cells were then analyzed using a fluorescence‐activated cell sorting (FACS) can cytometer using CELLQUEST software (Becton Dickinson, San Jose, CA, USA).
Subcellular fractionation. Mitochondrial and nuclear fractions were prepared by suspending cells in three volumes of lysis buffer (10 mM 4‐(2‐hydroxyethyl)‐1piperazineethanesulfonic acid [HEPES], pH 7.5, 5 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride [PMSF], 50 µg/mL aprotinin, 50 µg/mL leupeptin and 50 µg/mL antipain). The cells were homogenized using a tight‐fitting Dounce homogenizer. To isolate mitochondria, the lysate was mixed with 12 mL of lysis buffer containing 0.25 M sucrose and the mixture was centrifuged at 500 × g for 10 min at 4°C to pellet the nuclei. The supernatant was then recentrifuged at 10 000 × g for 10 min at 4°C and the resulting pellet was used as the mitochondrial fraction. Purity of the cytosolic, mitochondrial and nuclear fractions was confirmed by Western blot analysis using monoclonal antibodies against heat shock protein 70 (HSP70), Mn‐SOD and histon H1, respectively.
Isolation of DNA and RNA. DNA and RNA in total cells, the nuclear fraction and the mitochondrial fraction were extracted using DNeasy and RNeasy Kits (Qiagen, Valencia, CA, USA), respectively. The DNA and RNA were quantified by measuring the optical density at 260 nm and were used for analyses of 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG) levels, mitochondrial DNA deletion and expression of Tfam mRNA.
Analysis of 8‐OHdG levels. The levels of 8‐OHdG in nuclear and mitochondrial DNA were determined by automated coupled‐column high‐performance liquid chromatography with electrochemical detection.( 25 ) The 8‐OHdG value was calculated as 8‐OHdG/106 dG, which was simultaneously assayed with a UV detector.
Detection of mtDNA deletion. To detect the mitochondrial common deletion (ΔmtDNA4977), primer sets described by Prithivirajsingh et al.( 14 ) were commercially obtained from PE Applied Biosystems (Foster City, CA, USA). The prepared mtDNA was added to a PCR mixture containing 200 M deoxyribonucleotide triphosphates (dNTP), 1 × reaction buffer and 2.5 U of Taq polymerase (Roche Applied Science, Indianapolis, IN, USA). Amplification was accomplished by an initial denaturation at 94°C for 3 min followed by 30 cycles of template denaturation at 94°C for 1 min, primer–template annealing at 50°C for 1 min and primer‐extension at 72°C for 1 min. A final extension was performed at 72°C for 10 min. PCR was performed in a GeneAmp PCR system 9700 (Perkin–Elmer, Boston, MA, USA) using 0.2‐mL microcentrifuge tubes. PCR products were electrophoresed on a 1% agarose gel and stained with ethidium bromide.
Reverse transcription‐polymerase chain reaction (RT‐PCR). The prepared total RNA was reverse‐transcribed into complementary DNA (cDNA) using dNTP plus random hexamer primers and SuperScript II reverse transcriptase (Invitrogen, San Diego, CA, USA). PCR amplification was carried out in a total volume of 25 µL containing 0.6 U/µL Taq DNA polymerase (Invitrogen) and sequence‐specific oligonucleotide primers for Tfam.( 26 ) Amplification of the β‐actin gene was used as a reference value. PCR products were separated on a 1.2% agarose gel and were detected under UV transillumination after ethidium bromide staining.
Western blot analysis. Cells were solubilized with ice‐cold lysis buffer containing 1% (v/v) Triton X‐100, 50 mM NaCl, 25 mM HEPES (pH 7.4), 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM ethyleneglycoltetraacetic acid (EGTA), 1 mM PMSF, 20 µg/mL aprotinin, 10 µg/mL leupeptin and 1 µg/mL pepstatin. To isolate total cellular fraction, the lysates were centrifuged at 12 500 × g for 15 min at 4°C and the clarified supernatants were used as total cellular extracts. Cytosolic and nuclear extracts were isolated by using a ProteoExtract Kit (Calbiochem, San Diego, CA, USA) according to the vendor's protocol. Total cellular, cytosolic and nuclear extracts were snap‐frozen and stored at −80°C until use. Protein concentrations were determined using a bicinchoninic acid (BCA) protein assay kit (Pierce Biotechnology, Rockford, IL, USA). Extracted proteins (50 µg/lane) were separated by sodium dodecylsulfate (SDS)–polyacrylamide gel electrophoresis and transferred onto Immobilon‐P membranes (Immobilon, Millipore Corporation, Bedford, MA, USA). Blocking was performed in Tris‐buffered saline containing 5% (w/v) skim milk powder and 0.1% (v/v) Tween‐20. The membranes were probed with the following diluted antibodies: anti‐OGG1 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:500, anti‐POLG polyclonal antibody (Santa Cruz Biotechnology) at 1:200, anti‐Nrf1 polyclonal antibody (Santa Cruz Biotechnology) at 1:200, anti‐Akt polyclonal antibody (Santa Cruz Biotechnology) at 1:200, antiphosphorylated Akt polyclonal antibody (Santa Cruz Biotechnology) at 1:200, anti‐PI‐3K polyclonal antibody (Santa Cruz Biotechnology) at 1:200, antiphosphorylated PI‐3K polyclonal antibody (Santa Cruz Biotechnology) at 1:200, anticleaved poly(ADP‐ribose)polymerase (PARP) polyclonal antibody (Cell Signaling Technology, Beverly, MA, USA) at 1:1000, anti‐HSP70 monoclonal antibody (Santa Cruz Biotechnology) at 1:200, antiâ‐actin antibody (Santa Cruz Biotechnology) at 1:200, anti‐Mn‐SOD monoclonal antibody (Chemicon, Temecula, CA, USA) at 1:1000 and antihistone H1 monoclonal antibody (Upstate, Temecula, CA, USA) at 1:1000. Detection was performed using an enhanced chemiluminescence (ECL) system (Amersham, Piscataway, NJ, USA).
RNA interference (RNAi). Small interfering RNAs (siRNAs) for Tfam, POLG and OGG1 and control siRNA were obtained from PE Applied Biosystems. OSC‐2 and OSC‐5 cells were plated at a concentration of 2 × 105 cells/dish in 35‐mm dishes. siRNAs were transfected into the cells using oligofectamine reagents (Invitrogen) according to the manufacturer's recommendations. After cultivation for 24 h, the transfected cells were used for further experiments.
POLG activity assay. POLG activity was measured by an RNA‐dependent DNA polymerase (reverse transcriptase) assay.( 27 ) Briefly, freshly isolated mitochondria were lyzed in 25 mM HEPES–KOH, pH 8.0, 100 mM NaCl, 1%(v/v) Triton X‐100. After incubation on ice for 30 min and centrifugation at 16 000 × g for 1 min at 4°C, activity was assayed in 10‐µg aliquots of lysate in a final volume of 50 µL containing 25 mM HEPES–KOH, pH 8.0, 100 mM NaCl, 2.5 mM 2‐mercaptoethanol, 10 µg/mL acetylated bovine serum albumin (BSA), 0.5 mM MgCl2, 2.5 µg/mL poly(rA)·oligo(dT) (Pharmacia) and 50 µg/mL aphidicolin. The lysates were preincubated for 5 min at room temperature before the addition of 5 µL of a mixture of [α‐32P] 2′‐deoxythymidine 5′‐triphosphate (dTTP; Amersham, 3000–6000 Ci/mmol) diluted 0.6:10 with 100 µM dTTP. The reaction mixtures were then incubated for 15 min at 37°C, after which 10 µL was spotted on a glass fibre cartridge (GFC) filter, air‐dried, washed three times with 5%(w/v) trichloroacetic acid (TCA) then once with 70%(v/v) ethanol, air‐dried and counted using liquid scintillation counting. For each lysate a control assay without template was used to measure the background TCA‐precipitable counts. Activity (arbitrary units; AU) was normalized to template‐dependent 32P incorporation in extracts from control cells.
OGG1 activity assay. OGG1 activity was assayed using an Ogg1 Assay Kit (Sigma). This assay is based on OGG1 glycosylase activity that recognizes and removes an oxidized guanine base (8‐OH‐G). Briefly, the 23‐mer oligonucleotide (CTCTCCCTTC[8‐OH‐G] CTCCTTTCCTCT) was labeled at its 5′ end using γ32P‐adenine triphosphate (ATP) (Amersham) and T4 polynucleotide kinase. The 32P‐labeled strand was hybridized with the complementary oligonucleotide (AGAGGAAAGGAGCGAAGGGAGAG) by incubation at 95°C for 1 min and 37°C for 5 min followed by slow cooling to room temperature. The assay mixtures (100 µL‐final volume) contained 25 fmoles of 32P‐labeled DNA duplex and mitochondrial extracts (4 µg of protein) in reaction buffer (50 mM HEPES, pH 7.5, 50 mM KCl, 0.025%(v/v) Triton X‐100, 1 µg/mL BSA). The reaction was performed at 37°C for 60 min and the radiolabeled substrate and product were extracted using phenol‐chloroform‐isoamyl alcohol (25:24:1). The extracted products were separated by 15% denaturing polyacrylamide gel electrophoresis in the presence of 7 M urea. The gels were developed and analyzed by autoradiography.
Results
Different susceptibility of oral squamous cell carcinoma cell lines to γ‐rays. Susceptibility of OSC‐1 to OSC‐6 cells against 30 Gy of γ‐ray‐irradiation was examined by MTT assay and flow cytometry. Inhibition of mitochondrial activities and cell death were more strongly induced by γ‐ray‐irradiation in OSC‐2, OSC‐3 and OSC‐6 cells than those in OSC‐1, OSC‐4 and OSC‐5 cells (Fig. 1a). Apoptosis was more strongly induced by γ‐ray‐irradiation in OSC‐2 cells than in OSC‐5 cells (Fig. 1b).
Figure 1.
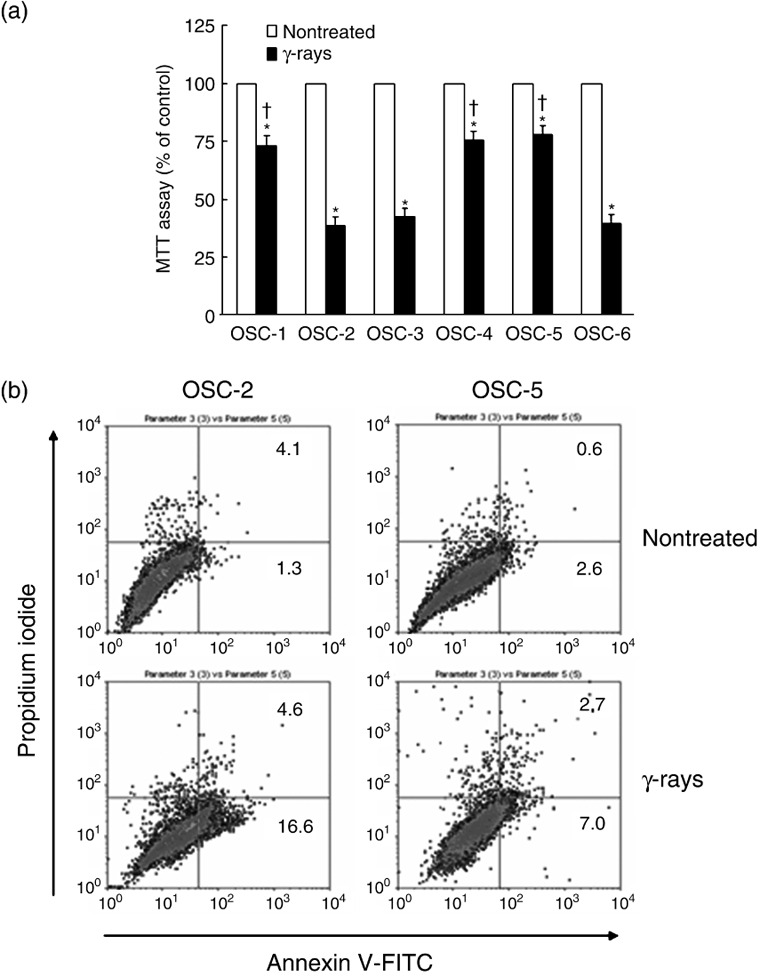
Susceptibility of oral squamous cell (OSC) carcinoma cell lines to γ‐rays. After OSC cells were irradiated at a dose of 30 Gy, the cell viability (a) and apoptosis induction (b) were determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐2H‐tetrazolium bromide (MTT) assay and FACscan using propidium iodide and fluoroscein isothiocyanate (FITC)‐conjugated annexin V staining at 48 h and 24 h, respectively. *P < 0.05 compared with each non‐treated cell and † P < 0.05 compared with γ‐ray‐treated OSC‐2 cells, by Mann–Whitney's U‐test.
8‐OHdG and ΔmtDNA accumulation by ionizing radiation. Accumulation of 8‐OHdG was observed in both nuclear DNA and mtDNA after the irradiation, and the levels of 8‐OHdG were higher in both the nuclear and mtDNA from OSC‐2 cells than in those from OSC‐5 cells (Fig. 2a). The levels of 8‐OHdG of OSC‐3 and OSC‐6 in both the nuclear and mtDNA after the irradiation were similar to OSC‐2 cells and those of OSC‐1 and OSC‐4 were similar to OSC‐5 cells (data not shown). ΔmtDNA was present at a higher level in OSC‐2 cells than in OSC‐5 cells after the irradiation (Fig. 2b). Similarly, ΔmtDNA levels were higher in OSC‐3 and OSC‐6 cells than in OSC‐1 and OSC‐4 cells after the irradiation (data not shown). These results suggest that sensitivity to γ‐rays was correlated with mtDNA damage.
Figure 2.
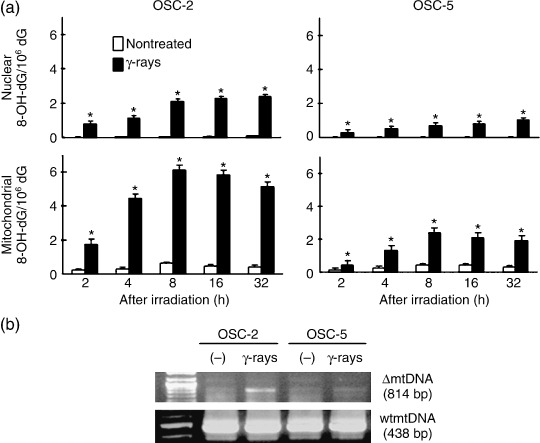
8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG) and ΔmtDNA accumulation in γ‐ray‐treated OSC cells. (a) The levels of 8‐OHdG in nuclear DNA and mtDNA were analyzed at the indicated times after 30 Gy irradiation. *P < 0.05 compared with each non‐treated cell, by Mann–Whitney's U‐test. (b) ΔmtDNA accumulation was determined at 24 h after 30 Gy irradiation.
Influence of ionizing radiation on the expression of DNA repairing‐associated molecules, Akt‐phosphorylation and activity of POLG and OGG1. We then assessed the relationship between the sensitivity to γ‐irradiation and intracellular molecules associated with the repair of mtDNA. The levels of Tfam–mRNA were increased after the irradiation in OSC cell lines, but higher Tfam–mRNA levels were observed in OSC‐1, OSC‐4 and OSC‐5 cells 6 h after irradiation (Fig. 3a). Moreover, the nuclear translocation of Nrf1 after treatment with γ‐rays was more prominent in OSC‐1, OSC‐4 and OSC‐5 cells than OSC‐2, OSC‐3 and OSC‐6 cells (Fig. 3b). The expression of POLG was also stronger in the irradiated OSC‐1, OSC‐4 and OSC‐5 cells (Fig. 3c). The expression of OGG1 was stronger in the OSC‐1, OSC‐4 and OSC‐5 cells than in the OSC‐2, OSC‐3 and OSC‐6 cells before the irradiation (Fig. 3c). In parallel with the expression levels of POLG, the activity in OSC cell lines was increased at 12 h after the irradiation and the activity in OSC‐5 cells was approximately 1.5‐fold higher than that in OSC‐2 cells (Fig. 3d). Similarly, POLG activity was higher in OSC‐1 and OSC‐4 cells than in OSC‐3 and OSC‐6 cells after the irradiation (data not shown). Although OGG1 activity was not influenced by γ‐ray‐irradiation, the activity in OSC‐5 cells was approximately 1.5‐fold higher than in OSC‐2 cells. Similarly, OGG1 activity was higher in OSC‐1 and OSC‐4 cells than in OSC‐3 and OSC‐6 cells (data not shown). The upstream of Nrf1 is regulated by PI‐3K and Akt, so we assessed PI‐3K and Akt activities. After γ‐ray treatment, phosphorylation of Akt and PI‐3K was detected more strongly in OSC‐5 cells than in OSC‐2 cells (Fig. 3e). Similarly, phosphorylation of Akt and PI‐3K was higher in OSC‐3 and OSC‐6 cells than in OSC‐1 and OSC‐4 cells after the irradiation (data not shown).
Figure 3.
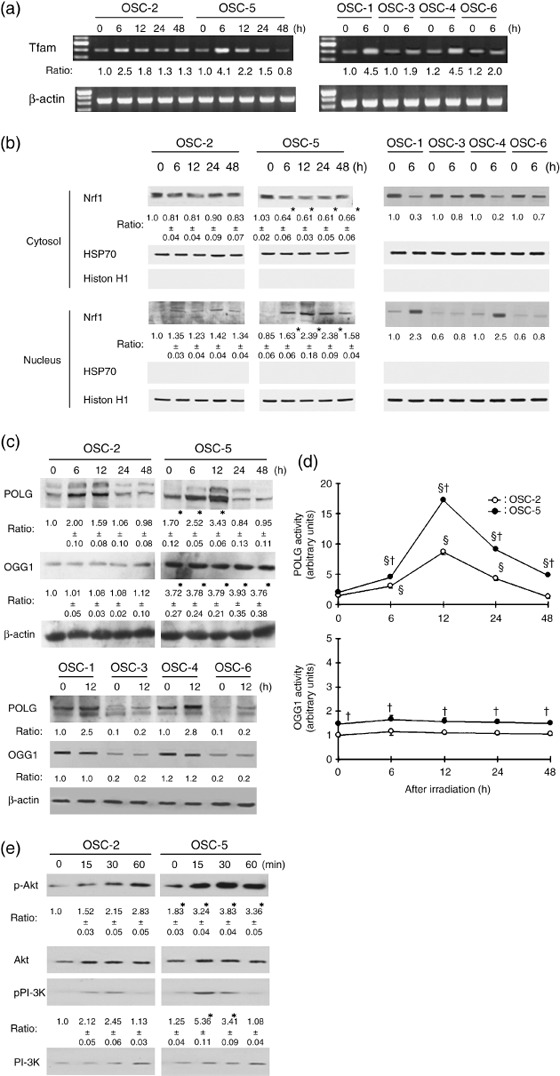
Influence of ionizing irradiation on the expression and activity of DNA repair‐associated molecules and on PI‐3K/Akt signaling in oral squamous cells (OSC). OSC were irradiated at a dose of 30 Gy and the expression of mitochondrial transcription factor A (Tfam) mRNA (a), Nrf1 (b), DNA polymerase γ (POLG) and 8‐hydroxyguanine DNA glycosylase (OGG1) (c), the activity of POLG and OGG1 (d) and the phosphorylation level of PI‐3K and Akt (e) were analyzed as described in Materials and Methods. Each blot is representative of three independent experiments. HSP70, histone H1 and β‐actin were used as loading control. The average results ± SE from three separate experiments are shown as relative ratios. *P < 0.05 compared with non‐treated OSC‐2 cells, § P < 0.05 compared with each non‐treated cell and † P < 0.05 compared with OSC‐2 cells, by Mann–Whitney's U‐test.
Influence of inhibitors of PI‐3K and Akt on the expression of DNA repairing‐associated molecules, ΔmtDNA and 8‐OHdG accumulation and cell viability after ionizing radiation. To further assess the association of the PI‐3K/Akt pathway with Nrf1 nuclear translocation, we next examined the influence of inhibitors of both PI‐3K and Akt. Pretreatment with LY 294002 or Inhibitor VI, inhibitors of PI‐3K and Akt, respectively, down‐regulated expression of Tfam–mRNA and POLG proteins in both OSC‐2 and OSC‐5 cells (Fig. 4a). In OSC‐5 cells, γ‐irradiation alone did not induce deletion of mtDNA, but the pretreatment of the cells with both γ‐rays and the PI‐3K or Akt inhibitor clearly induced ΔmtDNA (Fig. 4a). In OSC‐2 cells, the pretreatment of cells with both γ‐rays and the PI‐3K or Akt inhibitor induced more obvious ΔmtDNA. In addition, pretreatment of these cells with PI‐3K or Akt inhibitor increased 8‐OHdG accumulation after γ‐irradiation, and apoptosis of these cells was also increased (Fig. 4b,c).
Figure 4.
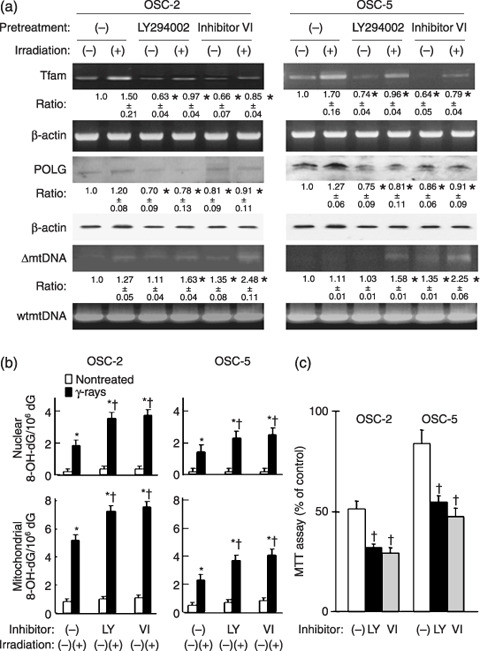
Influence of inhibitors of PI‐3K and Akt on the expression of DNA repair‐associated molecules, ΔmtDNA accumulation, 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG) synthesis and cell viability in γ‐ray‐irradiated oral squamous cells (OSC). After pretreatment with LY294002 (50 µM) or Akt inhibitor VI (50 µM) for 1 h, OSC were exposed to 30 Gy of γ‐rays. The expression of mitochondrial transcription factor A (Tfam) mRNA, DNA polymerase γ (POLG) protein, ΔmtDNA accumulation (a) and 8‐OHdG accumulation (b) and cell viability of OSC‐2 and ‐5 cells (c) were determined 48 h after the irradiation. The average results ± SE from three separate experiments are shown as relative ratios. *P < 0.05 compared with each non‐treated cell and † P < 0.05 compared with without inhibitors, by Mann–Whitney's U‐test.
Influence of siRNA of Tfam, OGG1 and POLG on apoptosis by ionizing radiation. To examine the involvement of mitochondrial DNA repair proteins, we assessed the influence of siRNA on Tfam, OGG1 and POLG. Figure 5a shows that each siRNA down‐regulated the corresponding protein expression. Transfection of the siRNAs increased apoptosis of the irradiated OSC‐5 cells (Fig. 5b). Similar up‐regulated cleavage of PARP was obtained in these siRNAs‐transfected OSC‐2 cells after irradiation (data not shown). Transfection of these siRNAs down‐regulated cell viability in both irradiated OSC‐2 and OSC‐5 cells (Fig. 5c).
Figure 5.
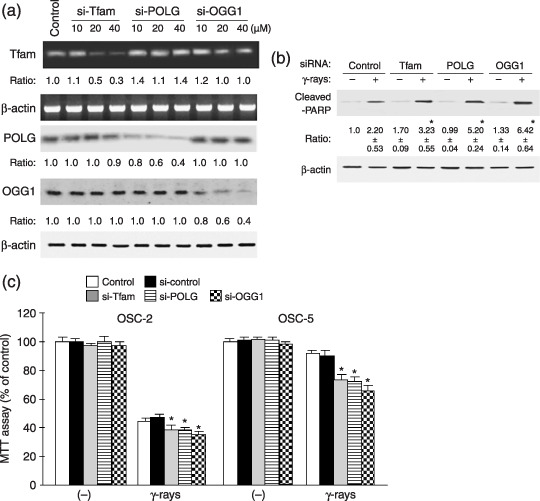
Influence of small interfering RNA (siRNA) of mitochondrial transcription factor A (Tfam), 8‐hydroxyguanine DNA glycosylase (OGG1) and DNA polymerase γ (POLG) on apoptosis by ionizing radiation. OSC‐2 and OSC‐5 cells were transiently transfected with each concentration of control, Tfam, POLG and OGG1 siRNAs. The expression of Tfam mRNA, POLG and OGG1 in OSC‐5 (a), the cleavage of poly(ADP‐ribose)polymerase (PARP) in siRNA‐transfected OSC‐5 cells exposed to 30 Gy of γ‐rays (b) and cell viability in siRNA‐transfected OSC‐2 and OSC‐5 cells (c) were determined 48 h after the irradiation. The average results ± SE from three separate experiments are shown as relative ratios. *P < 0.05 compared with control siRNA, by Mann–Whitney's U‐test.
Discussion
Mitochondria play a crucial role in many cellular functions, including energy production, respiration, heme synthesis, lipid synthesis and metabolism of amino acids and nucleotides, as well as the maintenance of intracellular homeostasis of inorganic ions.( 28 , 29 ) Mitochondria also regulate cell proliferation and apoptosis. These mitochondrial functions depend on mitochondrial DNA and proteins with the cooperation of some cellular proteins. During mitochondrial oxidative phosphorylation, large amounts of ROS are generated, which can cause mitochondrial and nuclear DNA damage.( 30 , 31 , 32 ) Since mtDNA lacks protection by histones and because the mtDNA repair system is insufficient, the mutation frequency of mtDNA is 10‐ to 1000‐fold higher than that of nuclear DNA.( 2 ) All genes in the mtDNA are essential for the biogenesis and energic function of mitochondria, and therefore mutations of mtDNA may result in malfunction of the altered gene products.
γ‐rays are known to induce cellular activation and also cellular impairment by direct interaction of the hydroxy radicals generated.( 33 ) Therefore, it seems likely that γ‐rays cause extensive damage to mtDNA and that they induce apoptosis of cancer cells through mitochondrial damage when the mtDNA repairing capacity of the cancer cells is weak. In accord with this possibility, the γ‐ray‐sensitive cancer cell lines OSC‐2, OSC‐3 and OSC‐6 showed more prominent mtDNA damage than the γ‐ray‐resistant cell lines OSC‐1, OSC‐4 and OSC‐5.
Mitochondria possess some mtDNA repairing proteins.( 17 , 18 , 20 ) Of these, Tfam, OGG1 and POLG have recently been explored and their roles have gradually been clarified.( 34 , 35 , 36 , 37 ) According to previous reports, Tfam is essential for mitochondrial gene expression and maintenance of intact mtDNA and is associated with repair of mtDNA indirectly.( 38 ) It was also shown that Tfam recognizes cisplatin‐damaged and oxidized DNA and binding of Tfam to cisplatin‐modified DNA was significantly enhanced by the p53 tumor suppressor gene product.( 26 ) As reported previously, OGG1 is involved in the base excision repair of 8‐oxoguanine.( 39 ) In addition, POLG is the sole DNA polymerase in mitochondria and it plays an essential role in mtDNA replication and repair.( 19 ) In the present study, OSC‐1, OSC‐4 and OSC‐5 cells, which are γ‐ray resistant, showed a stronger induction of Tfam and POLG and up‐regulation of POLG activity by treatment with γ‐rays than γ‐ray‐sensitive OSC‐2, OSC‐3 and OSC‐6 cells. This present result is in agreement with other investigations that cancer cells with markedly reduced DNA‐repairing capacity were highly sensitive to ionizing irradiation and chemotherapeutic drugs.( 22 , 23 )
It is well known that Nrf1 and Nrf2 are important in the regulation of Tfam expression and that Nrf1 activation is regulated by the PI‐3K/Akt pathway.( 21 ) Although the regulatory system of OGG1 and POLG expression has not well been investigated, the OGG1 promoter region contains the Nrf2 and SP‐1 binding sites and the POLG promoter region has a DNA element that is potentially recognized by Nrf1.( 37 , 40 ) Therefore, the PI‐3K/Akt pathway is thought to be involved in the up‐regulation of OGG1 and POLG. Hance et al. reported that elevation of the Tfam level during mammalian embryogenesis increased POLG expression via a positive feedback mechanism.( 37 ) According to recent reports, OGG1 suppresses the radiosensitivity of cancer cells and inhibits oxidative‐stress‐induced apoptosis.( 22 , 23 ) In agreement with these reports, higher expression and activity of OGG1 were observed in radioresistant OSC‐1, OSC‐4 and OSC‐5 cells. In the present study, PI‐3K and Akt were strongly activated in OSC‐5 cells and Nrf1 was more strongly translocated into the nuclei in these cells than in OSC‐2 cells. In parallel with these results, strong expression of Tfam, OGG1 and POLG and higher activity of POLG were observed in OSC‐1, OSC‐4 and OSC‐5 cells after treatment with γ‐rays. In addition, inhibitors of PI‐3K and Akt down‐regulated the γ‐ray‐induced expression of Tfam and POLG. From these results, it is concluded that PI‐3K and Akt efficiently up‐regulate the expression of POLG, which results in repair of damaged mtDNA and suppression of apoptosis. This conclusion appears to be supported by our finding that siRNAs against OGG1, POLG and Tfam induced strong increases of apoptosis of cancer cells induced by γ‐rays. Therefore, the down‐regulation of expression of these molecules and PI‐3K/Akt signaling appears likely to be useful for cancer treatment in combination with anticancer drugs and radiation. In the mtDNA repairing system, PI‐3K and Akt act upstream of POLG, regulating Tfam expression and Nrf1 activation, and the inhibition of PI‐3K/Akt signaling by appropriate approaches may be useful in cancer treatment.
In the present study, we demonstrated that the inhibition of mtDNA repair proteins sensitized radioresistant OSC cells to ionizing radiation. We also demonstrated that the expression of Tfam and POLG was down‐regulated by inhibitors of PI‐3K/Akt signaling. These results indicate that the inhibition of mtDNA repair capacity by PI‐3K/Akt signal inhibitors in cancer cells may be a useful strategy for not only chemo‐/radiosensitive but also chemo‐/radioresistant cancer cells. Many signal inhibitors affecting Akt pathways are currently under development.( 41 , 42 , 43 , 44 ) It is likely that the greatest benefit of PI‐3K/Akt signaling inhibitors and OGG1 down‐regulators will be in combination with ionizing irradiation and chemotherapeutic drugs.
References
- 1. Kim R, Emi M, Tanabe K. The role of apoptosis in cancer cell survival and therapeutic outcome. Cancer Biol Ther 2006; 5: 1429–42. [DOI] [PubMed] [Google Scholar]
- 2. Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 2006; 125: 1241–52. [DOI] [PubMed] [Google Scholar]
- 3. Panka DJ, Atkins MB, Mier JW. Targeting the mitogen‐activated protein kinase pathway in the treatment of malignant melanoma. Clin Cancer Res 2006; 12: 2371s–5s. [DOI] [PubMed] [Google Scholar]
- 4. Kim HJ, Hawke N, Baldwin AS. NF‐κB and IKK as therapeutic targeted in cancer. Cell Death Differ 2006; 13: 738–47. [DOI] [PubMed] [Google Scholar]
- 5. Foster DA. Targeting mTOR‐mediated survival signals in anticancer therapeutic strategies. Expert Rev Anticancer Ther 2004; 4: 691–701. [DOI] [PubMed] [Google Scholar]
- 6. Murphy FJ, Seery LT, Hayes I. Therapeutic approaches to the modulation of apoptosis. Essays Biochem 2003; 39: 131–53. [DOI] [PubMed] [Google Scholar]
- 7. Ueta E, Yoneda K, Yamamoto T, Osaki T. Manganese superoxide dismutase negatively regulates the induction of apoptosis by 5‐fluorouracil, peplomycin and γ‐rays in squamous cell carcinoma cells. Jpn J Cancer Res 1999; 90: 555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pattison DI, Dean RT, Davies MJ. Oxidation of DNA, proteins and lipids by DOPA, protein‐bound DOPA, and related catechol(amine)s. Toxicology 2002; 177: 23–37. [DOI] [PubMed] [Google Scholar]
- 9. Fujino G, Noguchi T, Takeda K, Ichijo H. Thioredoxin and protein kinases in redox signaling. Semin Cancer Biol 2006; 16: 427–35. [DOI] [PubMed] [Google Scholar]
- 10. Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol 2006; 16: 420–6. [DOI] [PubMed] [Google Scholar]
- 11. Li D, Ueta E, Kimura T, Yamamoto T, Osaki T. Reactive oxygen species (ROS) control the expression of Bcl‐2 family proteins by regulating their phosphorylation and ubiquitination. Cancer Sci 2004; 95: 644–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ueta E, Yoneda K, Kimura T et al . Mn‐SOD antisense upregulates in vitro apoptosis of squamous cell carcinoma cells by anticancer drugs and γ‐rays regulating expression of the Bcl‐2 family proteins, cox‐2 and p21. Int J Cancer 2001; 94: 545–50. [DOI] [PubMed] [Google Scholar]
- 13. Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1999; 1410: 103–23. [DOI] [PubMed] [Google Scholar]
- 14. Prithivirajsingh S, Story MD, Bergh SA et al . Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett 2004; 571: 227–32. [DOI] [PubMed] [Google Scholar]
- 15. Park SY, Chang I, Kim JY et al . Resistance of mitochondrial DNA‐depleted cells against cell death: role of mitochondrial superoxide dismutase. J Biol Chem 2004; 279: 7512–20. [DOI] [PubMed] [Google Scholar]
- 16. Suematsu N, Tsutsui H, Wen J et al . Oxidative stress mediates tumor necrosis factor‐α‐induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003; 18: 1418–23. [DOI] [PubMed] [Google Scholar]
- 17. Stevnsner T, Thorslund T, de Souza‐Pinto NC, Bohr VA. Mitochondrial repair of 8‐oxoguanine and change with aging. Exp Gerontol 2002; 37: 1189–96. [DOI] [PubMed] [Google Scholar]
- 18. Pinz KG, Bogenhagen DF. The influence of the DNA polymerase gamma accessory subunit on base excision repair by the catalytic subunit. DNA Repair 2006; 5: 121–8. [DOI] [PubMed] [Google Scholar]
- 19. Yakubovskaya E, Chen Z, Carrodeguas JA, Kisker C, Bogenhagen DF. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J Biol Chem 2006; 281: 374–82. [DOI] [PubMed] [Google Scholar]
- 20. Gaspari M, Larsson NG, Gustafsson CM. The transcription machinery in mammalian mitochondria. Biochim Biophys Acta 2004; 1659: 148–52. [DOI] [PubMed] [Google Scholar]
- 21. Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem 2006; 281: 324–33. [DOI] [PubMed] [Google Scholar]
- 22. Hyun JW, Cheon GJ, Kim HS et al . Radiation sensitivity depends on OGG1 activity status in human leukemia cell lines. Free Radic Biol Med 2002; 32: 212–20. [DOI] [PubMed] [Google Scholar]
- 23. Jeong HG, Youn CK, Cho HJ et al . Metallothionein‐III prevents γ‐ray‐induced 8‐oxoguanine accumulation in normal and hOGG1‐depleted cells. J Biol Chem 2004; 279: 34 138–49. [DOI] [PubMed] [Google Scholar]
- 24. Pekkola‐Heino K, Servomaa K, Kiuru A, Grenman R. Sublethal damage repair capacity in carcinoma cell lines with p53 mutations. Head Neck 1998; 20: 298–303. [DOI] [PubMed] [Google Scholar]
- 25. De Martinis BS, de Lourdes Pires Bianchi M. Methodology for urinary 8‐hydroxy‐2′‐deoxyguanosine analysis by HPLC with electrochemical detection. Pharmacol Res 2002; 46: 129–31. [DOI] [PubMed] [Google Scholar]
- 26. Yoshida Y, Izumi H, Torigoe T et al . p53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res 2003; 63: 3729–34. [PubMed] [Google Scholar]
- 27. Spelbrink JN, Toivonen JM, Hakkaart GAI et al . In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J Biol Chem 2000; 275: 24 818–28. [DOI] [PubMed] [Google Scholar]
- 28. Modica‐Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion 2004; 4: 755–62. [DOI] [PubMed] [Google Scholar]
- 29. DiMauro S. Mitochondrial medicine. Biochim Biophys Acta 2004; 1659: 107–14. [DOI] [PubMed] [Google Scholar]
- 30. Cai S, Xu Y, Cooper RJ et al . Mitochondrial targeting of human O6‐methylguanine DNA methyltransferase protects against cell killing by chemotherapeutic alkylating agents. Cancer Res 2005; 65: 3319–27. [DOI] [PubMed] [Google Scholar]
- 31. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 2004; 7: 97–110. [DOI] [PubMed] [Google Scholar]
- 32. Rachek LI, Grishko VI, Ledoux SP, Wilson GL. Role of nitric oxide‐induced mtDNA damage in mitochondrial dysfunction and apoptosis. Free Radic Biol Med 2006; 40: 754–62. [DOI] [PubMed] [Google Scholar]
- 33. Xiao J, Biaglow JE, Chae‐Park HJ et al . Role of hydroxyl radicals in radiation‐induced activation of lyn tyrosine kinase in human B‐cell precursors. Leuk Lymphoma 1996; 22: 421–30. [DOI] [PubMed] [Google Scholar]
- 34. Kang D, Hamasaki N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: overview of its multiple roles. Ann NY Acad Sci 2005; 1042: 101–8. [DOI] [PubMed] [Google Scholar]
- 35. Klungland A, Bjelland S. Oxidative damage to purines in DNA: role of mammalian Ogg1. DNA Repair 2007; 6: 481–8. [DOI] [PubMed] [Google Scholar]
- 36. Hudson G, Chinnery PF. Mitochondrial DNA polymerase‐γ and human disease. Hum Mol Genet 2006; 15: R244–52. [DOI] [PubMed] [Google Scholar]
- 37. Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucl Acids Res 2002; 30: 2817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hance N, Ekstrand MI, Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum Mol Genet 2005; 14: 1775–83. [DOI] [PubMed] [Google Scholar]
- 39. Kang MK, Kim RH, Shin KH, Zhong W, Faull KF, Park NH. Senescence‐associated decline in the intracellular accumulation of hOGG1‐α and impaired 8‐oxo‐dG repair activity in senescing normal human oral keratinocytes in vivo . Exp Cell Res 2005; 310: 186–95. [DOI] [PubMed] [Google Scholar]
- 40. Dhenaut A, Boiteux S, Radicella JP. Characterization of the hOGG1 promotor and its expression during the cell cycle. Mutat Res 2000; 461: 109–18. [DOI] [PubMed] [Google Scholar]
- 41. Kim IA, Bae SS, Fernandes A et al . Selective inhibition of Rss, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res 2005; 65: 7902–10. [DOI] [PubMed] [Google Scholar]
- 42. Lee CM, Fuhrman CB, Planelles V et al . Phosphatidylinositol 3‐kinase inhibition by LY 294002 radiosensitizes human cervical cancer cell lines. Clin Cancer Res 2006; 12: 250–6. [DOI] [PubMed] [Google Scholar]
- 43. Nakamura JL, Karlsson A, Arvold ND et al . PKB/Akt mediates radiosensitivity by the signaling inhibitor LY294002 in human malignant gliomas. J Neurooncol 2005; 71: 215–22. [DOI] [PubMed] [Google Scholar]
- 44. Toulany M, Dittmann K, Kruger M, Baumann M, Rodemann HP. Radioresistance of K‐Ras mutated human tumor cells is mediated through EGFR‐dependent activation of PI3K‐AKT pathway. Radiother Oncol 2005; 76: 143–50. [DOI] [PubMed] [Google Scholar]