

Abstract
The 3‐hydroxy‐3‐methylglutaryl (HMG)‐CoA reductase inhibitors, also called statins, are commonly used as lipid‐lowering drugs that inhibit cholesterol biosynthesis. An anticancer effect, as a pleiotropic function of certain statins, has been hypothesized. In the present study, we investigated the effect of simvastatin, one of the natural statins, on cell proliferation, cell cycle, invasive activity, and molecular expressions associated with cell–extracellular matrix adhesion, signal transduction, and DNA synthesis in Tu167 and JMAR cells from head and neck squamous cell carcinoma. The addition of simvastatin resulted in a dose‐dependent inhibition of cell growth and migration into the extracellular matrix. Considerable morphological changes occurred after treatment with simvastatin, demonstrating loss of cell adhesion and disruption of actin filaments in cytoplasm. The inhibitory effect of simvastatin on cell proliferation seemed to be associated with cell cycle arrest and increased expression of p21, p27, and activated caspase‐3. The expression of β1‐integrin, a counter adhesion for the extracellular matrix, phosphorylated FAK, and phosphorylated ERK was decreased by treatment with simvastatin. The proapoptotic effect of simvastatin was inhibited by treatment with mevalonate. cDNA microarray assay demonstrated that molecular changes resulting from treatment with simvastatin included the up‐regulation of cell cycle regulators and apoptosis‐inducing factors and the down‐regulation of integrin‐associated molecules and cell proliferation markers. Of down‐regulated genes induced by simvastatin treatment, a significant depletion of thymidylate synthase was confirmed using western blot analysis. These results imply that simvastatin has the potential to be effective for the prevention of the growth and metastasis of cancer cells. (Cancer Sci 2007; 98: 890–899)
Simvastatin, one of the 3‐hydroxy‐3‐methylglutaryl (HMG)‐CoA reductase inhibitors, derived from fungal fermentation, is currently used widely as a safe and effective therapeutic agent in the treatment of hypercholesterolemia, contributing to the reduction in morbidity and mortality of atherosclerosis and coronary artery disease.( 1 ) HMG‐CoA reductase inhibitors are also commonly referred to as the statins. In addition to their original role in lowering serum cholesterol levels, statins exert antiproliferative and proapoptotic effects in cancer cells by causing cell cycle arrest at the G1‐S phase.( 2 ) An anticancer effect with in vitro simvastatin treatment has been reported in several human malignancies, including multiple myeloma, malignant lymphoma, small cell lung carcinoma, and nasopharyngeal undifferentiated carcinoma.( 3 , 4 , 5 , 6 ) However, the efficacy and the molecular mechanism of simvastatin on tumor progression has yet to be clarified.
Statins target mevalonate, one of the cholesterol precursors, which is catalyzed by HMG‐CoA reductase. Overexpression of mevalonate has been reported to be associated with cell survival and proliferation of cancer cells.( 7 ) Another mechanism that plays a role in the anticancer properties of simvastatin may be related to the down‐regulation of integrin‐associated signaling. Integrin family proteins are receptors that connect cells to the extracellular matrix (ECM). Integrins function as heterodimeric transmembrane cell surface receptors consisting of α‐ and β‐subunits that bind several ECM proteins such as collagen, fibronectin, and laminin at the extracellular domain.( 8 ) Integrin‐associated signaling factors have been demonstrated to directly regulate the apoptotic machinery, as well as invasion. The apoptotic mechanism associated with loss of attachment to the ECM is also called anoikis, which is also controlled by integrin‐mediated signaling in cooperation with cell cycle regulator and proapoptotic proteins.( 9 ) Among the integrin family members, β1‐integrin has been shown to play an important role in the invasiveness, metastasis formation, and drug resistance of cancer cells.( 10 , 11 , 12 ) In addition, the integrin–ECM interactions trigger signaling cascades such as ERK, Akt, and Rho family GTPases.( 13 )
Head and neck squamous cell carcinoma (HNSCC) is the fifth most common cancer worldwide and accounts for approximately 400 000 new cases annually.( 14 ) Despite considerable developments in surgery, chemotherapy, radiotherapy, and combination treatment modalities, the long‐term survival has remained constant at approximately 50% over the past three decades.( 15 ) This discrepancy appears to be mainly the result of the failure to treat for distant metastasis and locoregional recurrence.( 16 ) To improve the long‐term outcome for patients with HNSCC, novel therapeutic approaches for preventing metastasis and recurrence are needed. In the present study, we investigated the antiproliferative and anti‐invasive effects of simvastatin, the most widely prescribed statin, in HNSCC cell lines in order to assess its potency as a therapeutic agent.
Materials and Methods
Cell culture and culture conditions. Two oral squamous cell carcinoma cell lines, Tu167 and JMAR, were grown in DMEM supplemented with 10% fetal bovine serum (FBS), L‐glutamine, penicillin, and streptomycin and maintained at 37°C in a humidified atmosphere with 95% air and 5% CO2. It has been reported that both cell lines have mutant p53 as part of their genetic background and that JMAR cells obtain more aggressive and anoikis‐resistant phenotypes.( 17 )
Reagents and antibodies. Simvastatin was obtained from Calbiochem (Darmstadt, Germany). For in vitro administration, simvastatin was dissolved in dimethylsulfoxide (DMSO; Sigma, St Louis, MO, USA) at a concentration of 20 mM and stored at –20°C as stock solution. In each experiment, the final concentration of DMSO was 0.05%, which did not affect the cytotoxicity in either HNSCC cell line tested. (+/–)‐Mevalonic acid lactone (Sigma) was dissolved in phosphate‐buffered saline (PBS) at a concentration of 2 M as stock solution. Antibodies for immunoblotting were obtained from the following sources: rabbit polyclonal anti‐ERK1/2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); mouse monoclonal anti‐phospho‐ERK1/2 (Santa Cruz Biotechnology); rabbit polyclonal anti‐β1‐integrin (Chemicon International Inc., Temecula, CA, USA); rabbit polyclonal anti‐FAK (Santa Cruz Biotechnology); rabbit polyclonal anti‐phosphorylated FAK (Y397; Santa Cruz Biotechnology); mouse monoclonal anti‐p21 (Santa Cruz Biotechnology); mouse monoclonal anti‐p27 (Santa Cruz Biotechnology); mouse monoclonal anti‐thymidylate synthase (Chemicon); rabbit polyclonal anti‐active‐caspase‐3 (BioVision, Mountain View, CA, USA); and mouse monoclonal anti‐β‐actin (Abcam, Cambridge, MA, USA).
Cell viability assay. The growth inhibition of simvastatin on HNSCC cells was determined by counting viable cells using Cell Counting Kit‐8 (Dojindo, Kumamoto, Japan). An equal number of cells (5000 cells/well) in 100 µL of culture medium was seeded into a 96‐well microplate and incubated for 24 h. Then the cells were treated with DMSO, (+/–)‐mevalonic acid lactone or various concentrations of simvastatin. After incubation for 48 h, 10 µL of Cell Counting Kit‐8 solution were added to each well and the plates were further incubated for 4 h at 37°C. Spectrophotometric absorbance at 450 nm was measured with absorbance at 590 nm for reference. Culture medium contained 10% FBS.
Colony formation assay. To assess the effect of simvastatin on cell numbers, we plated 1 × 105 cells into 6‐well plates and incubated them for 12 h using the conditions described above for standard cell culture maintenance. The cells were subsequently exposed to simvastatin at various concentrations for 48 h. The medium was removed from 6‐well plates and adherent cells were fixed using methanol and stained with 0.1% Giemsa for 1 min. The plates were washed with distilled water and dried. The images of plates were captured using Fluor‐STM MultiImager (Bio‐Rad, Hercules, CA, USA) and the number of colonies was counted using Quantity One software (Bio‐Rad).
Cell cycle analysis. Cells were plated at a density of 2 × 105 cells per well in 6‐well plates and maintained for 12 h before treatment. After 48 h of treatment with vehicle or simvastatin, cells were washed using PBS three times, and collected by treatment with EDTA/trypsin solution. Collected cells were then fixed with 70% cold ethanol, incubated with RNaseA (2 mg/mL in PBS), and stained with 50 µg/mL of propidium iodide (PI; Sigma). Cell cycle data were acquired using FACScan equipped with Cell Quest software (Becton Dickinson, San Jose, CA, USA).
Detachment‐induced apoptosis (anoikis) assay. To evaluate apoptosis in suspension culture condition, cells were cultured in 60‐mm plastic dishes previously coated with polyHEMA (Sigma). PolyHEMA prevents the cells from attaching to the plastic bottom of the dishes, ensuring suspension of the cells in the cultures. For coating culture dishes with polyHEMA, 1 mL of 10% solution of polyHEMA in 95% ethanol was added to 60‐mm culture dishes and allowed to dry overnight under a tissue culture hood. HNSCC cells in 10% FBS‐containing medium were cultured at a density of 5 × 106 cells in polyHEMA‐coated dishes with or without simvastatin for 24 h in standard conditions. After incubation, suspended cells were collected and fixed for flow cytometry analysis. Apoptosis was measured as a sub‐G1 population using PI staining.
Matrigel invasion assay. The in vitro invasive potential in the presence or absence of simvastatin was assessed using 6‐well Matrigel invasion chambers and non‐coated control chambers (BD Biosciences, Bedford, MA, USA). Cell suspension (2 mL; 5 × 104/mL, supplemented with 10% FBS) with simvastatin (5 and 10 µM) or DMSO was added to the upper chambers of the Matrigel invasion chambers. The same medium without simvastatin was placed in the lower chambers. Cell suspension was also added to non‐coated control chambers without simvastatin. After seeding, the cells were incubated for 22 h in a humidified tissue culture incubator. Thereafter, non‐invading cells were removed from the top of membranes with a cotton‐tipped swab. Invading cells at the bottom of membranes were fixed in methanol, stained with 1% Toluidine blue, and counted in 10 random fields under light microscope at ×200 magnification. The percentage invasion was calculated as the ratio of the number of cells invading through the Matrigel insert membrane to the number of cells migrating through the control insert membrane.
Reverse transcription‐polymerase chain reaction (RT‐PCR) analysis. Cells were incubated at a density of 2 × 105 cells in 6‐well plates for 12 h and then incubated with or without simvastatin (5 and 10 µM) for 48 h. After removing the medium and washing with PBS, total RNA from cell lines was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and one microgram of total RNA was applied to one‐step RT‐PCR using the Gene Amp Gold RNA PCR reagent kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's protocol. Oligomer primers were synthesized for β1‐integrin (sense 5′‐CCCTTGCACAAGTGAACAGA‐3′ and antisense 5′‐ACA T TCCTCCAGCCAATCAG‐3′) and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; sense 5′‐CGAGATCCC TCC AAA ATCA A‐3′ and antisense 5′‐GTCTTCTGG GTGGCAGTGAT‐3′) (Sigma Genosys, Hokkaido, Japan). One‐step cDNA synthesis and PCR amplification were performed at 25°C for 10 min; 42°C for 12 min; 95°C for 12 min; 35 cycles at 94°C for 20 s, 60°C for 30 s, and 72°C for 30 s; and at 72°C for 7 min as a final extension. PCR products were electrophoresed on 2% agarose gels.
Western blot analysis. Cells were plated onto 6‐well plates at a concentration of 2 × 105 cells/well and then incubated for 12 h. After 12 h, cells were incubated with media containing agents or DMSO alone for control. After washing with PBS, the cells were then scraped with lysis buffer containing 20 mM Tris‐HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X, 2.5 mM sodium pyrophosphate, 1 mM β‐glycerophosphate, 1 mM Na3VO4, and 1 µg/mL leupeptin. After centrifugation, the supernatant was harvested as the total protein extract and stored at –80°C. Protein concentrations were measured using a protein assay reagent (Bio‐Rad). Equal amounts of protein (10 µg) were separated using gradient sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE; Atto, Tokyo, Japan) and electrophoretically transferred to polyvinylidene fluoride (PVDF) membrane (Atto). The membrane was blocked with 5% skim milk in PBS‐0.1% Tween 20 at room temperature for 1 h. The membrane was incubated with primary antibody at room temperature for 1 h. Immunoreactivity was detected using sequential incubation with horseradish peroxidase‐conjugated secondary antibody (Santa Cruz). Peroxidase activity was visualized using the enhanced chemiluminescence detection system (Amersham Biosciences, Buckinghamshire, UK). For quantitative analysis, bands were detected and evaluated densitometrically with the NIH Image program, normalized for β‐actin density.
Microarray hybridization and data analysis. Total RNA was collected from JMAR cells incubated with vehicle (DMSO) or 10 µM simvastatin for 48 h and then purified using RNeasy micro kit (Qiagen, Tokyo, Japan). 500 ng of total RNA was labeled using the Low RNA Fluorescent Linear Amplification Kit (Agilent Technologies, Palo Alto, CA, USA) and labeled RNA was further purified using RNeasy mini spin columns (Qiagen). The cRNA was fragmented and hybridized to Agilent Human 1 A ver.2 OligoMicroarray (Agilent Technologies) for 17 h at 60°C. The microarray was washed in 6 × SSC with 0.005% Triton X‐102 for 10 min at room temperature, followed by a 5‐min wash in 0.1 × SSC with 0.005% Triton X‐102 at 4°C. The microarrays were scanned in Agilent Technologies Microarray Scanner and signal intensities were analyzed using Feature Extraction software (Agilent).
Alexa fluorescein‐labeled phalloidin staining of actin filaments. HNSCC cells were seeded on fibronectin‐coated 8‐well cell culture slides (BD Bioscience, San Jose, CA, USA) at a density of 1000 cells/well. After incubation for 12 h, the cells were treated for 24 h with 0.05% DMSO or 5 µM simvastatin. They were then fixed in 3.7% formaldehyde solution in PBS for 10 min, treated with 0.1% Triton X‐100 for 5 min, and stained with 0.1 µg/mL Alexa‐Fluor 488‐phalloidin (Invitrogen). After repeated rinsing with PBS, the slides were mounted on Vectashield (Vector Laboratories, Burlingame, CA, USA) and observed using a confocal microscope.
Statistical analysis. Statistical analyses were performed using unpaired Student's t‐test with StatView statistical software (version 5.0; SAS, Cary, NC, USA). All of the statistics were performed as two‐sided tests. P < 0.05 was considered as significant. Each experiment was repeated at least three times.
Results
Simvastatin induced morphological and cytoskeletal changes in HNSCC cells. Simvastatin treatment rendered morphological changes of HNSCC cells in vitro. Cells started floating approximately 12 h after the start of the treatment. After 48 h, cells were considerably detached from the dishes, exhibiting shrinking, round, and spindle morphology or loss of cell–cell attachment (Fig. 1a). Such degenerative changes induced by simvastatin were also observed at a low concentration (1 µM). These morphological changes led us to hypothesize that simvastatin affected the cytoskeletal structure of cancer cells. Phalloidin staining showed that simvastatin treatment (5 µM for 24 h) led to the disruption or fragmentation of cytoplasmic actin stress fibers (Fig. 1b).
Figure 1.
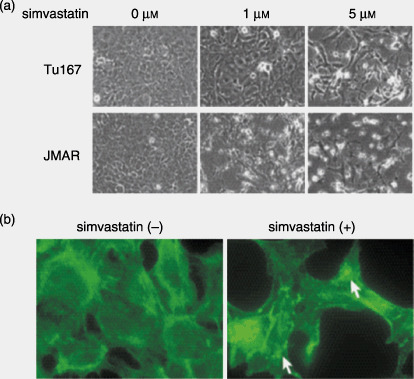
(a) Effect of simvastatin on cell morphology. Tu167 and JMAR cells were plated on 6‐well plates for 12 h and then treated with various concentrations of simvastatin for 48 h. Photographs were taken and representative fields are shown. (b) Effect of simvastatin on cytoskeletal structure of head and neck squamous cell carcinoma cells. Tu167 cells were seeded on fibronectin‐coated culture slides for 12 h and treated with or without 5 µM simvastatin for 24 h. Cells were then fixed and stained with phalloidin as described in the Materials and Methods section. Images were examined and photographed using confocal microscopy. Compared with control cells, simvastatin‐treated cells showed a dramatic disruption or fragmentation of actin stress fibers (→).
Simvastatin inhibited cell proliferation and induced cell cycle arrest at the G1 phase in adherent and suspended HNSCC cells. We next examined the effects of simvastatin on cell viability and proliferation of adherent HNSCC cells. The viability of treated cell lines was determined as the ratio between viable simvastatin‐treated cells and viable control cells treated with vehicle. Simvastatin‐treated HNSCC cell lines exhibited significant growth inhibition at concentrations of 5 and 10 µM (P < 0.01; Fig. 2a). Colony formation assay also showed that simvastatin significantly inhibited cell growth of both cell lines on dish bottoms at a high concentration (Fig. 2b). To assess the effect on the cell cycle, HNSCC cells were incubated for 48 h with 5 and 10 µM simvastatin or vehicle (DMSO), collected from culture dishes using EDTA/trypsin solution, and stained with PI. Treatment with simvastatin induced a dose‐dependent accumulation of G1 phase and a decrease in total S‐phase population in Tu167 cells and JMAR cells (Fig. 2c). The simvastatin‐induced decrease in the S‐phase appeared to be associated with an increase in the proportion of cells in the G1 phase fraction, suggesting that simvastatin induced G1 arrest in both cell lines. The cells were then deprived of their anchorage by suspension culture in polyHEMA‐coated dishes, which inhibits cell attachment. The population in the sub‐G1 phase (apoptotic cells), expressed as an average percentage of the total population, was significantly increased in both cell lines treated with 5 and 10 µM simvastatin for 24 h (P < 0.01; Fig. 2d).
Figure 2.
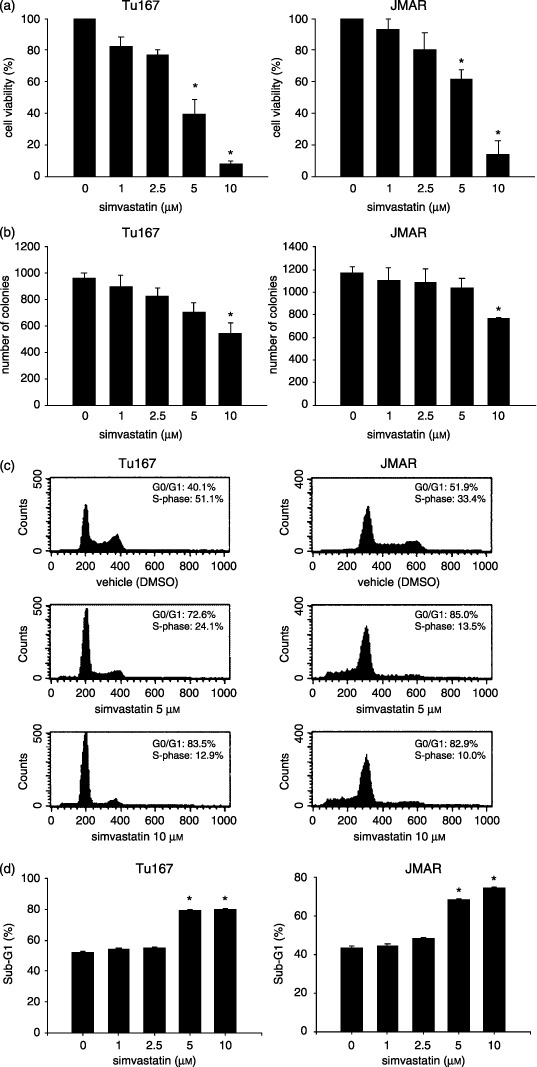
Inhibitory effects of simvastatin on the cell growth and cell cycle of head and neck squamous cell carcinoma cell lines. Cells were seeded on plates for 12 h and then treated with various concentrations of simvastatin for 48 h. Cell growth conditions were evaluated using (a) Cell Counting Kit‐8 (b) and colony formation assay, and the distribution of cell cycle fractions was determined using (c) propidium iodide staining as described in the Materials and Methods section. (d) The cells were deprived of their anchorage by suspension culture in polyHEMA‐coated dishes with different concentrations of simvastatin and apoptosis (anoikis) was determined as sub‐G1 phase population. The results were expressed as the mean ± SD. *P < 0.01, significantly different from control.
Effect of simvastatin on caspase‐3 expression. To assess whether the antiproliferative effect of simvastatin is involved with the activity of caspase family proteases, the expression of active caspase‐3 was evaluated using western blotting with specific antibody. The effect of simvastatin on caspase‐3 activation appeared to be dependent on concentration and time. After treatment for 48 h at different concentrations (5 and 10 µM), the expression of active caspase‐3 was significantly up‐regulated in both cell lines, showing stronger expression in cells treated with 10 µM simvastatin (Fig. 3a). When HNSCC cells were treated with 10 µM simvastatin at intervals of 24 h, caspase‐3 was highly activated in cells treated for 24 or 48 h (Fig. 3b).
Figure 3.
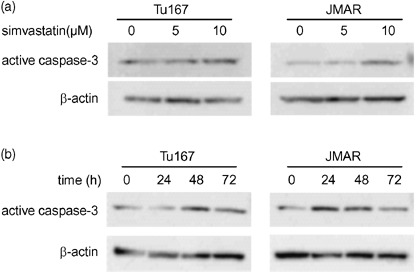
Simvastatin induced up‐regulation of active caspase‐3 expression. (a) Western blot analysis of active caspase‐3 expression in head and neck squamous cell carcinoma (HNSCC) cells treated with simvastatin (5 and 10 µM) or DMSO for 48 h. (b) Western blot analysis of active caspase‐3 expression in HNSCC cells treated with 10 µM (0, 24, 48, and 72 h).
Effect of mevalonate in combination with simvastatin on cell survival and caspase‐3 expression. Next, to study whether mevalonate, a precursor of cholesterol, prevents caspase‐dependent apoptosis and degenerative changes that were caused from simvastatin, cells were cultured with simvastatin in combination with mevalonic acid lactone (mevalonate), and then changes were analyzed using morphology, cell viability assay, and western blot for active caspase‐3. The protective effect of mevalonate was morphologically demonstrable. Cell–cell attachment seemed to be stronger in cells treated with both simvastatin and mevalonate than in cells treated with single simvastatin (Fig. 4a). Reduced cell viability from simvastatin was restored by the addition of 2 mM mevalonate (Fig. 4b). When cells were cultured with simvastatin and mevalonate, the increased expression of active caspase‐3 induced by simvastatin was significantly suppressed in both cell lines (Fig. 4c).
Figure 4.
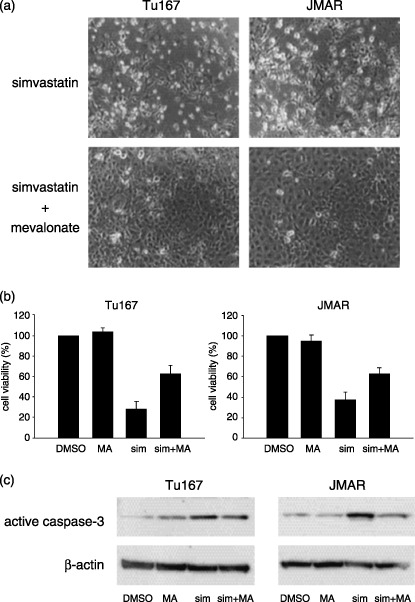
Effect of mevalonate in combination with simvastatin on morphology, cell viability, and active caspase‐3 expression. (a) Photographs were taken 48 h after treatment with simvastatin (10 µM) or combination of simvastatin (10 µM) and mevalonate (2 mM). (b) Cell viability was measured 48 h after treatment with vehicle, 2 mM mevalonate (MA), 10 µM simvastatin (sim), and combination of mevalonate and simvastatin (sim + MA). (c) Western blot analysis for active caspase‐3 was performed in samples from head and neck squamous cell carcinoma cells which were treated with vehicle, 2 mM mevalonate (MA), 10 µM simvastatin (sim), or a combination of mevalonate and simvastatin (sim + MA) for 48 h.
Simvastatin inhibited migration into the ECM and invasive activity. We further investigated whether simvastatin blocked migration or invasion in HNSCC cells. In vitro invasion assay using Matrigel invasion chambers was used to examine the anti‐invasive effect of simvastatin. The number of cells invading through the chamber was significantly decreased by simvastatin treatment at high concentration (P < 0.01 at 10 µM; Fig. 5).
Figure 5.
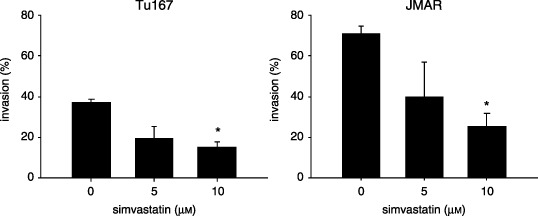
Effect of simvastatin on invasion or cell migration into the extracellular matrix. The mean value of percentage invasion was calculated as a ratio of the number of cells migrating through Matrigel‐coated membrane and the number of untreated cells migrating through control insert membrane. The results were expressed as the mean ± SD. *P < 0.01, significantly different from control.
Simvastatin down‐regulated the expression of β1‐integrin and phosphorylated FAK. The results of Matrigel invasion assay led us to the hypothesis that simvastatin may alter the expression status of molecules associated with cell–ECM adhesion. We therefore focused on the expression of β1‐integrin as a potential target of simvastatin, since β1‐integrin has been known to be commonly associated with the biological acquisition of invasive and metastatic potentials in various types of cancers. Although RT‐PCR and western blot analyses showed an inhibitory effect of simvastatin on β1‐integrin expression, its pattern was different in and between cell lines. RT‐PCR analysis showed that simvastatin induced decreased expression of β1‐integrin transcripts at concentrations of 5 and 10 µM and its transcript expression was more reduced at higher concentration in Tu167 cells (Fig. 6a). In contrast, protein expression seemed to be significantly blocked by treatment with 10 µM simvastatin (Fig. 6b). In JMAR cells, protein expression of β1‐integrin was suppressed in treatment with both 5 and 10 µM simvastatin (Fig. 6b), but an evident decrease of mRNA transcripts was observed when cells were incubated with 10 µM simvastatin (Fig. 6a). Next, the expression status of FAK was evaluated, since FAK is linked to β1‐integrin as a down‐stream molecule. Western blot analysis for phosphorylated‐FAK (Y397) showed that FAK was inactivated by simvastatin treatment in parallel with suppression of β1‐integrin expression, despite stable expression of total FAK (Fig. 6b).
Figure 6.
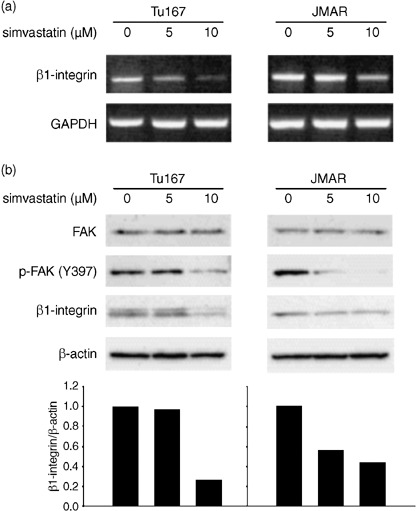
(a) Effect of simvastatin on expressions of β1‐integrin mRNA transcript and protein. Analysis of β1‐integrin mRNA expression using reverse transcription‐polymerase chain reaction (RT‐PCR). Head and neck squamous cell carcinoma cells were incubated for 48 h with 5 or 10 µM simvastatin and compared with untreated cells (incubated with vehicle). (b) Analysis of β1‐integrin, FAK, and p‐FAK (Y397) expressions using western blot. The condition of cell culture was identical with that of RT‐PCR analysis. The intensity of each band of western blot was determined using NIH Image program and the ratio of β1‐integrin and β‐actin was calculated for each treatment. The β1‐integrin/β‐actin ratio in untreated cells was set as 1. GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
Simvastatin attenuated the phosphorylation of ERK and up‐regulated cyclin dependent inhibitors, p21 and p27. To investigate whether simvastatin affects the signaling pathway, we focused on ERK, a down‐stream effecter of β1‐integrin‐associated signaling. Western blot analysis demonstrated that the phosphorylated forms of ERK1/2 were down‐regulated by treatment with simvastatin in Tu167 and JMAR cells (Fig. 7). As described above, simvastatin treatment led to cell cycle arrest at G1/S phase. Thus, we examined the effect of simvastatin on the expression of cell cycle regulators, such as p21 and p27. Western blot analysis demonstrated increased expressions of p21 and p27 by treatment with 5 and 10 µM simvastatin in both cell lines (Fig. 7).
Figure 7.
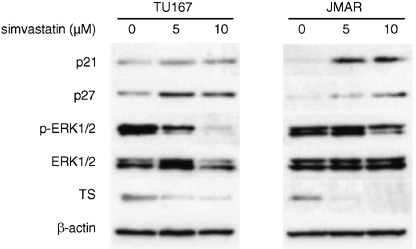
Western blot analyses of the effect of simvastatin on the expression levels of p21, p27, ERK1/2, phospho‐ERK1/2 and thymidylate synthase (TS). β‐actin was used as an internal control. Tu167 and JMAR cells were incubated for 48 h with the indicated concentration of simvastatin. The expression status was compared with protein samples extracted from untreated cells (incubated with vehicle). The entire cell lysates were subjected to western blotting.
Differential gene expression in simvastatin‐treated cells. To identify potential and novel target genes responsive to the anticancer effect in simvastatin‐treated HNSCC cells, we examined global changes in gene expression in JMAR cells after treatment with 10 µM simvastatin for 48 h. Of 18 000 genes analyzed, 389 had a twofold increase in simvastatin‐treated cells, whereas 543 decreased by a similar amount. The representative genes found to be up‐ and down‐regulated by 10 µM simvastatin treatment are summarized in 1, 2, respectively. The biological functions of the genes identified by the microarray analysis were diverse and included cell cycle, apoptosis, DNA synthesis, and cell adhesion as well as genes involved in the cell–ECM interaction and mevalonate pathway. Up‐regulated genes included cyclin dependent kinase inhibitors such as p57 (5.3‐fold) and p21 (4.5‐fold). The increased expression of p21 with simvastatin treatment was detected in both cell lines using western blot analysis (Fig. 7). In contrast, genes associated with cell proliferation, including activator of S‐phase kinase (0.24‐fold), PCNA (0.29‐fold), cyclin A2 (0.31‐fold), and cyclin D1 (0.42‐fold), were significantly down‐regulated. In addition, the expressions of certain integrin family genes including integrin‐β‐like‐1 (0.36‐fold) and integrin‐β3 binding protein (0.39‐fold) were also decreased. These molecular changes support the antiproliferative, proapoptotic, and anti‐invasive activity induced by simvastatin in HNSCC cells. To corroborate gene expression data from cDNA microarray analysis, we focused on the expression of thymidylate synthase (TS), one of the down‐regulated genes from simvastatin treatment in global gene expression analysis (Table 2). Western blot analysis demonstrated that simvastatin treatment (5 and 10 µM for 48 h) significantly inhibited the protein expression of TS in HNSCC cells (Fig. 7).
Table 1.
Up‐regulated genes after treatment with simvastatin in JMAR cells. Potential target genes were categorized according to their functions
| Function | Examples | Change (‐fold) |
|---|---|---|
| Signal transduction | calmodulin‐like 3 (CALML3) | 11.2 |
| connexin 26 (GJB2) | 4.6 | |
| phospholipase C, gamma 2 (PLCG2) | 4.3 | |
| regulator of G‐protein signaling 2 (RGS2) | 4.1 | |
| insulin‐like growth factor binding protein 6 (IGFBP6) | 4.1 | |
| Cell cycle | cyclin‐dependent kinase inhibitor 1C (p57) (CDKN1C) | 5.3 |
| cyclin‐dependent kinase inhibitor 1 A (p21) (CDKN1A) | 4.5 | |
| Apoptosis | BCL2 binding component 3 (BBC3) | 7.6 |
| BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) | 5.6 | |
| Transcriptional factor | Kruppel‐like factor 2 (KLF2) | 26.3 |
| activating transcription factor 3 (ATF3) | 6.1 | |
| Kruppel‐like factor 6 (KLF6) | 4.3 | |
| Metabolism | aldo‐keto reductase family 1, member C1 (AKR1C1) | 9.5 |
| aldehyde dehydrogenase 3 family, member A1 (ALDH3A1) | 7.1 | |
| Matrix metalloproteinase | matrix metalloproteinase 10 (MMP10) | 17.2 |
| matrix metalloproteinase 28 (MMP28) | 8.8 | |
| matrix metalloproteinase 1 (MMP1) | 7.9 | |
| matrix metalloproteinase 12 (MMP12) | 6.9 | |
| matrix metalloproteinase 13 (MMP13) | 6.1 | |
| Calcium binding protein | S100 calcium binding protein A8 (S100A8) | 17.3 |
| S100 calcium binding protein A6 (S100A6) | 6.9 | |
| S100 calcium binding protein A9 (S100A9) | 6.5 | |
| Extracellular matrix | spondin 2 (SPON2) | 8.6 |
| fibronectin type III domain containing 6 (MGC34923) | 5.0 | |
| collagen, type VII, alpha 1 (COL7A1) | 4.3 | |
| Cytokeratin | keratin 13 (KRT13) | 7.5 |
| keratin 15 (KRT15) | 7.0 | |
| keratin 17 (KRT17) | 4.3 |
Table 2.
Down‐regulated genes after treatment with simvastatin in JMAR cells. Potential target genes were categorized according to their functions
| Function | Examples | Change (‐fold) |
|---|---|---|
| Signal transduction | lymphotoxin beta (LTB) | 0.20 |
| AXL receptor tyrosine kinase (AXL) | 0.22 | |
| Rho GTPase activating protein 29 (PARG1) | 0.23 | |
| annexin A3 (ANXA3) | 0.36 | |
| Rac GTPase activating protein 1 (RACGAP1) | 0.40 | |
| serine/threonine kinase 6 (STK6) | 0.40 | |
| Cell cycle | CDC cell division cycle 6 homolog (CDC6) | 0.20 |
| MAD2 mitotic arrest deficient‐like 1 (MAD2L1) | 0.22 | |
| activator of S phase kinase (ASK) | 0.25 | |
| protein kinase, membrane associated tyrosine/threonine 1 (PKMYT1) | 0.25 | |
| E2F transcription factor 1 (E2F1) | 0.27 | |
| cell division cycle 2, G1 to S and G2 to M (CDC2) | 0.27 | |
| proliferating cell nuclear antigen (PCNA) | 0.29 | |
| cyclin A2 (CCNA2) | 0.31 | |
| cyclin D1 (CCND1) | 0.42 | |
| DNA replication | DNA replication factor (CDT1) | 0.18 |
| CDC45 cell division cycle 45‐like (CDC45L) | 0.20 | |
| primase, polypeptide 1 (PRIM1) | 0.22 | |
| MCM4 minichromosome maintenance deficient 4 (MCM4) | 0.23 | |
| thymidylate synthase (TS) | 0.24 | |
| replication factor C (activator 1) 5 (RFC5) | 0.25 | |
| Cell adhesion | catenin (cadherin‐associated protein), alpha‐like 1 (CTNNAL1) | 0.17 |
| thrombospondin 1 (THBS1) | 0.19 | |
| cysteine‐rich, angiogenic inducer, 61 (CYR61) | 0.25 | |
| integrin, beta‐like 1 (ITGBL1) | 0.35 | |
| integrin beta 3 binding protein (ITGB3 BP) | 0.39 |
Discussion
The present study showed that simvastatin could effectively suppress cell survival and the invasive activity of HNSCC cells and that these activities were associated with the inactivation of cell adhesion molecules and the signal transduction pathway, and the activation of cyclin‐dependent kinase (CDK) inhibitors and caspase‐3. The current in vitro concentrations showing antiproliferative effect are consistently identical to those of previous descriptions in other malignancies.( 5 , 6 ) It has been recently reported that treatment with more than 10 µM simvastatin induces apoptosis with down‐regulation of phospho‐Akt and such growth inhibitory effects are inhibited by additional treatment with cholesterol.( 18 ) We found that simvastatin inhibited the cell proliferation and invasive activities of HNSCC cell lines at a concentration of less than 10 µM. Cell viability was also inactivated in a concentration‐dependent manner (Fig. 2a).
Simvastatin induced significant morphological changes in HNSCC cell lines, demonstrating shrinking round‐shaped cells and loss of cell adhesion. Such degenerative changes were observed in cells treated with 1 µM simvastatin (Fig. 1a). Simvastatin has been reported to induce similar changes at concentration of 5 µM in nasopharyngeal carcinoma cells.( 6 ) In this report, electron microscopic analysis illustrated that the treatment of 5 µM for 5 days induced significant vacuolization of cytoplasm and partial chromatin condensation of nuclei. In the present study, Matrigel invasion assay demonstrated a remarkable inhibition of cell migration into the ECM. These results led us to consider that simvastatin may reduce the expression status of molecules associated with cell–ECM attachment. Integrin signaling stimulates these small G‐proteins and promotes cell contractility through the activation of ERK.( 19 , 20 , 21 ) In this context, we demonstrated that simvastatin inhibited β1‐integrin and ERK signaling, which is regarded as a down‐stream effecter of integrin signaling. There have been some evidences that β1‐integrin regulates apoptotic machinery induced by loss of cell‐ECM attachment, referred to as anoikis. Inhibition of β1‐integrin expression by its monoclonal antibody results in apoptosis with morphological changes in breast cancer cells,( 22 ) whereas overexpression of β1‐integrin has been shown to protect hepatoma cells from apoptosis induced by anoikis‐inducing cytotoxic agents such as cisplatin and docetaxel.( 23 ) Malignant cells including HNSCC frequently resist anoikis to various degrees and this property has been supposed to contribute to invasiveness and metastasis.( 24 ) These observations indicate that overexpressed β1‐integrin plays a central role in the acquisition of the anoikis‐resistant phenotype of cancer cells. Overall, β1‐integrin inhibition may be a viable therapeutic target in the treatment of HNSCC. In addition, the discrepancy in expression levels of β1‐integrin by simvastatin treatment was noted between protein and mRNA transcript levels, suggesting that simvastatin may inactivate β1‐integrin expression through both transcriptional and post‐transcriptional mechanisms. β1‐integrin may have a certain involvement as a molecular target of simvastatin, although β1‐integrin was not listed as a significantly up‐regulated gene in cDNA microarray analysis. To further investigate the involvement of β1‐integrin‐mediated signaling as a target of simvastatin, FAK inactivation by simvastatin treatment was confirmed using western blot analysis. It has been known that the cytoplasmic tail of β1‐integrin facilitates FAK activation through autophosphorylation at Y397.( 25 ) Therefore, the present observation of FAK inactivation supports an inhibitory effect of simvastatin on β1‐integrin‐mediated cell signaling. In the present study, we selected caspase‐3, a critical downstream protease in the caspase cascade, as a molecular marker for apoptosis or anoikis. Caspase‐3 activation has been linked to FAK degradation caused by detachment from ECM,( 26 ) which implies an association between FAK and anoikis.
Our PI staining analysis revealed that the treatment of adherent HNSCC cells with simvastatin resulted in the accumulation of cells with G1 accumulation and the reduction of cells with total S‐phase content and simvastatin yielded apoptosis (anoikis) in suspended cells demonstrating increased sub‐G1 phase population. Intriguingly, western blot analysis indicated that the cell cycle arrest induced by simvastatin is involved with the substantial elevation of CDK inhibitors, such as p21 and p27. Previous investigations have shown that other statins, such as pravastatin and lovastatin, yield to the up‐regulation of p21 and p27 through promoter activation.( 27 , 28 ) We were the first to show that simvastatin also increased expressions of these CDK inhibitors in HNSCC cells. Low levels of p21 and p27 expression have shown to be significantly associated with unfavorable clinicopathological factors in laryngeal squamous cell carcinoma.( 29 ) It has been reported that cell attachment to ECM leads to the down‐regulation of p21 and p27 and such antiapoptotic property associated with anchorage to the ECM is mediated by small G‐proteins including Rho, Cdc42 and Rac1.( 27 , 28 , 30 ) Our present data showed that simvastatin might overcome anoikis resistance by improving the expression of p21 and p27 in HNSCC cells. These CDK inhibitors are likely to be common targets for the anticancer effect of statins. Thus, this supports the effect of simvastatin on cell cycle regulation.
Despite the complexity of the anticancer mechanism of simvastatin, our data suggested that the blocking of ERK phosphorylation appeared to be one of the key mechanisms of overall potency. Constitutive ERK activation has been shown to play a pivotal role in the progression of tumorigenesis in different cancer types including HNSCC.( 31 ) The down‐regulation of constitutive ERK activity may be a common mechanism of action of statin‐induced apoptosis, because statins are known to regulate small G‐proteins and Ras/Raf/MEK/ERK signaling.( 32 ) A sustained activation of ERK signaling increases cyclin D1 expression in adherent, but not suspended cells, and this effect is also mediated by β1‐integrin expression.( 33 ) In contrast, the biological implication of simvastatin underlying the mechanisms of cell signaling has been controversial in the cancer field. Fluvastatin inhibited cell growth, migration, and angiogenesis of renal cancer cells and such effects were involved with the activation of p53 and its down‐stream effector, p21.( 34 ) Highly concentrated simvastatin attenuated phosphorylation of Akt in prostate cancer cells.( 18 ) In contrast, it has been reported that simvastatin contributes to maintenance of cell survival and angiogenesis of endothelial cells through the dose‐dependent activation of Akt, and a low concentration of simvastatin inactivates the function of p53 and p21 in hepatoma cells.( 35 , 36 ) These results imply that the statins may simultaneously exert variable and occasionally opposite effects on cell survival signals, depending on cell types and their concentrations.
Screening JMAR cells with cDNA microarray for gene expression modified by 10 µM simvastatin revealed that numerous genes related to cell proliferation, DNA replication, and cell adhesion were altered. Of altered genes associated with cell cycle regulation, CDK inhibitors including p57 and p21 were up‐regulated, whereas the cell cycle accelerators or cell proliferation markers such as cyclin D1, cyclin A2, and PCNA were significantly down‐regulated by simvastatin treatment. The CDK inhibitors including p21, p27, and p57 belong to the Cip/Kip family, which shares a common N‐terminal domain for inhibiting the kinase activity of CDK–cyclin complexes. Of these genes, p21 and p57 have been shown to bind to PCNA and consequently contribute to the inhibition of DNA replication.( 37 ) Increased expression of p21 with simvastatin treatment was validated by western blot analysis. Our observations of PI staining also supported the effects of simvastatin on cell cycle arrest and apoptosis. In contrast, cDNA microarray analysis demonstrated up‐regulation of several members of the matrix metalloproteinase (MMP) family and the S100 calcium‐binding protein family in simvastatin‐treated JMAR cells. Statins have been shown to induce up‐regulation of S100 in primitive neuroectodermal tumor cells and down‐regulation of MMP‐2 in endothelial cells.( 38 , 39 ) Although these results implied that these family genes had certain involvement as the pharmaceutical targets of statins, further investigation is needed to clarify the molecular implications.
The present data from cDNA microarray and western blot analysis showed that simvastatin treatment significantly suppressed the expression of TS. TS is known to be a target for 5‐fluorouracil (5‐FU), which has been widely prescribed as a chemotherapeutic agent to treat patients with HNSCC. Although the mechanisms of cellular resistance to 5‐FU are not fully understood, several mechanisms are attributed to an increased TS expression.( 40 ) In addition, several immunohistochemical investigations have shown that HNSCC patients with low TS levels are expected to respond better to 5‐FU‐based chemotherapy than patients with a high TS expression.( 41 , 42 ) In an experimental investigation, the induction of the wild‐type TS gene to mammalian cells, but not the mutant type, gave rise to a transformation into the malignant phenotype, whereas the blockade of TS expression by siRNA inhibited cell proliferation and survival.( 43 ) These observations imply that the TS gene potentially plays a role as a therapeutic target and as an oncogene associated with cell growth and proliferation. Because simvastatin exerts antiproliferative and anti‐invasive effects with a down‐regulation of TS expression, simvastatin may overcome drug resistance to conventional chemotherapeutic agents in patients with refractory disease in the near future.
Our present study demonstrated that the addition of mevalonate inhibited caspase‐dependent apoptosis caused by simvastatin. One of the mechanisms of its effect may be associated with the activation of cell survival signaling mediated by stabilized lipid rafts, since statins including simvastatin have been shown to lead to lipid raft disruption as a result of their cholesterol‐lowering activity.( 18 ) In addition, a previous report showing that mevalonate promotes proliferation of human cancer cells supports an antiapoptotic effect of mevalonate.( 7 ) We speculate that mevalonate may maintain cell survival and protect simvastatin‐induced cell death through the recruitment of lipid raft. Another putative mechanism is supposed to involve a reduction in the cellular uptake of simvastatin. Assessment of whether mevalonate alters the uptake of simvastatin into cancer cells is needed. However, these current observations imply an important role of the mevalonate pathway in the cancer field.
In conclusion, we have shown that simvastatin treatment of HNSCC cell lines leads to the inhibition of cell growth and invasiveness along with cell cycle arrest and anoikis and that these effects are associated with down‐regulation of β1‐integrin and ERK signaling. These negative effects on cell adhesion and subsequent survival indicate a certain role in cancer treatment.
Acknowledgments
This work was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan (S. Maruya). We also thank Dr Kazunori Futai for his assistance in the preparation of figures and Dr Manabu Ichinohe for his constructive advice.
References
- 1. Lennernas H, Fager G. Pharmacodynamics and pharmacokinetics of the HMG‐CoAreductase inhibitors. Similarities and differences. Clin Pharmacokinet 1997; 32: 403–25. [DOI] [PubMed] [Google Scholar]
- 2. Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res 1991; 51: 3602–9. [PubMed] [Google Scholar]
- 3. Cafforio P, Dammacco F, Gernone A, Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 2005; 26: 883–91. [DOI] [PubMed] [Google Scholar]
- 4. Katano H, Pesnicak L, Cohen JI. Simvastatin induces apoptosis of Epstein‐Barr virus (EBV)‐transformed lymphoblastoid cell lines and delays development of EBV lymphomas. Proc Natl Acad Sci USA 2004; 101: 4960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khanzada UK, Pardo OE, Meier C, Downward J, Seckl MJ, Arcaro A. Potent inhibition of small‐cell lung cancer cell growth by simvastatin reveals selective functions of Ras isoforms in growth factor signaling. Oncogene 2006; 25: 877–87. [DOI] [PubMed] [Google Scholar]
- 6. Pioche‐Durieu C, Keryer C, Souquere S et al. In nasopharyngeal carcinoma cells, Epstein‐Barr virus LMP1 interacts with galectin 9 in membrane raft elements resistant to simvastatin. J Virol 2005; 79: 13 326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duncan RE, El‐Sohemy A, Archer MC. Mevalonate promotes the growth of tumors derived from human cancer cells in vivo and stimulates proliferation in vitro with enhanced cyclin‐dependent kinase‐2 activity. J Biol Chem 2004; 279: 33 079–84. [DOI] [PubMed] [Google Scholar]
- 8. Giancotti FG, Ruoslahti E. Integrin signaling. Science 1999; 285: 1028–32. [DOI] [PubMed] [Google Scholar]
- 9. Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol 2001; 13: 555–62. [DOI] [PubMed] [Google Scholar]
- 10. Van Golen CM, Soules ME, Grauman AR, Feldman EL. N‐Myc overexpression leads to decreased β1 integrin expression and increased apoptosis in human neuroblastoma cells. Oncogene 2003; 22: 2664–73. [DOI] [PubMed] [Google Scholar]
- 11. Seales EC, Jurado GA, Brunson BA, Wakefield JK, Frost AR, Bellis SL. Hypersialylation of β1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up‐regulating cell motility. Cancer Res 2005; 65: 4645–52. [DOI] [PubMed] [Google Scholar]
- 12. Sethi T, Rintoul RC, Moore SM et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo . Nat Med 1999; 5: 662–8. [DOI] [PubMed] [Google Scholar]
- 13. Brakebusch C, Bouvard D, Stanchi F, Sakai T, Fassler R. Integrins in invasive growth. J Clin Invest 2002; 109: 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA-Cancer J Clin 1999; 49: 33–64. [DOI] [PubMed] [Google Scholar]
- 15. Greenlee RT, Hill‐Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA-Cancer J Clin 2001; 51: 15–36. [DOI] [PubMed] [Google Scholar]
- 16. Greenberg JS, El‐Naggar AK, Mo V, Roberts D, Myers JN. Disparity in pathologic and clinical lymph node staging in oral tongue carcinoma. Implication for therapeutic decision making. Cancer 2003; 98: 508–15. [DOI] [PubMed] [Google Scholar]
- 17. Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS‐354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non‐small cell lung cancer cells. Clin Cancer Res 2005; 11: 6924–32. [DOI] [PubMed] [Google Scholar]
- 18. Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest 2005; 115: 959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clark EA, King WG, Brugge JS, Symons M, Hynes RO. Integrin‐mediated signals regulated by members of the Rho family of GTPases. J Cell Biol 1998; 142: 573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J 2000; 19: 2008–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manohar A, Shome SG, Lamar J et al. α3β1 integrin promotes keratinocyte cell survival through activation of a MEK/ERK signaling pathway. J Cell Sci 2004; 117: 4043–54. [DOI] [PubMed] [Google Scholar]
- 22. Park CC, Zhang H, Pallavicini M et al. β1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimension cultures and in vivo . Cancer Res 2006; 66: 1526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang H, Ozaki I, Mizuta T et al. β1‐integrin protects hepatoma cells from chemotherapy induced apoptosis via a mitogen‐activated protein kinase dependent pathway. Cancer 2002; 95: 896–906. [DOI] [PubMed] [Google Scholar]
- 24. Zhu Z, Sanchez‐Sweatman O, Huang X et al. Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res 2001; 61: 1707–16. [PubMed] [Google Scholar]
- 25. Toutant M, Costa A, Studler JM, Kadare G, Carnaud M, Girault JA. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol Biol Cell 2002; 22: 7731–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wen LP, Fahrni JA, Troie S, Guan JL, Orth K, Rosen GD. Cleavage of focal adhesion kinase by caspases during apoptosis. J Biol Chem 1997; 272: 26 056–61. [DOI] [PubMed] [Google Scholar]
- 27. Lee SJ, Ha MJ, Lee J et al. Inhibition of the 3‐hydroxy‐3 methylglutaryl‐coenzyme A reductase pathway induces p53‐independent transcriptional regulation of p21 (WAF1/CIP1) in human prostate carcinoma cells. J Biol Chem 1998; 273: 10 618–23. [DOI] [PubMed] [Google Scholar]
- 28. Hirai A, Nakamura S, Noguchi Y et al. Geranylgeranylated rho small GTPase (s) are essential for the degradation of p27Kip1 and facilitate the progression for G1‐S phase in growth‐stimulated rat FRTL‐5 cells. J Biol Chem 1997; 272: 13–16. [PubMed] [Google Scholar]
- 29. Pruneri G, Pignataro L, Carboni N et al. Clinical relevance of expression of the CIP/KIP cell‐cycle inhibitors p21 and p27 in laryngeal cancer. J Clin Oncol 1999; 17: 3150–9. [DOI] [PubMed] [Google Scholar]
- 30. Bao W, Thullberg M, Zhang H, Onischenko A, Stromblad S. Cell attachment to the extracellular matrix induces proteasomal degradation of p21CIP1 via Cdc42/Rac1 signaling. Mol Cell Biol 2002; 22: 4587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mishima K, Inoue K, Hayashi Y. Overexpression of extracellular‐signal regulated kinases on oral squamous cell carcinoma. Oral Oncol 2002; 38: 468–74. [DOI] [PubMed] [Google Scholar]
- 32. Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin‐induced apoptosis. Cancer Res 2004; 64: 6461–8. [DOI] [PubMed] [Google Scholar]
- 33. Roovers K, Davey G, Zhu X, Bottazzi ME, Assoian RK. α5β1 integrin controls cyclin D1 expression by sustaining mitogen‐activated protein kinase activity in growth factor‐treated cells. Mol Biol Cell 1999; 10: 3197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Horiuchi A, Sumitomo M, Asakuma J, Asano T, Asano T, Hayakawa M. 3‐Hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitor, fluvastatin, as a novel agent for prophylaxis of renal cancer metastasis. Clin Cancer Res 2004; 10: 8648–55. [DOI] [PubMed] [Google Scholar]
- 35. Kureishi Y, Luo Z, Shiojima I et al. The HMG‐CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 2000; 6: 1004–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paajarvi G, Roudier E, Crisby M, Hogberg J, Stenius U. HMG‐CoA reductase inhibitors, statins, induce phosphorylation of Mdm2 and attenuate the p53 response to DNA damage. FASEB J 2005; 19: 476–8. [DOI] [PubMed] [Google Scholar]
- 37. Watanabe H, Pan ZQ, Schreiber‐Agus N, Depinho RA, Hurwitz J, Xiong Y. Suppression of cell transformation by the cyclin‐dependent kinase inhibitor p57KIP2 requires binding to proliferating cell nuclear antigen. Proc Natl Acad Sci USA 1998; 95: 1392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim JS, Pirnia F, Choi YH et al. Lovastatin induces apoptosis in a primitive neuroectodermal tumor cell line in association with RB down‐regulation and loss of the G1 checkpoint. Oncogene 2000; 19: 6082–90. [DOI] [PubMed] [Google Scholar]
- 39. Vincent L, Chen W, Hong L et al. Inhibition of endothelial cell migration by cerivastatin, an HMG‐CoA reductase inhibitor: contribution to its anti‐angiogenic effect. FEBS Lett 2001; 495: 159–66. [DOI] [PubMed] [Google Scholar]
- 40. Longley DB, Harkin DP, Johnston PG. 5‐fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3: 330–8. [DOI] [PubMed] [Google Scholar]
- 41. Johnston PG, Mick R, Recant W et al. Thymidylate synthase expression and response to neoadjuvant chemotherapy in patients with advanced head and neck cancer. J Natl Cancer Inst 1997; 89: 308–13. [DOI] [PubMed] [Google Scholar]
- 42. Shiga H, Heath EI, Rasmussen AA et al. Prognostic value of p53, glutathione S‐transferase π, and thymidylate synthase for neoadjuvant cisplatin‐based chemotherapy in head and neck cancer. Clin Cancer Res 1999; 12: 4097–104. [PubMed] [Google Scholar]
- 43. Rahman L, Voeller D, Rahman M et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell 2004; 5: 341–51. [DOI] [PubMed] [Google Scholar]