

Abstract
We previously reported that the telomere‐targeting drug telomestatin induces apoptosis accompanied by G‐tail reduction and dissociation of binding protein TRF2 from telomeres in cancer cell lines but not normal or human telomerase reverse transcriptase (hTERT)‐immortalized cells. Because telomere‐targeting drugs induce growth arrest in normal cells at higher doses, their development is dependent on the ability to predict toxicity before in vivo use, but no models for this are available. Here, we established two new cell lines, telomerase immortalized human fetal hepatocytes, Hc3716‐hTERT, and telomerase immortalized hepatic stellate cells, NPC‐hTERT. Examinations showed that Hc3716‐hTERT maintained normal mammalian cell morphology, cell growth, albumin expression, and wild‐type p53 responsiveness, whereas NPC‐hTERT maintained hepatic stellate‐like morphology, expression of hepatic stellate markers, α‐smooth muscle actin, and secretion of type I collagen, an extracellular matrix protein. Given our finding that telomere G‐tail length in Hc3716 cells was decreased in senescence and increased by hTERT infection, we next examined the effect of high‐dose telomestatin‐induced telomere dysfunction and G‐tail shortening on cellular functions in Hc3716‐hTERT cells. Interestingly, telomestatin decreased expression of cytochrome P450 (CYP) family members CYP3A3/4, CYP3A5, and CYP3A7, mRNA and induced albumin expression at both mRNA and protein levels. These gene expression responses to telomestatin were similar to those of the normal parental cell Hc3716. These established cell lines thus represent the first model for predicting the side‐effects of telomere‐targeting drugs in normal cells, and should be powerful tools in the development of these drugs. (Cancer Sci 2010)
Telomeres are special structures at the end of eukaryotic chromosomes that play a role in chromosome end protection.( 1 , 2 ) The first G‐rich telomere repeat sequences, 5′‐(TTGGGG)n‐3′, were discovered in Tetrahymena by Blackburn.( 3 ) Telomeric DNA in humans consists of 5′‐(TTAGGG)n‐3′ repeats, followed by a G‐rich single‐stranded 3′‐overhang, the so‐called telomere G‐tail.( 4 ) The telomere is gradually shortened with cell division due to problems with end‐replication. Telomere repeat sequences are synthesized by telomerase, a cellular ribonucleoprotein reverse transcriptase.( 5 ) Expression of the human telomerase catalytic subunit gene, hTERT, correlates with the presence of telomerase activity in human cells,( 6 , 7 ) and introduction of the hTERT gene alone into normal cells is sufficient to induce telomerase activity, followed by telomere elongation and cell immortalization, without damage to the genome.( 8 , 9 ) The status of changes in the G‐tail has been controversial, with one study reporting shortening at replicative senescence,( 10 ) and another reporting the maintenance of length in human fibroblasts at senescence.( 11 ) Our method to determine G‐tail length using the G‐tail telomere hybridization protection assay (HPA)( 12 ) showed that G‐tail length in normal fibroblasts and HUVEC cells gradually shortens with cell division, but that introduction of the hTERT gene induces G‐tail length elongation in these hTERT‐infected cells.( 13 ) However, little is known about G‐tail length alterations in human hepatocytes during replicative senescence or after hTERT infection.
G‐quadruplex structures are special secondary structures consisting of guanine‐rich DNA sequences such as telomeres and other important regulatory regions. Telomere G‐tails can adopt a G‐quadruplex conformation both in vitro and in vivo, and many leading compounds targeting G‐quadruplex structures have been reported to have anticancer effects. Telomestatin, originally isolated as a highly potent telomerase inhibitor from Streptomyces anulatus 3533‐SV4,( 14 ) enhances the stabilization of telomeric G‐quadruplex DNA,( 15 ) and has 70‐fold higher selectivity for G‐quadruplex DNA over duplex DNA. Telomestatin induces telomere dysfunction through t‐loop destruction, and might therefore be useful in determining G‐tail function.
We previously reported that telomestatin rapidly induces apoptosis in cancer cells at concentrations that do not cause normal cells to die, and that this process is accompanied by dissociation of telomere‐binding protein TRF2 from telomeres in cancer cells.( 16 ) These characteristics may suggest the potential of telomestatin as a telomere‐targeting anticancer drug. Although little is known about its effects in normal cells, higher doses induce cell growth arrest. Given that the telomere capping structure, the t‐loop, is essential to chromosome maintenance as well as genomic stability in both normal and cancer cells, the ongoing development of telomere‐targeting anticancer drugs depends on the availability of new cellular models able to evaluate the characteristics of normal cells under the induction of telomere dysfunction using higher doses of G‐quadruplex DNA stabilizers, such as telomestatin. The many types of normal cultured cells now available are not suitable for this use because of their relatively rapid mortality and accompanying loss of normal functions, and new models have thus been sought.
Cultured human hepatocytes have broad research and clinical potential. Although several published works have reported long‐term culture, this is generally considered difficult in both rodent and human hepatocytes.( 17 , 18 ) Several recent studies succeeded in establishing immortalized human hepatocytes by introduction of the hTERT gene.( 19 , 20 ) However, the relation between cell proliferation and G‐tail length and cellular function in hTERT‐immortalized human hepatocytes is still unknown. Furthermore, little is known about the effect of telomere‐targeting drugs on the cellular function of these cell lines and their usefulness for toxicity testing, which may assist the prediction of side‐effects in vivo.
Here, we established immortalized fetal hepatocyte and hepatic stellate cell lines using a retroviral vector expressing only hTERT. We then investigated the cellular function of these hTERT immortalized hepatocytes, as well as the effect of telomestatin as a model telomere‐targeting drug on cellular function, including cell growth and hepatic functions such as expression of the cytochrome P450 (CYP) superfamily and albumin (ALB).
Materials and Methods
Cell culture and drug treatment. Human fetal hepatocytes (Hc3716) were obtained from Applied Cell Biology Research Institute (Kirkland, WA, USA) (Data S1).
hTERT gene transduction by retroviral method. To produce retrovirus supernatants, pMSCV‐puro‐hTERT or pFB‐neo‐hTERT retrovirus constructs were transfected into the PT67 packaging cell line (Takara Bio USA, Madison, WI, USA) with the FuGENE6 transfection reagent (Roche Diagnostics, Mannheim, Germany). After 2 days, the supernatants were collected and passed through a 0.22 μm filter (Millipore, Billerica, MA, USA) after adding polybrene at a final concentration of 6 μg/mL. Filtered supernatants were then used to infect the target cell. Retrovirus supernatants from pMSCV‐puro‐hTERT were used to infect human fetal Hc3716 hepatocytes. Retrovirus supernatants from pFB‐neo‐hTERT were used to infect non‐parenchymal cells (NPC). After 24 h of incubation with these viruses, the medium was replaced with fresh complete medium containing puromycin (0.7 μg/mL) or G‐418 (600 μg/mL).
Telomerase assay. Telomerase activity was measured by modified telomere repeat amplification protocol (TRAP) as described previously Data S1.( 21 )
Telomere length assay by Southern blot analysis. Genomic DNA was purified from cells using phenol–chloroform extraction. Purified DNA was then digested with Hinf I restriction enzyme, and run on 0.9% agarose gel with 0.5 × TBE buffer. Telomere length was measured using the TeloTAGGG telomere length assay kit according to the manufacturer’s instructions (Roche Diagnostics).
Quantification of telomere length and telomere G‐tail length by G‐tail telomere hybridization protection assay. G‐tail length was measured using the G‐tail telomere hybridization protection assay.( 12 ) Quantification of total telomere length (both double‐stranded and single‐stranded) was carried out using telomere HPA methods as described previously.( 22 ) For the telomere G‐tail assay, 5 μg non‐denatured DNA was used to measure the G‐tail, and 0.5 μg denatured DNA was used to measure total telomere length. To normalize the luminescence of each sample, we took 1 μL from each sample tube and measured the DNA amount using NanoDrop (ND‐1000; Thermo Fisher Scientific Inc., Waltham, MA, USA). Probes for AE‐labeling of telomeres were supplied by Fujirebio (Tokyo, Japan).
RNA isolation and RT‐PCR. Total RNA was extracted using TRIzol reagent (Life Technologies Co., Carlsbad, CA, USA) in accordance with the manufacturer’s instructions Data S1.
Western blot analysis. Monoclonal antibodies against human proteins and their dilution were as follows: ALB (1:1000) (Sigma, Aldrich Co., St Louis, MO, USA); α‐fetoprotein (AFP; 1:1000) (Sigma); cytokeratin8 (CK8; 1:1000) (Sigma); cytokeratin18 (CK18; 1:4000) (Sigma); α‐smooth muscle actin (αSMA; 1:1000) (Sigma); p53 (1:400) (Upstate Biotechnology, Lake Placid, NY, USA); p21 (1:2000) (Upstate Biotechnology, Millipore, Billerica, MA, USA); and β‐actin (1:10 000) (Sigma). The polyclonal antibody was cyclin dependent kinase4 (CDK4) (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) Data S1.
Results
Establishment of hTERT‐infected human hepatocytes and NPC. To establish hTERT immortalized hepatocytes and NPC, hTERT retrovirus was infected into human Hc3716 hepatocytes at nine population doubling levels (PDL), and NPC cells at eight PDL. The resulting antibiotic‐resistant cells were obtained as hTERT hepatocytes and hTERT‐NPC cells. These hTERT‐infected cells, Hc3716‐hTERT and NPC2‐hTERT, showed high levels of hTERT mRNA using RT‐PCR (Fig. 1a). Evaluation of telomerase activity by a modified TRAP assay (Fig. 1b) showed that control Hc3716 (six PDL) cells had low telomerase activity, whereas hTERT‐infected cells showed strong activity. Having established that hTERT infection successfully reconstituted telomerase activity in Hc3716‐hTERT and NPC‐hTERT cells, we assessed telomere length by Southern blot analysis (Fig. 1c). Terminal restriction fragment (TRF) length analysis indicated that telomeres were lengthened in the hTERT‐infected cells compared with non‐infected normal cells (Fig. 1c). Although telomere length is known to decrease with cellular senescence in human hepatocytes, technical difficulties have prevented close investigation of G‐tail length, and to our knowledge no studies have been reported. We therefore measured telomere G‐tail length using our novel, previously developed method, G‐tail telomere HPA.( 12 ) Results showed that telomere G‐tail length decreased with senescence, but was elongated in hTERT‐infected Hc3716 cells (Fig. 1d).
Figure 1.
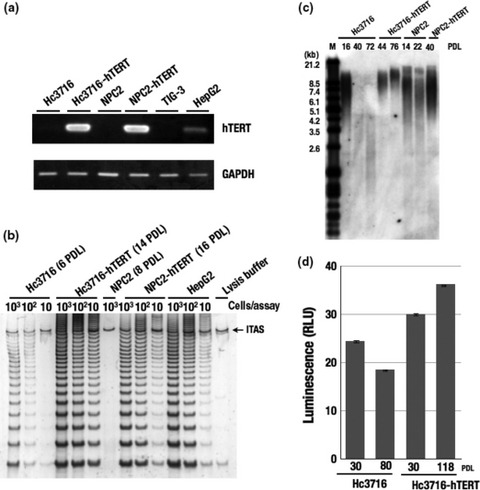
Telomerase activity and human telomerase reverse transcriptase (hTERT) mRNA expression in hTERT‐infected cells. (a) Expression of hTERT mRNA by RT‐PCR in parental cells and hTERT‐infected cells. GAPDH was used as an internal control. Hc3716, human fetal hepatocytes; HepG2, a telomerase‐positive hepatoma cell line used as a control; NPC2, non‐parenchymal cells; TIG‐3, telomerase‐negative normal human fetal fibroblasts. (b) Telomerase activity was measured by telomere repeat amplification protocol assay. ITAS, internal telomerase assay standard. (c) Telomere length analysis in hTERT‐infected cells. A Southern blot analysis for terminal‐restriction fragment TRF was carried out on parental and hTERT‐infected cells at different population doubling levels (PDL). M, marker. (d) Telomere G‐tail length in Hc3716 and Hc3716‐hTERT cells. Hc3716 (30 PDL) was used for young cells, and Hc3716 (80 PDL) for senescent cells. RLU, relative light unit.
Growth characteristics and cell morphology in hTERT hepatocytes and hTERT NPC cells. We first cultured human fetal hepatocytes with a Hepatocyte Medium BulletKit (Cambrex Co., Charles City, IA, USA) containing 10% FBS but without human serum (HS). These divided for several passages only and then rapidly stopped proliferating, before finally detaching from the dish. After optimization for O2 concentration, culture dish, feeder layer culture, and serum species, we found that human serum was the key factor for the maintenance of human hepatocytes. From these trials we formulated a culture medium that effectively supports the growth of human hepatocytes, which we termed the “hepatocytes medium kit” containing 5% FBS and 10% HS.
To examine the ability of Hc3716‐hTERT and NPC‐hTERT to bypass replicative senescence, we cultured control Hc3716, Hc3716‐hTERT, control NPC, and NPC‐hTERT with 5% FBS and 10% HS culture medium. After 86–90 PDL, cell proliferation virtually ceased in control Hc3716 with entry into replicative senescence (Fig. 2a). When cultured without 10% HS, the proliferation potential of Hc3716 and Hc3716‐hTERT dramatically decreased, and finally the cells stopped growing (data not shown). In contrast, Hc3716‐hTERT grew at a similar rate to Hc3716 and actively doubled to the last count of the study at day 800 (population doubling approximately every 2.5 days).
Figure 2.
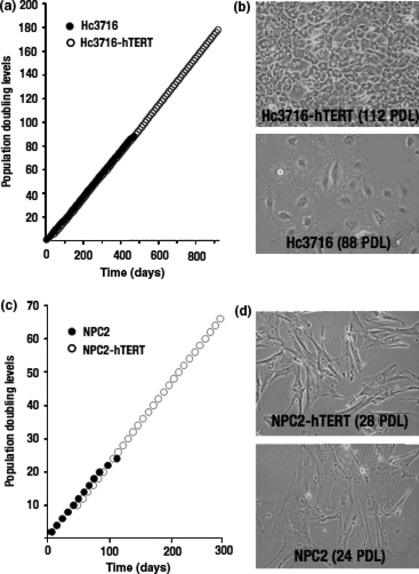
Morphology and growth curve of parental cells and human telomerase reverse transcriptase (hTERT)‐infected cells. (a) Cumulative growth curve in Hc3716 human fetal hepatocytes and Hc3716‐hTERT. (b) Morphology of Hc3716 [88 population doubling levels (PDL); senescent state] and Hc3716‐hTERT (112 PDL). (c) Cumulative growth curve in normal non‐parenchymal cells (NPC2) and NPC2‐hTERT. (d) Morphology of NPC2 (24 PDL; senescent state) and NPC2‐hTERT (28 PDL).
Hc3716 showed a typical cobblestone‐like morphology when in a confluent monolayer (Fig. 2b). The cells became flattened and enlarged as the number of passages increased, particularly after 86 PDL. Hc3716‐hTERT showed a similar morphology in a confluent monolayer and appeared to be in a state of contact inhibition of growth, because they did not pile up on each other.
Cell proliferation virtually ceased in control NPC after 30 PDL. In NPC‐hTERT, however, proliferation continued for more than 50 PDL (approximately one population doubling every 4 days) (Fig. 2c). NPC‐hTERT appeared closely similar to non‐infected cells and NPC cells and NPC‐hTERT showed a typical spindled morphology (Fig. 2d). Taken together, these characteristics, including cell growth, contact inhibition, and cell morphology, showed that the Hc3716‐hTERT and NPC‐hTERT lines were normal hepatocytes and NPC‐like cells, respectively, with normal cell‐like characteristics.
Hc3716‐hTERT expressed ALB but not AFP. To determine whether Hc3716‐hTERT had the functional properties of hepatocytes, we examined the expression of several hepatic marker proteins, ALB, AFP, CK8, and CK18 by Western blot analysis. AFP and ALB are the first secreted proteins produced by the embryonic liver, whereas CK8 and CK18 are expressed in adult hepatocytes. To examine ALB expression, cells were washed with PBS three times to remove human ALB derived from HS, cultured in media without HS for 3 days, and detected by Western blot analysis. Hc3716‐hTERT (40 PDL) expressed much more ALB than control Hc3716 (40 PDL) (Fig. 3a). AFP expression was not detected in either Hc3716‐hTERT or Hc3716, whereas the hepatic cancer cell line HepG2 expressed AFP at high levels (Fig. 3b). Hc3716‐hTERT (40 PDL) as well as control Hc3716 also expressed either CK8 or CK18 but not AFP, a marker of immature or tumorous hepatocytes such as HepG2. NPC‐hTERT did not express CK8 or CK18 (Fig. 3c).
Figure 3.
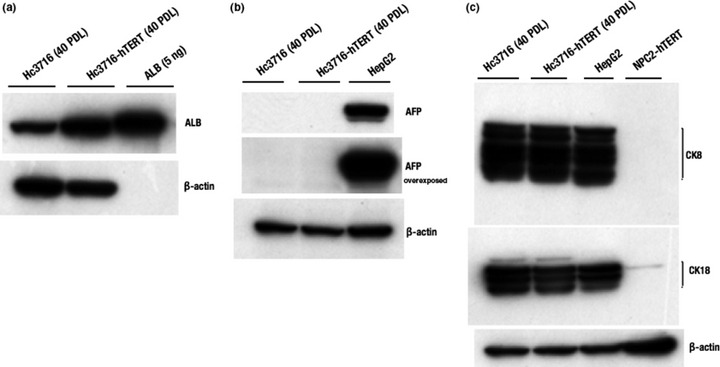
Characterization of human fetal hepatocytes (Hc3716) and telomerase immortalized human fetal hepatocytes (Hc3716‐hTERT). (a) Protein expression of albumin (ALB) in Hc3716 and Hc3716‐hTERT were examined by Western blot analysis. Purified ALB (5 ng) was used as a positive control. (b) Protein expression of α‐fetoprotein (AFP) in Hc3716 and Hc3716‐hTERT was examined by Western blot analysis. HepG2, hepatoma cell line used as a positive control. (c) Protein expression of cytokeratin8 (CK8) and cytokeratin18 (CK18) in Hc3716 and Hc3716‐hTERT were examined by Western blot analysis. β‐actin was used as a loading control.
We also examined the expression of hepatocyte nuclear factor (HNF)4 and CYP3A7 mRNA by RT‐PCR (Fig. 4a). Hepatocyte nuclear factor 4 is well known as a liver‐enriched differentiation factor, whereas CYP3A7, originally isolated from fetal liver,( 23 ) accounts for up to 50% of total fetal hepatic CYP content.( 24 ) Although expression of HNF4 and CYP3A7 was not detected in proliferating states, both mRNA were induced when the cells reached confluence (Fig. 4a). Moreover, CYP3A4 and CYP3A7 are known to be induced by nifedipine and rifampicin. Induction of CYP3A4 and CYP3A7 by these compounds was partial in normal Hc3716 cells, but both CYP3A4 and CYP3A7 were upregulated in Hc3716‐hTERT (Fig. 4b), suggesting that Hc3716‐hTERT maintains normal hepatocyte characteristics.
Figure 4.
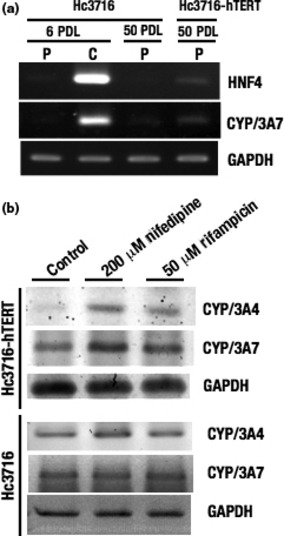
(a) mRNA expression of hepatocyte nuclear factor 4 (HNF4) and cytochrome P450 (CYP)/3A7 in normal human fetal hepatocytes (Hc3716) and those infected with human telomerase reverse transcriptase (Hc3716‐hTERT) were examined by RT‐PCR. C, confluent monolayers; P, proliferative phase. (b) Induction of CYP/3A4 and CYP/3A7 after treatment with nifedipine and rifampicin in Hc3716 and Hc3716‐hTERT was examined by RT‐PCR. GAPDH was used as an internal control. Control, no treatment.
Characterization of NPC‐hTERT. To determine the characteristics of NPC‐hTERT, we examined the expression of αSMA, a marker for activated hepatic stellate cells (HSC) by Western blot analysis. Human HSC strain LI90, established from a mesenchymal liver tumor of a 55‐year‐old Japanese woman,( 25 ) was used as the positive control in the present study. NPC‐hTERT (26 PDL) as well as untransduced NPC (14 PDL) also expressed αSMA, whereas Hc3716‐hTERT (40 PDL) did not (Fig. 5a).
Figure 5.
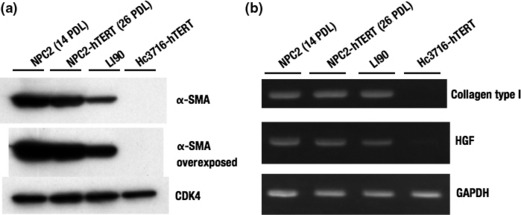
(a) Characterization of normal non‐parenchymal cells (NPC2) and those infected with human telomerase reverse transcriptase (NPC2‐hTERT). Protein expression of α‐smooth muscle actin (α‐SMA) in NPC2 and NPC2‐hTERT were examined by Western blot analysis. Cyclin dependent kinase 4 (CDK4) was used as an internal control. LI90, hepatic stellate cell line derived from mesenchymal liver tumor. (b) mRNA expression of collagen type I and hepatocyte growth factor (HGF) in NPC2 and NPC2‐hTERT was examined by RT‐PCR. GAPDH was used as an internal control.
Hepatic stellate cells constitute a major cell type responsible for liver fibrosis following their activation into fibrogenic myofibroblast‐like cells.( 26 , 27 , 28 , 29 ) Activated HSC are considered the major source of extracellular matrix in hepatic fibrosis.( 30 ) Following liver injury, HSC undergo an activation process in which quiescent vitamin A storing cells transform into proliferative, smooth muscle actin‐positive myofibroblast‐like cells that secrete extracellular matrix proteins, especially type I collagen.( 26 ) We examined the expression of collagen type I mRNA and HGF mRNA in NPC‐hTERT (26 PDL) by RT‐PCR and compared it with NPC (14 PDL) (Fig. 5b). Expression levels of collagen type I mRNA and HGF mRNA in NPC‐hTERT (26 PDL) were similar to those of NPC (14 PDL), whereas Hc3716‐hTERT did not express collagen type I mRNA. From these results, we identified NPC and NPC‐hTERT as HSC.
HC3716‐hTERT cells maintain p53‐dependent DNA damage signals, similar to normal Hc3716 hepatocytes. To study the molecular basis of DNA damage response to topoisomerase poison, normal and hTERT‐infected cells were treated with the indicated concentrations of adriamycin for 24 h. p53 proteins were accumulated in response to adriamycin (a topoisomerase II inhibitor) in a dose‐dependent manner in both normal and hTERT‐infected cells, including Hc3716‐hTERT and NPC‐hTERT (Fig. 6). As expected, the p53 target, cyclin‐dependent kinase inhibitor p21waf1/cip1/sdi1, was also induced by adriamycin in hTERT‐infected cells (Fig. 6). We found that Hc3716‐hTERT and NPC‐hTERT maintained wild‐type p53.
Figure 6.
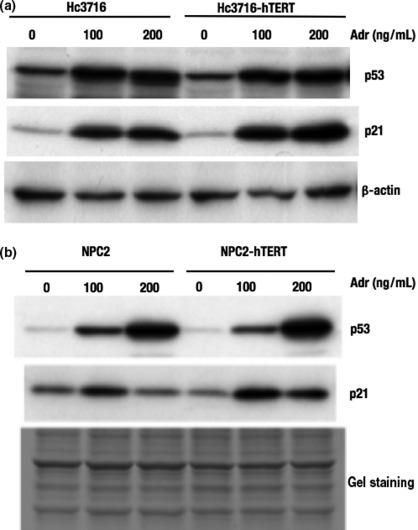
DNA damage response to topoisomerase poison in human telomerase reverse transcriptase (hTERT)‐infected cells. (a) Accumulation of p53 and induction of p21 in normal (Hc3716) and infected (Hc3716‐hTERT) human fetal hepatocytes were examined by Western blot analysis. Adr, adriamycin. (b) Accumulation of p53 and induction of p21 in normal (NPC2) and infected (NPC2‐hTERT) non‐parenchymal cells were examined by Western blot analysis. Gel staining, loading control.
Telomestatin inhibits cell growth in Hc3716‐hTERT cells. Telomestatin is known to induce telomere dysfunction by destruction of the telomere t‐loop structure through binding to the G‐quadruplex structure of the G‐tail. We reported that cancer cell lines were sensitive to apoptosis by telomestatin treatment.( 16 ) Although the mechanism of this difference in sensitivity between normal cells and a cancer cell line is still unclear, higher doses of telomestatin treatment might induce telomere dysfunction, accompanied by G‐tail reduction. To examine the effect of telomere dysfunction on cellular functions of Hc3716‐hTERT, we used telomestatin as an inducer of telomere dysfunction through G‐tail reduction. To determine the effects of telomestatin on cell growth, Hc3716‐hTERT cells were cultured in the presence of telomestatin at concentrations of 0, 5, and 25 μM for 5 days. When Hc3716‐hTERT cells were treated with telomestatin at 25 μM, the cells ceased growing and cell death was induced. G‐tail lengths were examined by G‐tail telomere HPA (Fig. 7a). Total telomere length was also examined using HPA assay with the denaturation of genomic DNA before assay (Fig. 7b). When Hc3716‐hTERT cells were treated with telomestatin at 25 μM for 5 days, a remarkable reduction in G‐tails was observed (Fig. 7a). We also measured total telomere length using the telomere HPA. Results showed no significant difference in total telomere length after treatment with telomestatin for 5 days (Fig. 7b), which is similar to the findings with cancer cells. Telomestatin did not affect total telomere length, but G‐tail lengths were decreased by treatment with telomestatin at 25 μM in Hc3716‐hTERT cells, suggesting that high doses of telomestatin can induce telomere dysfunction through G‐tail reduction in Hc3716‐hTERT.
Figure 7.
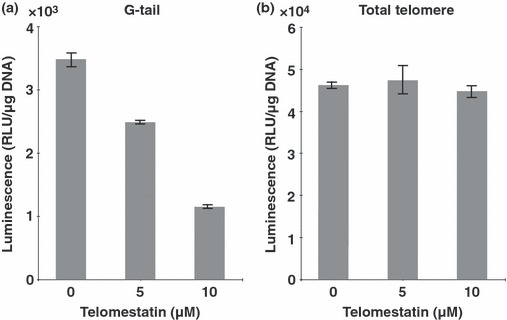
Telomere G‐tail length and total telomere length after treatment with telomestatin in telomerase immortalized human fetal hepatocytes (Hc3716‐hTERT). (a) Telomere G‐tail length in Hc3716‐hTERT cells were examined by G‐tail telomere hybridization protection assay. Each chemiluminescent value was calibrated with DNA content used in the assay. RLU, relative light unit. (b) Total telomere length in Hc3716‐hTERT cells was examined using the telomere hybridization protection assay method. Denatured DNA (0.5 μg) was used in each assay.
Hc3716‐hTERT cells showed similar characteristics of CYP expression in response to telomestatin. To determine whether G‐tail length affects the function of Hc3716‐hTERT cells, expression of ALB and the CYP family was examined by RT‐PCR and Western blot analysis after treatment with telomestatin at 0, 5, and 25 μM. Unexpectedly, the ALB expression level increased with increasing telomestatin concentration at both the mRNA and protein levels (Fig. 8a). This ALB induction was also observed in parental Hc3716 cells at the mRNA level, suggesting that ALB induction by telomestatin is not due to introduction of the hTERT gene or elongation of telomere length. We examined CYP family expression after treatment with telomestatin in Hc3716 and Hc3716‐hTERT cells, including CYP1A2, CYP2B6, CYP2C, CYP2D6, CYP2E1, CYP3A3/4, CYP3A5, and CYP3A7. Interestingly, mRNA of CYP1A2, CYP2B6, CYP2C, CYP2D6, and CYP2E1 was strongly induced in telomestatin‐treated Hc3716‐hTERT cells (Fig. 8b, left). In contrast, expression of CYP3A3/4, CYP3A5, and CYP3A7 was decreased in these cells (Fig. 8b, left), which is opposite to the effects of treatment with nifedipine or rifampicin (Fig. 4b). This CYP family response to telomestatin is similar to that in telomestatin‐treated Hc3716 parental cells (Fig. 8b, right). Further examination of the molecular mechanism of these responses of the CYP family is required. CYP expression was clearly altered by telomestatin treatment, and CYP responses to telomestatin were similar to those of parental Hc3716 cells. Although concerns have been expressed that hTERT expression might affect the metabolism of compounds, no other immortalized cell line bearing characteristics of normal mammalian cells are available for use in the prediction of drug cytotoxicity. Taken together, these results suggest that our established cell line, Hc‐3716‐hTERT, represents a suitable tool for the prediction of drug cytotoxicity in the development of telomere‐targeting anticancer drugs.
Figure 8.
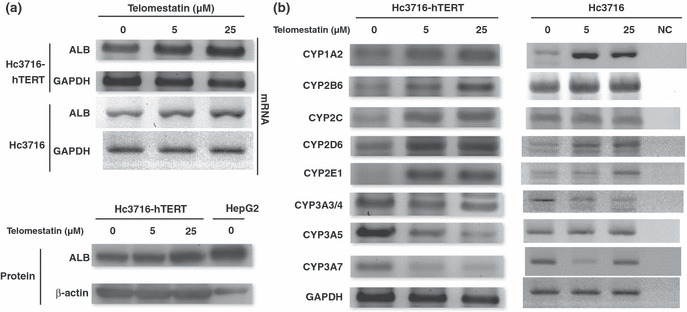
(a) Expression of albumin (ALB) after treatment with telomestatin in normal (Hc3716) and telomerase immortalized (Hc3716‐hTERT) human fetal hepatocytes examined by RT‐PCR. Albumin protein levels in Hc3716‐hTERT were examined by Western blot analysis. HepG2, hepatoma cell line used as a positive control. (b) Cytochrome P450 (CYP) induction after treatment with telomestatin in Hc3716‐hTERT and HC3716 cells was examined by RT‐PCR. GAPDH was used as an internal control. NC, no RNA sample as negative control.
Discussion
In this study, we established human hepatocyte immortalized cells, HC3716‐hTERT, and human hepatic satellite cells, NPC‐hTERT, which bear characteristics of normal cells. In HC3716‐hTERT, the telomere‐targeting anticancer drug telomestatin decreased CYP3A3/4, CYP3A5, and CYP3A7 expression and induced ALB expression at both mRNA and protein levels, similar to the responses seen in the normal parental cell Hc3716. These findings suggest that the combination of telomestatin and Hc3716‐hTERT might represent a useful new cellular model to evaluate the characteristics of normal cells, and to predict the toxicity of drugs under induction of telomere dysfunction.
Given that inappropriate culture induces senescence programming in human cells, we first optimized the culture medium for the growth and maintenance of human fetal hepatocytes by varying the concentration of FBS. The resulting culture medium contained 5% FBS and 10% HS, and effectively supported the growth of human fetal hepatocytes. Results clearly showed that normal fresh HS promoted the growth of human fetal hepatocytes and extended their lifespan to over 80 PDLs, compared to 10 PDLs with normal culture conditions (FBS alone). Hepatocytes finally reached senescence at 80 PDLs, at which time analysis of TRF and G‐tail HPA length showed shortening of both. The average TRF in senescent Hc3716 cells with optimal culture medium was approximately 2.5 kb, which appears to be the minimum length required for proper chromosome maintenance.
We combined the use of these optimal culture conditions with forced expression of hTERT and subsequent telomerase activity to establish and immortalize the Hc3716‐ and NPC‐based cell lines Hc3716‐hTERT and NPC‐hTERT. After several population doublings, TRF length and HPA analysis showed that the telomeres and G‐tails in these hTERT‐transduced cells were lengthened (Fig. 1c). This finding indicated that these cells were suitable for the evaluation of telomere‐targeting drugs.
Although these hTERT‐infected cells have an immortal phenotype, several important phenotypic characteristics of the parental cell were retained. For example, expression of CK8, CK18, CYP family, and ALB were seen in both Hc3716 and Hc3716‐hTERT cells, but not in NPC‐hTERT. The hTERT‐infected cells did not show typical oncogenic phenotype traits such as anchorage‐independent growth, contact inhibition, pile‐up, or expression of mutant p53 protein. Cellular functions examined through the gene expression of hepatic markers such as CYP, CK, and ALB were more strongly maintained in the Hc3716‐hTERT cells than in the parental cells, which was expected given that hepatic differential function generally decreases with proliferative aging in normal hetatocytes. Hc3716‐hTERT maintained the cellular functioning of young normal hepatocytes regarding CYP, CK, and ALB expression.
NPC‐hTERT showed the differential functions of HSC, including expression of collagen type I and HGF mRNA (Fig. 5b). Co‐culture of Hc3716‐hTERT with NPC‐hTERT will help to maintain the differentiated phenotype of hepatocytes in vitro, for example, with regard to ALB and CYP.( 31 ) NPC‐hTERT cells can therefore be used for basic research into hepatic fibrosis.
The clinical use of telomere‐targeting drugs in cancer therapy requires an understanding of their effects in normal cells, but to our knowledge, no such study or model has been reported. In this paper, we examined cell growth and hepatic function, including ALB and CYP expression, with or without telomestatin treatment, in Hc3716‐hTERT cells. At higher concentrations of telomestatin, Hc3716‐hTERT cells ceased proliferation, accompanied by G‐tail reduction. Interestingly, total telomere length was not affected by treatment with telomestatin, suggesting that its antiproliferative effect in normal hepatocytes was induced by telomere t‐loop destruction accompanied by G‐tail reduction. Although we expected that ALB expression, a hallmark of hepatocytes, would be decreased by the telomere dysfunction induced by higher doses of telomestatin, we unexpectedly found that expression was increased with higher doses in hTERT hepatocytes. Although we did not undertake an in vivo study, we speculate that if telomestatin and other telomere‐targeting drugs induce ALB expression in normal hepatocytes and secretion into blood, then the ALB level might be a candidate marker of telomere dysfunction of normal heptocytes.
The CYP family of proteins is large and ubiquitous. The proteins catalyze a multitude of reactions, including oxidation, hydroxylation, and conjugation. Depending on the specific CYP and compound, these CYP‐dependent reactions may convert active compounds to the inactive state, or vice versa; for example, a pro‐carcinogen to an active carcinogen, or a pro‐drug to an active drug. Therefore, even though anticancer drugs significantly inhibit the growth of cancer, CYP‐induced changes should be examined before in vivo application. The two models developed here might be helpful for this purpose. For example, more than 30 known pharmaceutical drugs are known to be metabolized by CYP2D6; if telomestatin is metabolized by CYP2D6, and thereby loses its antitumor activity, telomestatin might not suitable for in vivo application, owing to the CYP2D6 expression it induces (Fig. 8b).
Several limitations of this study warrant mention. First, CYP expression varies with race and sex, but we did not investigate these variables in the development of these models. Second, the effects of hTERT infection are not completely predictable. This unpredictability might hamper efforts to replicate our present results. Finally, the response of genes in these in vitro cultured cells are different from in vivo responses. Thus, further investigation is required to establish conclusively the usefulness of these cell lines in evaluating the toxicity of telomere‐targeting drugs.
In conclusion, these new predictive models of toxicity are promising tools in the development of new telomere‐targeting anticancer drugs before application to in vivo studies.
Acknowledgments
This work was partially supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan, with a Grant‐in‐Aid for Scientific Research on Priority Areas (Grant No. 20014015), and the Takeda Science Foundation.
Supporting information
Data S1. Material and methods.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
References
- 1. De Lange T. T‐loops and the origin of telomeres. Nat Rev Mol Cell Biol 2004; 5: 323–9. [DOI] [PubMed] [Google Scholar]
- 2. De Lange T. Protection of mammalian telomeres. Oncogene 2002; 21: 532–40. [DOI] [PubMed] [Google Scholar]
- 3. Szostak JW, Blackburn EH. Cloning yeast telomeres on linear plasmid vectors. Cell 1982; 29: 245–55. [DOI] [PubMed] [Google Scholar]
- 4. Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 1997; 88: 657–66. [DOI] [PubMed] [Google Scholar]
- 5. Greider CW. Telomere length regulation. Annu Rev Biochem 1996; 65: 337–65. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura TM, Morin GB, Chapman KB et al. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997; 277: 955–9. [DOI] [PubMed] [Google Scholar]
- 7. Nakayama J, Tahara H, Tahara E et al. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet 1998; 18: 65–8. [DOI] [PubMed] [Google Scholar]
- 8. Carney SA, Tahara H, Swartz CD et al. Immortalization of human uterine leiomyoma and myometrial cell lines after induction of telomerase activity: molecular and phenotypic characteristics. Lab Invest 2002; 82: 719–28. [DOI] [PubMed] [Google Scholar]
- 9. Bodnar AG, Ouellette M, Frolkis M et al. Extension of life‐span by introduction of telomerase into normal human cells. Science 1998; 279: 349–52. [DOI] [PubMed] [Google Scholar]
- 10. Stewart SA, Ben‐Porath I, Carey VJ, O’Connor BF, Hahn WC, Weinberg RA. Erosion of the telomeric single‐strand overhang at replicative senescence. Nat Genet 2003; 33: 492–6. [DOI] [PubMed] [Google Scholar]
- 11. Chai W, Shay JW, Wright WE. Human telomeres maintain their overhang length at senescence. Mol Cell Biol 2005; 25: 2158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tahara H, Kusunoki M, Yamanaka Y, Matsumura S, Ide T. G‐tail telomere HPA: simple measurement of human single‐stranded telomeric overhangs. Nat Methods 2005; 2: 829–31. [DOI] [PubMed] [Google Scholar]
- 13. Anno K, Hayashi A, Takahashi T, Mitsui Y, Ide T, Tahara H. Telomerase activation induces elongation of the telomeric single‐stranded overhang, but does not prevent chromosome aberrations in human vascular endothelial cells. Biochem Biophys Res Commun 2007; 353: 926–32. [DOI] [PubMed] [Google Scholar]
- 14. Shin‐ya K, Wierzba K, Matsuo K et al. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J Am Chem Soc 2001; 123: 1262–3. [DOI] [PubMed] [Google Scholar]
- 15. Tauchi T, Shin‐Ya K, Sashida G et al. Activity of a novel G‐quadruplex‐interactive telomerase inhibitor, telomestatin (SOT‐095), against human leukemia cells: involvement of ATM‐dependent DNA damage response pathways. Oncogene 2003; 22: 5338–47. [DOI] [PubMed] [Google Scholar]
- 16. Tahara H, Shin‐Ya K, Seimiya H, Yamada H, Tsuruo T, Ide T. G‐Quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric overhang in cancer cells. Oncogene 2006; 25: 1955–66. [DOI] [PubMed] [Google Scholar]
- 17. Lazaro CA, Croager EJ, Mitchell C et al. Establishment, characterization, and long‐term maintenance of cultures of human fetal hepatocytes. Hepatology 2003; 38: 1095–106. [DOI] [PubMed] [Google Scholar]
- 18. Hino H, Tateno C, Sato H et al. A long‐term culture of human hepatocytes which show a high growth potential and express their differentiated phenotypes. Biochem Biophys Res Commun 1999; 256: 184–91. [DOI] [PubMed] [Google Scholar]
- 19. Wege H, Le HT, Chui MS et al. Telomerase reconstitution immortalizes human fetal hepatocytes without disrupting their differentiation potential. Gastroenterology 2003; 124: 432–44. [DOI] [PubMed] [Google Scholar]
- 20. Tsuruga Y, Kiyono T, Matsushita M et al. Establishment of immortalized human hepatocytes by introduction of HPV16 E6/E7 and hTERT as cell sources for liver cell‐based therapy. Cell Transplant 2008; 17: 1083–94. [DOI] [PubMed] [Google Scholar]
- 21. Tahara H, Nakanishi T, Kitamoto M et al. Telomerase activity in human liver tissues: comparison between chronic liver disease and hepatocellular carcinomas. Cancer Res 1995; 55: 2734–6. [PubMed] [Google Scholar]
- 22. Nakamura Y, Hirose M, Matsuo H, Tsuyama N, Kamisango K, Ide T. Simple, rapid, quantitative, and sensitive detection of telomere repeats in cell lysate by a hybridization protection assay. Clin Chem 1999; 45: 1718–24. [PubMed] [Google Scholar]
- 23. Kitada M, Kamataki T, Itahashi K, Rikihisa T, Kato R, Kanakubo Y. Purification and properties of cytochrome P‐450 from homogenates of human fetal livers. Arch Biochem Biophys 1985; 241: 275–80. [DOI] [PubMed] [Google Scholar]
- 24. Wrighton SA, Vandenbranden M. Isolation and characterization of human fetal liver cytochrome P450HLp2: a third member of the P450III gene family. Arch Biochem Biophys 1989; 268: 144–51. [DOI] [PubMed] [Google Scholar]
- 25. Murakami K, Abe T, Miyazawa M et al. Establishment of a new human cell line, LI90, exhibiting characteristics of hepatic Ito (fat‐storing) cells. Lab Invest 1995; 72: 731–9. [PubMed] [Google Scholar]
- 26. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000; 275: 2247–50. [DOI] [PubMed] [Google Scholar]
- 27. Rockey DC. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin Liver Dis 2001; 21: 337–49. [DOI] [PubMed] [Google Scholar]
- 28. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis 2001; 21: 351–72. [DOI] [PubMed] [Google Scholar]
- 29. Xu L, Hui AY, Albanis E et al. Human hepatic stellate cell lines, LX‐1 and LX‐2: new tools for analysis of hepatic fibrosis. Gut 2005; 54: 142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maher JJ, McGuire RF. Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest 1990; 86: 1641–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bhatia SN, Balis UJ, Yarmush ML, Toner M. Effect of cell‐cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells. FASEB J 1999; 13: 1883–900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Material and methods.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item