

Abstract
Cell migration is a critical step in tumor invasion and metastasis, and regulation of this process will lead to appropriate therapies for treating cancer. Cancer cells migrate in various ways, according to cell type and degree of differentiation. The different types of cell migration are regulated by different mechanisms. Reorganization of the actin cytoskeleton is the primary mechanism of cell motility and is essential for most types of cell migration. Actin reorganization is regulated by Rho family small GTPases such as Rho, Rac, and Cdc42. These small GTPases transmit extracellular chemotactic signals to downstream effectors. Of these downstream effectors, Wiskott–Aldrich syndrome protein (WASP) family proteins are key regulators of cell migration. Activated WASP family proteins induce the formation of protrusive membrane structures involved in cell migration and degradation of the extracellular matrix. Inhibition of Rho family small GTPase signaling suppresses the migration and invasion of cancer cells. Thus, control of cell migration via the actin cytoskeleton provides the possibility of regulating cancer cell invasion and metastasis. (Cancer Sci 2005; 96: 379 – 386)
Cancer cells move within tissues during invasion and metastasis by their own motility, and control of cancer cell migration is an important problem in tumor treatment. Cell migration has been well studied in non‐neoplastic cells such as fibroblasts and epithelial cells, and the molecular mechanisms underlying cell migration are common to both non‐neoplastic cells and cancer cells. Cell migration involves multiple processes that are regulated by various signaling molecules.( 1 ) The actin cytoskeleton and its regulatory proteins are crucial for cell migration in most cells. During cell migration, the actin cytoskeleton is dynamically remodeled, and this reorganization produces the force necessary for cell migration.( 2 ) Because inhibition of these processes decreases cell motility, elucidation of the molecular mechanisms of actin reorganization is important for cancer therapeutics. Actin‐related protein (Arp) 2/3 complex‐dependent actin polymerization and its regulation are of particular interest.
Most studies of cell migration have been performed on 2‐D substrates. However, given that motile cells move throughout tissues and across tissue barriers, further understanding is required regarding migration within 3‐D matrices. Recent developments in imaging techniques in cell biology have enabled the investigation of invasive cancer cell migration in 3‐D matrices.( 3 ) Cancer cells in 3‐D matrices show different types of cell migration from those on 2‐D substrates. This review will focus on the migration of cancer cells and regulatory mechanisms involving the actin cytoskeleton.
Mechanisms of cell migration
Directed cell migration is a critical feature of various physiological and pathological processes, including development, wound healing, immunity, angiogenesis, and metastasis. Cell migration is triggered by gradients of chemotactic factors, for example platelet‐derived growth factor (PDGF).( 4 , 5 ) In the presence of chemoattractants, extracellular signals are transduced through cell‐surface receptors and induce various intracellular responses, for example polarization, reorganization of the cytoskeleton, and vesicular transport. These complex responses contribute cooperatively to cell migration. Directed cell movement on 2‐D substrates consists of cycles of four ordered processes: membrane protrusion at the leading edge, adhesion of the protrusions to the substrate, translocation of the cell body, and retraction of the trailing edge (Fig. 1).( 6 ) Reorganization of the actin cytoskeleton is involved in these processes and is regulated temporally and spatially by Rho family small GTPases.
Figure 1.
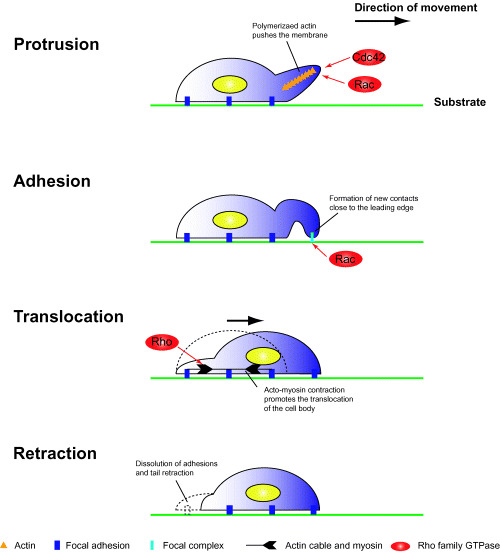
Cell migration on 2‐D substrates. Cell migration consists of the following four successive processes. Protrusion: when cells are polarized by extracellular stimuli, de novo actin polymerization occurs at the leading edge; polymerized actin filaments induce the formation of membrane protrusions such as filopodia and lamellipodia. Adhesion: protruded membranes contact the substrate and form novel integrin‐dependent cell‐substrate adhesions; newly formed adhesion complexes are stabilized and mature into focal adhesions. Translocation: the nucleus and cell body are translocated by acto‐myosin contractile forces. Retraction: cell‐substrate adhesive structures at the trailing edge are disassembled, and the trailing edge retracts.
Rho family small GTPases. During cell migration, Rho family small GTPases play pivotal roles in reorganization of the actin cytoskeleton.( 7 ) Rho family GTPases act as molecular switches and can cycle between GTP‐ and GDP‐bound states. When cells are stimulated by growth factors, bound GDP is exchanged for GTP. In the GTP‐bound state, Rho family small GTPases are active and interact with specific downstream effectors, leading to the translocation and activation of the effector, and induction of various intracellular responses. Among these responses, reorganization of the actin cytoskeleton has been studied most. Small GTPases of the Rho family, for example Cdc42, Rac, and Rho, are key regulators of actin assembly and control the formation of filopodia, lamellipodia, and stress fibers, respectively.( 8 )
Directional cell movement requires a defined cell polarity in which cytoskeletal components are differentially localized at two poles of a cell in response to directional signals. Recent studies in Dictyostelium cells, leukocytes, and fibroblasts indicate that phosphatidylinositide 3‐kinase (PI3K) plays a central role in the amplification of internal signaling asymmetry and thus helps to establish cell polarity and to define the leading edge of the cell.( 9 , 10 ) PI3K activation in response to chemoattractants leads to accumulation of phosphatidylinositol 3,4,5 triphosphate (PIP3), which stimulates small GTPases of the Rho family, for example Rac and Cdc42, inducing formation of a leading edge (Fig. 1).
Activated Rac and Cdc42 induce reorganization of the actin cytoskeleton at the leading edge.( 11 ) Localized actin polymerization at the leading edge pushes the membrane forward in finger‐like structures known as filopodia and in sheet‐like structures known as lamellipodia. These structures generate the locomotive force in migrating cells. Unlike Rac and Cdc42, Rho regulates the assembly of contractile acto‐myosin filaments.( 12 ) Rho regulates myosin‐mediated contractility through downstream effectors, such as ROCK/Rho kinase.( 13 ) The Rho‐mediated acto‐myosin contractile force promotes the locomotion of the cell body and the trailing edge (Fig. 1). Thus, during cell movement, Rac and Cdc42 stimulate formation of protrusions at the leading edge, and Rho induces retraction at the trailing edge. This coordinated reorganization permits cells to move toward a target.
Migration of cancer cells
Modes of cancer cell migration. Cancer cells have various modes of migration (Fig. 2).( 14 ) In most tumors, several types of cell migration are observed simultaneously. In the case of epithelial tumors, in which cell–cell junctions are present, cells move together in sheet‐like structures. This mode of migration is observed in epithelial cells during wound repair and in endothelial cells during angiogenesis.( 15 ) As dedifferentiation of epithelial cancer cells proceeds, the function of cadherin, a cell–cell junction protein, is suppressed and individual cells separate from the other cells and move as single cells.( 16 ) These modes of cell migration are called collective and individual cell migration, respectively. The transition from collective to individual migration is termed epithelial–mesenchymal transition and is a well‐studied indicator of tumor progression.( 17 ) Thus, cancer cells show distinct modes of cell migration according to differentiation state.
Figure 2.
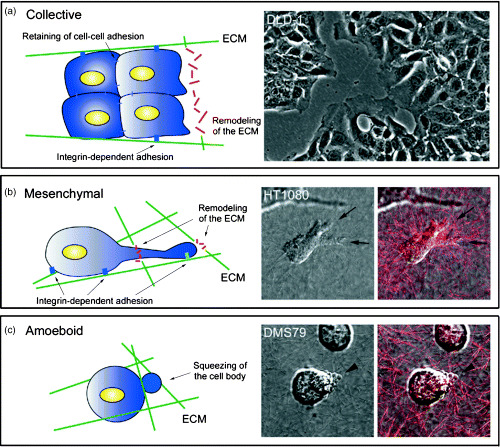
Modes of migration in cancer cells. Migration of cancer cells involves the following three processes. (a) Collective cell migration. Cells move as sheet‐like structures, maintaining cell–cell adhesions. The image shows migration of DLD‐1 cells on a 2‐D substrate. (b) Mesenchymal migration. In 3‐D matrices, cells form membrane protrusions at the leading edge and adhere to the substrate in an integrin‐dependent manner. By releasing extracellular matrix (ECM)‐degrading enzymes, cells remodel the ECM to form a path. The images show the migration of HT1080 cells in collagen gels. Arrows indicate membrane protrusions at the leading edge. The red signal indicates the backscatter of collagen fibers. (c) Amoeboid migration. Cell‐substrate adhesion is weak and is independent of integrin function. Cells move by acto‐myosin contractile force and move within the ECM by squeezing the cell body. The images show the migration of DMS79 cells in collagen gels. Arrowheads indicate membrane blebbing. The red signal indicates the backscatter of collagen fibers.
Cancer cell migration in 3‐D matrices. When cultured in 3‐D matrices, cancer cells, which undergo individual cell migration similar to that of dedifferentiated epithelial tumor cells and lymphoma cells, show two typical morphologies. One is an elongated morphology similar to that of fibroblasts and the other is a rounded morphology. These two types of morphology use different migratory mechanisms; elongated cells undergo mesenchymal migration and rounded cells undergo amoeboid migration.
With respect to mesenchymal migration, fibroblast‐like cells on 2‐D matrices use the same mechanisms described earlier.( 18 ) Their elongated morphology and mesenchymal migration are dependent on integrin‐mediated adhesion. In addition to the four processes observed on 2‐D substrates, mesenchymal migration in 3‐D matrices requires degradation of the extracellular matrix (ECM). Elongated cells have protruding membrane at the leading edge and form integrin‐dependent adhesions with the substrate. Matrix‐degrading proteases such as matrix metalloproteinase also accumulate in an integrin‐dependent manner at the leading edge of migrating cells, leading to localized proteolysis.( 19 ) By degradation of the ECM, elongated cells form a path and overcome tissue barriers. Multiple ECM‐degrading proteases can be upregulated and activated in cancer cells.
In contrast to mesenchymal migration, rounded cells move in an integrin‐independent manner. In these cells, cell‐substrate adhesions are weak; therefore, the cells show a rounded morphology. When rounded cells migrate through the ECM, they change shape and squeeze themselves into gaps in the ECM. This amoeboid migration does not require degradation of the ECM and is seen both in non‐neoplastic cells such as lymphocytes and neutrophils, and in neoplastic cells such as those of lymphoma and small‐cell lung carcinoma.( 20 , 21 , 22 )
Cancer cell migration can convert between the mesenchymal and amoeboid types under certain conditions. Whereas remodeling of the ECM is required for mesenchymal migration, in the presence of protease inhibitors, cancer cells change to amoeboid migration and pass through the ECM.( 19 , 23 ) This ability makes it difficult to repress cancer cell invasion by using protease inhibitors. To regulate cancer cell migration, both mesenchymal and amoeboid migration must be repressed simultaneously.
Regulation by Rho family small GTPases. Mesenchymal and amoeboid types of migration use different driving forces. Both types of migration are based on reorganization of the actin cytoskeleton, but their requirements for Rho and Rac signaling differ. With respect to mesenchymal migration, membrane protrusions at the leading edge are formed in a Rac‐dependent manner. Expression of a dominant–negative form of Rac in elongated cells, for example B16F10 melanoma cells, inhibits the formation of such protrusive membrane structures, causing decreased cell migration.( 24 ) In contrast, Rho signaling is not essential for mesenchymal migration.( 23 ) With respect to amoeboid migration, the actin cytoskeleton is reorganized along the plasma membrane, causing dynamic membrane blebbing along the cell surface.( 23 ) This cortical actin reorganization is dependent on Rho/ROCK signaling.( 25 ) Activated ROCK can induce further blebbing in rounded cells.( 23 ) Inhibitors of Rho/ROCK signaling suppress the formation of membrane blebs and inhibit amoeboid migration.
HT1080 fibrosarcoma cells show both mesenchymal and amoeboid types of cell migration. Inhibition of Rac signaling induces membrane blebs, indicating a transition from mesenchymal to amoeboid migration. In contrast, the inhibition of Rho/ROCK signaling induces an elongated morphology (S. Kurisu et al., unpublished data 2005). Thus, mesenchymal and amoeboid types of migration are dependent on Rac and Rho signaling, respectively. The balance between Rac and Rho signaling may determine the type of cell migration at any particular time.
Regulation of cancer cell migration by Wiskott–Aldrich syndrome protein family proteins
Wiskott–Aldrich syndrome protein family proteins. As described earlier, Rho family small GTPase‐induced reorganization of the actin cytoskeleton plays a central role in cell migration and invasion. Many recent studies have elucidated the molecular mechanisms underlying Rac‐ and Cdc42‐induced actin reorganization in the formation of filopodia and lamellipodia.( 2 ) In these membrane protrusions, monomeric actin is polymerized into filaments dependent on the Arp2/3 complex. The Arp2/3 complex consists of seven different proteins, including actin‐related proteins, which comprise a nucleation core of actin polymerization. Wiskott–Aldrich syndrome protein (WASP) family proteins are key regulators that link extracellular stimuli to actin reorganization and regulate cell migration through induction of membrane protrusions at the leading edge.( 26 , 27 ) WASP family proteins link Cdc42‐ and Rac‐dependent signals to filopodium and lamellipodium formation, respectively, by virtue of their ability to activate Arp2/3 complex‐mediated de novo actin polymerization.
To date, five mammalian WASP family proteins have been identified: WASP, neural WASP (N‐WASP), and WAVE (WASP‐family verprolin‐homologous proteins) 1, 2, and 3. These proteins share two main regions of homology: a central segment rich in proline residues and a carboxy‐terminal module comprising three characteristic domains, a verprolin‐homology (V) domain, a cofilin‐like (C) domain, and an acidic (A) domain (VCA) (Fig. 3). The V domain is a monomeric actin binding site, and the CA domain binds the Arp2/3 complex. When bound by the VCA domain, the Arp2/3 complex is activated and catalyzes actin polymerization. The amino‐terminal half of the molecule defines two functional groups on the basis of the presence of either a WASP homology 1/Ena VASP homology 1 (WH1/EVH1) domain (found in WASP and N‐WASP) or a WAVE homology domain suppressor of cAMP receptor (SCAR) homology domain (WHD/SHD), specific to WAVE isoforms.
Figure 3.
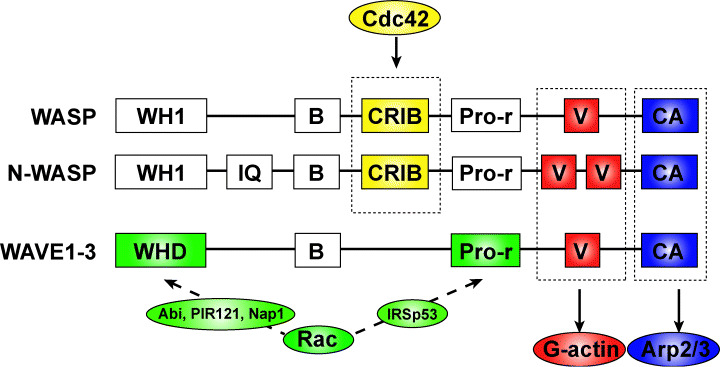
The Wiskott–Aldrich syndrome protein (WASP) family proteins. WASP family proteins consist of multiple functional domains. WASP and neural WASP (N‐WASP) are activated by direct binding to Cdc42 via the CRIB domain, whereas WAVE (WASP‐family verprolin‐homologous proteins) bind to Rac indirectly via various molecules. The C‐terminal verprolin‐homology, cofilin‐like, and acidic (VCA) domains are important for the induction of actin polymerization. The V and CA domains bind actin monomer (G‐actin) and the Arp2/3 complex, respectively. WASP family proteins activated by Rac and Cdc42 induce Arp2/3 complex‐dependent actin polymerization via the VCA domain. B, basic; CA, cofilin‐like and acidic; CRIB, Cdc42 and Rac interactive binding; IQ, IQ motif; Pro‐r, Proline‐rich; V, verprolin homology; WH1, WASP homology 1; WHD, WAVE homology domain.
The WASP family proteins have been identified as the causative gene products of Wiskott‐Aldrich syndrome (WAS), which is characterized by eczema, bleeding and recurrent infections. This protein is expressed exclusively in hematopoietic cells. Lymphocytes from WAS patients show impaired actin remodeling in response to stimulation as well as a reduction in the number of cell‐surface microvilli.( 28 , 29 ) WASP binds to Cdc42 via its GBD/CRIB (GTPase‐binding domain/Cdc42 and Rac interactive binding) domain, and overexpression of WASP induces the formation of actin clusters, suggesting that it has a role in actin polymerization.
Neural WASP was first purified as an Ash/Grb2‐binding protein from bovine brain and is expressed ubiquitously; it is particularly enriched in the brain.( 30 ) N‐WASP shares 50% homology with WASP. N‐WASP induces filopodium formation downstream of Cdc42 activation.( 31 ) Cdc42 binds directly to N‐WASP and induces a conformational change that releases the autoinhibited structure.( 32 ) Activated N‐WASP then binds to the Arp2/3 complex and induces rapid actin polymerization.
The WAVE proteins are identified as novel proteins that possess a V domain.( 33 ) Each WAVE isoform has a different tissue distribution.( 34 ) WAVE1 and WAVE3 are expressed predominantly in the brain, whereas WAVE2 is expressed ubiquitously, with particularly strong expression in the placenta, lung, and peripheral blood leukocytes. In Dictyostelium, SCAR, a WAVE homolog, was isolated in a genetic screen for proteins downstream of the chemotaxis receptor for cAMP.( 35 ) WAVE proteins possess a WHD/SHD motif, a basic domain, a proline‐rich domain, and a VCA domain. WAVE1 regulates membrane ruffle formation by activating the Arp2/3 complex downstream of Rac.( 33 ) Endogenous WAVE1 localizes to membrane ruffles induced by treatment with PDGF or by expression of constitutive‐active Rac. In contrast to WASP and N‐WASP, WAVE proteins do not contain binding sites for Rac or Cdc42 and do not bind directly to Rac.
IRSp53, a substrate of the insulin receptor with unknown function, is an intermediary protein linking Rac and WAVE2.( 36 , 37 ) Activated Rac binds to the amino terminus of IRSp53, and the carboxy‐terminal Src‐homology‐3 domain of IRSp53 binds to WAVE2 to form a trimolecular complex. IRSp53 does not bind WAVE1 or WAVE3.
Recently, Eden et al. showed that purified WAVE1 is constitutively active.( 38 ) They proposed a novel mechanism whereby WAVE1 is kept inactive, via its association with four proteins: Nap125, PIR121, Abi2 and HSPC300. This complex is unable to stimulate actin polymerization in in vitro assays, but the addition of purified, active Rac removes this inhibition. However, other groups have reported that this complex is stable in vivo and can activate the Arp2/3 complex in vitro.( 39 ) The functions of this WAVE‐regulatory complex remain to be determined.
Functions of WASP family proteins in cancer cell migration. The WASP family proteins have two physiological roles with respect to migration and invasion of cancer cells in 3‐D matrices. One is the regulation of mesenchymal cell migration, and the other is degradation of the ECM. WASP family proteins regulate these processes through the formation of various actin‐based protrusive membrane structures downstream of Rac and Cdc42 activation.
Mesenchymal migration. During cell migration, WAVE2 is concentrated at the tips of membrane protrusions formed in the direction of cell locomotion.( 40 , 41 ) The binding of basic residues in WAVE2 to PIP3 mediates recruitment to the leading edge.( 42 ) Downstream of Rac, WAVE2 induces Arp2/3‐mediated actin polymerization, causing the formation of lamellipodia,( 11 ) which are crucial for mesenchymal cell migration. Lamellipodia are thin, flattened structures that consist of highly branched actin networks.( 43 ) WAVE proteins activate the Arp2/3 complex, which binds pre‐existing actin filaments and induces new side‐branched actin polymerization. Of the WAVE proteins, WAVE2 is localized to the tips of the lamellipodial protrusions, predominantly in fibroblasts.( 41 ) In WAVE2‐deficient fibroblasts, lamellipodium formation is severely inhibited. Defective lamellipodium formation leads to decreased cell migration and invasion toward chemoattractants in WAVE2‐deficient cells. Thus, WAVE2 is important for directional movement in fibroblasts. WAVE2 also plays a role in the migration of B16F10 melanoma cells, which undergo mesenchymal migration in 3‐D matrices.( 24 ) Repression of WAVE2 expression by RNAi methods reduces cancer cell migration and metastasis into the lungs by B16F10 melanoma cells via regulation of membrane protrusions (Fig. 4). Thus, WAVE2 is essential for mesenchymal migration, and inhibition of WAVE2 function can inhibit the migration and invasion of cancer cells.
Figure 4.
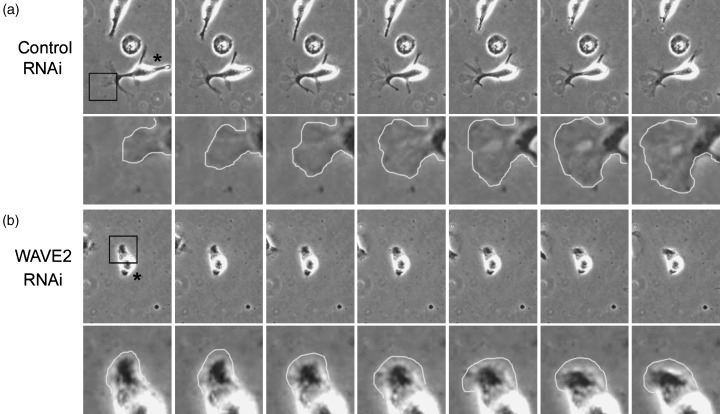
The WAVE2 (Wiskott–Aldrich syndrome protein family verprolin‐homologous protein 2) is required for cancer cell migration in 3‐D matrices. ( 24 ) Time‐lapse phase‐contrast images of RNA‐interference (RNAi)‐treated B16F10 cells in extracellular matrix‐containing gels. Asterisks represent transfected cells. Repression of WAVE2 expression by RNAi methods reduces membrane protrusions at the leading edge, which are important for migration and invasion of melanoma cells. Magnified images of boxed areas are shown in the lower panels.
Dorsal ruffles, podosomes and invadopodia. In some invasive cells, characteristic protrusive membrane structures are involved in ECM degradation, for example dorsal ruffles, podosomes, and invadopodia.( 44 ) Formation of these unique structures is also regulated by WASP family proteins.
Dorsal ruffles. When fibroblasts and epithelial cells are stimulated by growth factors, a drastic change in dorsal membrane structures is observed. These protrusive membrane structures are known as dorsal ruffles, but their physiological function is unknown. Recently, analysis of WAVE1‐deficient cells has shown that dorsal ruffles are involved in degradation of the ECM.( 41 ) When mouse embryonic fibroblasts (MEF) are stimulated by PDGF, formation of dorsal ruffles is induced, and the ECM‐degrading enzyme MMP‐2 accumulates at the dorsal ruffles. The formation of dorsal ruffles and of lamellipodial protrusions is dependent on Rac. However, WAVE1, not WAVE2, is required for dorsal ruffle formation. A WAVE1 deficiency decreases dorsal ruffle formation and subsequent MMP secretion. WAVE1‐deficient MEF show normal chemotactic migration toward PDGF, but chemotactic invasion into the ECM toward PDGF is decreased. Dorsal ruffles are formed only when cells are cultured on 2‐D substrates, and it is unknown what form they take in 3‐D matrices. The relationship between dorsal ruffle structure and chemotactic invasion remains to be determined. However, it is clear that WAVE1‐mediated protrusive membrane structures are crucial for migration within the ECM.
Podosomes and invadopodia. Podosomes are cell‐substrate adhesion sites that differ from focal adhesions, which are usually observed in adhesive cells.( 45 ) These structures are observed in limited cell types, for example osteoclasts and macrophages. WASP is involved in the formation of these structures,( 46 ) which are also termed invadopodia in oncogene‐transformed cells. The distinction between podosomes and invadopodia is obscure; because invadopodia persist longer than podosomes, podosomes are thought to be precursor structures of invadopodia. Podosomes consist of filamentous actin‐rich cores and surrounding ring structures containing adhesive proteins such as vinculin and talin. Podosomes and invadopodia are also involved in degradation of the ECM. When Src‐transformed cells are cultured on fibronectin (FN), an ECM protein, spot‐like degradation of FN is observed and these spots colocalize with podosomes. N‐WASP is recruited to podosome structures and is essential for podosome formation. Expression of a dominant‐negative form of N‐WASP, which cannot induce Arp2/3 complex‐dependent actin polymerization, inhibits podosome formation and subsequent proteolysis of the ECM. Recent studies with the use of RNA interference methods also show the importance of N‐WASP in invadopodia formation in metastatic carcinoma cells.( 47 )
Conclusions and future directions
Multiple genes involved in cell motility are deregulated in human cancers,( 48 ) and regulation of cancer cell migration is inhibited by various signals.( 49 ) Cancer cells show multiple types of cell migration. Because each type of cell migration has distinct molecular mechanisms, it is difficult to regulate the motility of all cancer cells by using a single strategy (Fig. 5). Reorganization of the actin cytoskeleton plays a central role in the migration of cancer cells. Indeed, inhibition of actin polymerization suppresses most types of cell migration. Rho family small GTPase signaling is also activated in cancer cells and is a good candidate for cancer therapy.( 50 ) Recently, new regulators of actin polymerization, in addition to the Arp2/3 complex, have been identified.( 51 , 52 ) To fully elucidate the details of cancer cell migration, it will be important to investigate more precisely the mechanisms underlying regulation of the actin cytoskeleton.
Figure 5.
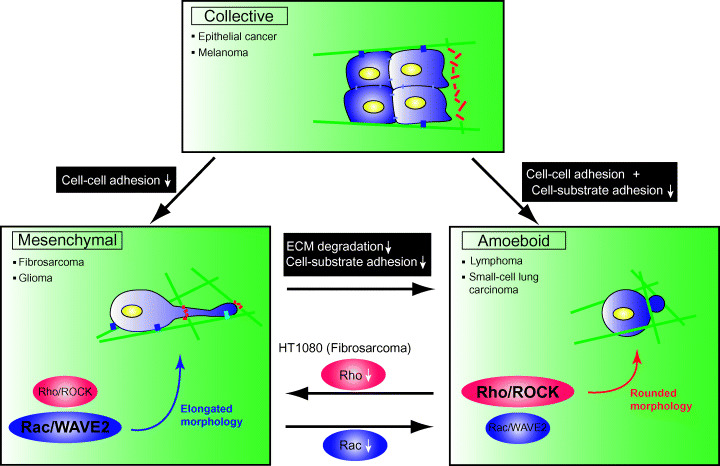
Transition of migration in cancer cells. When cell–cell adhesions are lost by inhibition of cadherin function, cancer cells that undergo collective migration begin to move separately (mesenchymal migration). When both cell–cell and cell–substrate adhesion are inhibited simultaneously, collective migration changes to amoeboid. Inhibition of extracellular matrix degradation and integrin‐dependent adhesion cause mesenchymal‐amoeboid transition (MAT). In HT1080 fibrosarcoma cells, which show both mesenchymal and amoeboid types of cell migration, inhibition of Rac signaling induces MAT. In contrast, inhibition of Rho/Rho‐kinase (ROCK) signaling induces an elongated morphology.
References
- 1. Ridley AJ, Schwartz MA, Burridge K et al. Cell migration: integrating signals from front to back. Science 2003; 302: 1704–9. [DOI] [PubMed] [Google Scholar]
- 2. Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003; 112: 453–65. [DOI] [PubMed] [Google Scholar]
- 3. Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer 2003; 3: 921–30. [DOI] [PubMed] [Google Scholar]
- 4. Kundra V, Escobedo JA, Kazlauskas A et al. Regulation of chemotaxis by the platelet‐derived growth factor receptor‐beta. Nature 1994; 367: 474–6. [DOI] [PubMed] [Google Scholar]
- 5. Huttenlocher A, Sandborg RR, Horwitz AF. Adhesion in cell migration. Curr Opin Cell Biol 1995; 7: 697–706. [DOI] [PubMed] [Google Scholar]
- 6. Lauffenburger DA. Cell motility. Making connections count. Nature 1996; 383: 390–1. [DOI] [PubMed] [Google Scholar]
- 7. Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol 2004; 265: 23–32. [DOI] [PubMed] [Google Scholar]
- 8. Etienne‐Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420: 629–35. [DOI] [PubMed] [Google Scholar]
- 9. Weiner OD et al. A PtdInsP(3)‐ and Rho GTPase‐mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol 2002; 4: 509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3) Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol 2002; 4: 513–8. [DOI] [PubMed] [Google Scholar]
- 11. Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol 2002; 12: 112–20. [DOI] [PubMed] [Google Scholar]
- 12. Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 1999; 144: 1235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alblas J, Ulfman L, Hordijk P, Koenderman L. Activation of Rhoa and ROCK are essential for detachment of migrating leukocytes. Mol Biol Cell 2001; 12: 2137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friedl P, Wolf K. Tumour‐cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 2003; 3: 362–74. [DOI] [PubMed] [Google Scholar]
- 15. Friedl P, Hegerfeldt Y, Tusch M. Collective cell migration in morphogenesis and cancer. Int J Dev Biol 2004; 48: 441–9. [DOI] [PubMed] [Google Scholar]
- 16. Lozano E, Betson M, Braga VM. Tumor progression: Small GTPases and loss of cell–cell adhesion. Bioessays 2003; 25: 452–63. [DOI] [PubMed] [Google Scholar]
- 17. Friedl P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol 2004; 16: 14–23. [DOI] [PubMed] [Google Scholar]
- 18. Tamariz E, Grinnell F. Modulation of fibroblast morphology and adhesion during collagen matrix remodeling. Mol Biol Cell 2002; 13: 3915–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wolf K, Mazo I, Leung H et al. Compensation mechanism in tumor cell migration: mesenchymal‐amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 2003; 160: 267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedl P, Borgmann S, Brocker EB. Amoeboid leukocyte crawling through extracellular matrix: lessons from the Dictyostelium paradigm of cell movement. J Leukoc Biol 2001; 70: 491–509. [PubMed] [Google Scholar]
- 21. Francis K, Palsson B, Donahue J, Fong S, Carrier E. Murine Sca‐1(+)/Lin(−) cells and human KG1a cells exhibit multiple pseudopod morphologies during migration. Exp Hematol 2002; 30: 460–3. [DOI] [PubMed] [Google Scholar]
- 22. Wang W, Wyckoff JB, Frohlich VC et al. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res 2002; 62: 6278–88. [PubMed] [Google Scholar]
- 23. Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 2003; 5: 711–9. [DOI] [PubMed] [Google Scholar]
- 24. Kurisu S, Suetsugu S, Yamazaki D, Yamaguchi H, Takenawa T. Rac‐WAVE2 signaling is involved in the invasive and metastatic phenotypes of murine melanoma cells. Oncogene 2005; 24: 1309–19. [DOI] [PubMed] [Google Scholar]
- 25. Worthylake RA, Lemoine S, Watson JM, Burridge K. RhoA is required for monocyte tail retraction during transendothelial migration. J Cell Biol 2001; 154: 147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci 2001; 114: 1801–9. [DOI] [PubMed] [Google Scholar]
- 27. Miki H, Takenawa T. Regulation of actin dynamics by WASP family proteins. J Biochem (Tokyo) 2003; 134: 309–13. [DOI] [PubMed] [Google Scholar]
- 28. Kenney D, Cairns L, Remold‐O'Donnell E, Peterson J, Rosen FS, Parkman R. Morphological abnormalities in the lymphocytes of patients with the Wiskott–Aldrich syndrome. Blood 1986; 68: 1329–32. [PubMed] [Google Scholar]
- 29. Molina IJ, Kenney DM, Rosen FS, Remold‐O'Donnell E. T cell lines characterize events in the pathogenesis of the Wiskott–Aldrich syndrome. J Exp Med 1992; 176: 867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miki H, Miura K, Takenawa T. N‐WASP, a novel actin‐depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2‐dependent manner downstream of tyrosine kinases. EMBO J 1996; 15: 5326–35. [PMC free article] [PubMed] [Google Scholar]
- 31. Miki H, Sasaki T, Takai Y, Takenawa T. Induction of filopodium formation by a WASP‐related actin‐depolymerizing protein N‐WASP. Nature 1998; 391: 93–6. [DOI] [PubMed] [Google Scholar]
- 32. Rohatgi R, Ma L, Miki H et al. The interaction between N‐WASP and the Arp2/3 complex links Cdc42‐dependent signals to actin assembly. Cell 1999; 97: 221–31. [DOI] [PubMed] [Google Scholar]
- 33. Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP‐family protein involved in actin reorganization induced by Rac. EMBO J 1998; 17: 6932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suetsugu S, Miki H, Takenawa T. Identification of two human WAVE/SCAR homologues as general actin regulatory molecules which associate with the Arp2/3 complex. Biochem Biophys Res Commun 1999; 260: 296–302. [DOI] [PubMed] [Google Scholar]
- 35. Bear JE, Rawls JF, Saxe CL3rd. SCAR, a WASP‐related protein, isolated as a suppressor of receptor defects in late Dictyostelium development. J Cell Biol 1998; 142: 1325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature 2000; 408: 732–5. [DOI] [PubMed] [Google Scholar]
- 37. Miki H, Takenawa T. WAVE2 serves a functional partner of IRSp53 by regulating its interaction with Rac. Biochem Biophys Res Commun 2002; 293: 93–9. [DOI] [PubMed] [Google Scholar]
- 38. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1‐induced actin nucleation by Rac1 and Nck. Nature 2002; 418: 790–3. [DOI] [PubMed] [Google Scholar]
- 39. Bompard G, Caron E. Regulation of WASP/WAVE proteins: making a long story short. J Cell Biol 2004; 166: 957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamazaki D, Suetsugu S, Miki H et al. WAVE2 is required for directed cell migration and cardiovascular development. Nature 2003; 424: 452–6. [DOI] [PubMed] [Google Scholar]
- 41. Suetsugu S, Yamazaki D, Kurisu S, Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell 2003; 5: 595–609. [DOI] [PubMed] [Google Scholar]
- 42. Oikawa T, Yamaguchi H, Itoh T et al. PtdIns (3,4,5)P3 binding is necessary for WAVE2‐induced formation of lamellipodia. Nat Cell Biol 2004; 6: 420–6. [DOI] [PubMed] [Google Scholar]
- 43. Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol 1999; 145: 1009–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol 2004; 5: 647–57. [DOI] [PubMed] [Google Scholar]
- 45. Linder S, Aepfelbacher M. Podosomes: adhesion hot‐spots of invasive cells. Trends Cell Biol 2003; 13: 376–85. [DOI] [PubMed] [Google Scholar]
- 46. Linder S, Nelson D, Weiss M, Aepfelbacher M. Wiskott–Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci USA 1999; 96: 9648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamaguchi H, Lorenz M, Kempiak S et al. Molecular mechanisms of invadopodium formation: the role of the N‐WASP‐Arp2/3 complex pathway and cofilin. J Cell Biol 2005; 168: 441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev 2005; 15: 87–96. [DOI] [PubMed] [Google Scholar]
- 49. Eccles SA. Targeting key steps in metastatic tumour progression. Curr Opin Genet Dev 2005; 15: 77–86. [DOI] [PubMed] [Google Scholar]
- 50. Walker K, Olson MF. Targeting Ras and Rho GTPases as opportunities for cancer therapeutics. Curr Opin Genet Dev 2005; 15: 62–8. [DOI] [PubMed] [Google Scholar]
- 51. Zigmond SH. Formin‐induced nucleation of actin filaments. Curr Opin Cell Biol 2004; 16: 99–105. [DOI] [PubMed] [Google Scholar]
- 52. Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD. Drosophila Spire is an actin nucleation factor. Nature 2005; 433: 382–8. [DOI] [PubMed] [Google Scholar]