

Abstract
Anti‐epidermal growth factor receptor (EGFR) monoclonal antibody, cetuximab, is a promising targeted drug for EGFR‐expressing tumors. Glioblastomas frequently overexpress EGFR including not only the wild type but also a deletion mutant form called ‘variant III (vIII)’, which lacks exon 2–7, does not bind to ligands, and is constitutively activated. In this study, we investigated the antitumor activity of cetuximab against malignant glioma cells overexpressing EGFRvIII. For this purpose, we transfected human malignant glioma cell lines with the retroviral vector containing cDNA for EGFRvIII, and analyzed the mode of cetuximab‐induced action on the EGFRvIII in the cells. Immunoprecipitation and immunofluorescence revealed binding of cetuximab to EGFRvIII. Notably, immunoblotting analyses showed that cetuximab treatment resulted in reduced expression levels of the EGFRvIII. However, cetuximab alone did not exhibit a growth‐inhibitory effect against the EGFRvIII‐expressing cells. On the other hand, an assay for antibody‐dependent cell‐mediated cytotoxicity (ADCC) demonstrated cetuximab‐induced cytolysis in the presence of human peripheral blood mononuclear cells in a dose‐dependent manner. These results suggest that deletion mutant EGFRvIII can be a target of cetuximab and that ADCC activity substantially contributes to the antitumor efficacy of cetuximab against the EGFRvIII‐expressing glioma cells. Thus, cetuximab could be a promising therapy in malignant gliomas that express EGFRvIII. (Cancer Sci 2008; 99: 2062–2069)
Long‐term survival of patients with malignant gliomas has not substantially improved despite aggressive multimodality treatments including cytoreductive surgery, radiotherapy, and cytotoxic chemotherapy. To efficiently suppress these tumors, additional therapeutic strategies are necessary. Understanding the molecular genetics, biology, and immunology of gliomas will enable the potential development of new adjuvant treatments for malignant glioma patients.
Malignant gliomas may arise via a heterogeneous process resulting from multiple genetic alterations.( 1 ) One of the well‐known molecular features of gliomas is amplification of the epidermal growth factor receptor (EGFR) gene, leading to overexpression of this receptor in approximately 40–60% of glioblastomas.( 2 , 3 ) High levels of EGFR expression have been shown to be correlated with malignant progression in gliomas and associated with a poor prognosis and resistance to therapies.( 4 ) Therefore, therapeutic strategies directed against the EGFR may have potential in these malignancies.
In the EGFR‐amplified tumors, multiple types of EGFR mutations can be detected as a result of intragene deletions.( 2 ) The most frequent mutation in malignant gliomas is EGFR variant III (EGFRvIII), characterized by a consistent and tumor‐specific in‐frame deletion of 801 base pairs from the coding sequence of the extracellular domain.( 5 , 6 ) This mutated gene encodes a protein with a ligand‐independent and constitutively active tyrosine kinase domain, which greatly enhances the tumorigenicity of the cells, mostly found in vivo, not in vitro.( 7 , 8 , 9 ) The deletion mutant EGFRvIII, which is clonally expressed on the cell surface of ~40% of glioblastomas, has been clinically correlated with increased glioma cell growth, proliferation, invasion, and angiogenesis.( 2 , 10 , 11 ) For the EGFR‐targeted strategies in malignant glioma therapy, EGFRvIII expression should be considered because of its highly malignant nature.
Methods of targeting the EGFR that have been developed and trialed include monoclonal antibodies (mAbs), synthetic tyrosine kinase inhibitors, conjugates of toxins to anti‐EGFR mAbs and ligands, and antisense gene therapy of EGFR.( 12 , 13 ) Small molecule tyrosine kinase inhibitors and mAbs are the most fully developed of these approaches.( 14 , 15 )
Cetuximab (Erbitux, IMC‐C225) is a recombinant, human‐murine chimeric mAb specifically targeting the EGFR.( 13 ) Cetuximab competes with endogenous ligands for binding to the extracelluar domain of EGFR and binding of cetuximab prevents stimulation of the receptor by ligands. Cetuximab‐binding also results in internalization of the antibody‐receptor complex which leads to down‐regulation of EGFR expression on the cell surface.( 13 ) Furthermore, this type of mAb including the human IgG1 Fc region may cause recruitment and activation of host immune‐effector cells (T cells, natural‐killer cells, and macrophages) or complements to induce antibody‐dependent cell‐mediated cytotoxicity (ADCC) or complement‐dependent cytotoxicity (CDC).( 16 , 17 )
A variety of human epithelial cancers expressing EGFR have been successfully treated by cetuximab with promising results and it was recently approved for use in treating advanced‐stage EGFR‐expressing colorectal, head, and neck cancers.( 13 , 18 ) However, little is known as to whether or not it is an effective therapy for the treatment of highly malignant tumors expressing deletion mutant EGFRvIII. Based on the encouraging results of cetuximab in the EGFR‐expressing cancers and the importance of EGFRvIII expression in the biology of glioblastomas, we investigated whether cetuximab would be capable of effectively targeting the EGFRvIII expressed in malignant glioma cell lines.
Materials and Methods
Cell culture. Human glioblastoma cell lines were obtained as follows: U‐251 MG, A‐172, SF126, and YH‐13, JCRB Cell Bank (Osaka, Japan); U‐118 MG, U‐87 MG, DBTRG‐05 MG, LN‐229, LN‐18, and M059K, ATCC (Manassas, VA, USA). These cell lines were maintained in RPMI‐1640 supplemented with 10% fetal bovine serum and cultured at 37°C in a humidified atmosphere containing 5% CO2.
Expression vector construction and cell transfection. Construction of expression vector was generously contributed by Dr Hideyuki Yokote (Wakayama, Japan). Full‐length cDNA of wild‐type (wt) EGFR was amplified by reverse transcription–polymerase chain reaction (RT‐PCR) from a human embryonal kidney cell line HEK293 using a High Fidelity RNA PCR Kit (TaKaRa, Shiga, Japan) and the following primer sets: forward, CGCTAGCGATGCGACCCTCCGGGAC; reverse, CCCCTGACTCCGTCCAGTATTGA. The PCR products were amplified using the following primer sets: forward, CGCTAGCGATGCGACCCTCCGGGAC; reverse, CGAAGCTTTGCTCCAATAAATTCACTGC. The amplified DNA included NheI‐ and HindIII‐cut cohesive ends at the 5′‐ and 3′‐ends, respectively. The product was subcloned into a pCR BluntII‐TOPO vector (Invitrogen, Carlsbad, CA, USA) and the sequences were confirmed. Oligonucleotides encoding the myc‐tag sequence (EQKLISEEDLN) were designed and synthesized as follows: forward, AGCTTGAACAGAAGCTGATCTCAGAGGAGGACCTGAATTGAC; reverse, TCGAGTCAATTCAGGTCCTCCTCTGAGATCAGCTTCTGTTCA. These oligos were annealed, and the ds‐oligos were generated including HindIII‐ and XhoI‐cut cohesive ends, at the 5′‐ and 3′‐ends, respectively. Wt EGFR and myc‐tag DNA fragments were cut out and transferred into a pQCXIX retroviral vector (BD Biosciences Clontech, San Diego, CA, USA) containing EGFP following internal ribosome entry site sequence. EGFRvIII was synthesized with the recombinant PCR method using the following primers: F1, CGCTAGCGATGCGACCCTCCGGGAC; R1, ATCTGTCACCACATAATTACCTTTCTTTTCCTCCAGAGCC; F2, GGCTCTGGAGGAAAAGAAAGGTAATTATGTGGTGACAGAT; R2, CGGTGGAGGTGAGGCAGATG. Two DNA fragments of wt EGFR were amplified using the F1/R1 and F2/R2 primer sets. The EGFR fragment deleting exon 2–7 was amplified using the two PCR products as templates and the F1/R2 primer set. After confirming the sequence, a wt EGFR fragment was substituted for the NheI‐ and EcoRI‐ cut recombinant PCR fragment. A pVSV‐G vector (BD Biosciences Clontech) and the pQCXIX constructs were cotransfected into the GP2‐293 cells (BD Biosciences Clontech) using a FuGENE6 transfection reagent (Roche Diagnostics, Basel, Switzerland). Briefly, 80% confluent cells cultured on a 10‐cm dish were transfected with 2‐mg pVSV‐G plus 6‐mg pQCXIX vectors. Forty‐eight h after transfection, culture medium was collected and the viral particles were concentrated by centrifugation. The viral pellet was resuspended in fresh medium. The titer of the viral vector was calculated by counting the EGFP‐positive cells which were infected by serial dilution of the virus‐containing medium and the multiplicity of infection was determined.
Chemicals. Cetuximab was kindly provided by Brystol‐Myers Squibb (Princeton, NJ, USA).
Antibodies. Abs were purchased as follows: antihuman EGFR mouse mAb cocktail, Biosource Corp. (Camarillo, CA, USA); antiphosphotyrosine mouse mAb, BD Biosciences; antiphospho‐Akt, phospho‐p44/42 mitogen activated protein kinase (MAPK), rabbit polyclonal, anti‐β‐actin rabbit monoclonal, and antimouse and rabbit IgG horseradish peroxidase–linked Abs, Cell Signaling Technology (Beverly, MA, USA).
Immunoblotting. Cells were seeded and grown to near confluency. Then, cells were washed with phosphate‐buffered saline (PBS) and culture medium with or without cetuximab was added. At the time of harvest, cells were washed and lyzed with lysis buffer (50 mM Hepes buffer, 1% Triton X‐100, 5 mM ethylenediaminetetraacetic acid [EDTA], 50 mM sodium chloride, 10 mM sodium pyrophosphate, 50 mM sodium fluoride, and 1 mM sodium orthovanadate) supplemented with protease inhibitors (Roche Diagnostics, Penzberg, Germany). Cell lysates were clarified by centrifugation, and equal concentrations of lysates were mixed with 4× sodium dodecyl sulfate (SDS)–gel sample buffer (0.25 mol/L Tris‐HCl, pH 6.8, 2% SDS, and 25% glycerol), denatured with 2‐mercaptoethanol. Equivalent amounts of protein were separated on SDS‐polyacrylamide gels by electrophoresis, and transferred onto polyvinylidene difluoride (PVDF) membranes by wet electroblotting. The PVDF membranes were blocked with 3% bovine serum albumin in PBS with 0.1% Tween‐20 and probed with primary Ab. The membranes were washed and incubated with secondary Ab. After the membranes were washed, immunoblotted proteins were detected using the ECL Western Blotting Detection System (GE Healthcare, Buckinghamshire, UK).
Immunoprecipitation. Cell lysates were extracted and treated with Ab overnight at 4°C. The lysates were mixed with protein A agarose and centrifuged. Immunoprecipitates were washed with lysis buffer and resuspended in 1.5× SDS‐sample buffer and denatured with 2‐mercaptoethanol. Samples were processed by immunoblotting.
Immunofluorescence. For immunofluorescence, cells were grown overnight, washed in PBS and fixed for 20 min in 4% paraformaldehyde at room temperature (RT). Cells were then washed twice in PBS and blocked for 1 h in 10% normal goat serum at RT. Cetuximab (10 mg/mL) was diluted in 1.5% normal goat serum and incubated overnight at 4°C. Cells were washed twice in PBS for 5 min and incubated with a secondary Ab for 45 min at RT. Secondary Abs included Alexa Fluor 546 Goat Anti‐Mouse IgG (2 mg/mL; Molecular Probes, Invitrogen) and Texas Red antihuman IgG (1.5 mg/mL; Vector, Burlingame, CA, USA). Cells were washed twice for 5 min in PBS and then examined under a microscope (Keyence Biozero, Osaka, Japan).
Growth‐inhibition assay (MTS assay). The growth‐inhibitory effect of cetuximab was evaluated using the CellTiter96 Aqueous One Solution Cell Proliferation Assay (Promega Corporation, Madison, WI, USA). MTS (3‐[4,5‐dimethylthiazol‐2‐yl]‐5‐[3‐carboxymethoxyphenyl]‐2‐[4‐sulfophenyl]‐2H‐tetrazolium, inner salt) provides a measure of mitochondrial dehydrogenase activity within the cell and thereby offers an indication of cellular proliferation status.( 19 ) Briefly, 3000 cells were seeded in 96‐well plates. The plates were incubated overnight to permit adherence. Cells were then exposed to different concentrations of cetuximab ranging from 0 to 100 mg/mL for 48 h. MTS reagent was added and incubated for 1 h. The plates were read on a microplate spectrophotometer (optical density, 490 nm). The percentage cell growth was calculated by comparison of the absorbance value reading obtained from treated samples versus controls.
Antibody‐dependent cell‐mediated cytotoxicity (ADCC). Peripheral blood mononuclear cells (PBMCs) were separated from whole blood of healthy volunteers using LSM Lymphocyte Separation Medium (Cappel, Aurora, OH, USA). 1 × 105 cells were seeded into each well of a 12‐well plate. After incubation for 24 h, human PBMCs alone (effector : target = 10:1), and/or cetuximab (10 mg/mL), control human IgG (10 mg/mL), or control medium were added to the wells. The cultures were then examined under a microscope (Keyence) 24‐h post treatment.
Propidium iodide (PI) nucleic acid stain for cetuximab‐mediated ADCC. This assay was performed for identifying dead cells in a population under the same experimental condition as described in the ‘ADCC’ section. A 4‐µM solution of Cellstain PI (1 mg/mL; Dojindo, Kumamoto, Japan) was made at a dilution rate of 1:3000 and added per well. After incubation in the dark for 15 min, cells were viewed under a microscope (Keyence).
MTS assay for cetuximab‐mediated ADCC. For analysis of the ADCC activity of cetuximab, we used the MTS assay.( 20 ) Briefly, 3000 cells per well were exposed to various concentrations of cetuximab in the presence of PBMCs at an effector/target (E/T) ratio of 10 for 48 h. Natural killer (NK) activity was calculated from the absorbance value of the cells cultured without cetuximab and PBMCs (A), the absorbance value of the cells cultured with PBMCs (B), the absorbance value of medium alone (C), and the absorbance value of PBMCs (D) as follows: {1 – [(B – C) –(D – C)]/[(A – C) – (D – C)]} × 100. ADCC was calculated from the values of A, D, the NK activity (E), and the absorbance value of the cells treated with cetuximab and PBMCs (F): {1 – (F – D)/(A – D)} × 100 – E.
Results
Initially, we determined the expression of endogenous EGFR protein in 10 human malignant glioma cell lines by immunoblotting (Fig. 1a). All of these malignant glioma cell lines displayed only low amounts of wt EGFR protein (170 kDa) but not EGFRvIII protein (145 kDa). These findings were consistent with the previous notion that because primary explants of human glioblastoma rapidly lose expression of amplified, rearranged receptors in culture, no existing glioblastoma cell lines exhibit such expression.( 22 ) Therefore, we introduced the EGFRvIII gene into malignant glioma cell lines and engineered them to express this mutant receptor protein in three cell lines: U‐251 MG, U‐87 MG, and LN‐229 (Fig. 1b). Malignant glioma cells that expressed the EGFRvIII protein were observed under fluorescence microscopy due to the coexpression of EGFP (Fig. 1c).
Figure 1.

(a) Expression of epidermal growth factor receptor (EGFR) in human malignant glioma cell lines. Ten malignant glioma cell lines were cultured and lyzed. Equal amounts of cell lysate were immunoblotted with anti‐EGFR antibody (Ab) to show endogenous expression of EGFR. The human epidermoid carcinoma A431 cell lines served as a reference marker for high expression of wild‐type (wt) EGFR.( 21 ) Beta‐actin showed protein loading. IB, immunoblotting. (b) Introduction of EGFRvIII into human malignant glioma cell lines. Three malignant glioma cell lines, into which the EGFRvIII gene was introduced, were cultured and lyzed. Equal amounts of cell lysates were immunoblotted with an anti‐EGFR Ab to recognize exogenous expression of EGFRvIII. (c) Microscopic images and immunofluorescence of malignant glioma cells expressing EGFRvIII. EGFRvIII‐expressing cells (EGFRvIII) were monitored by their coexpression of EGFP. Mock control (EGFP) expressed only EGFP. FM, fluorescent microscopy. EGFRvIII‐expressing cells were stained with anti‐EGFR Ab, then Alexa Fluor‐conjugated antimouse IgG secondary Ab (lower panels). Staining of A431 or EGFP served as a reference of endogeneously expressed wt EGFR. Scale bar represents 100 µm.
Cetuximab has a binding ability to EGFRvIII. We investigated the binding capability of cetuximab to EGFRvIII by immunoprecipitation. Cell lysates were precipitated with cetuximab and then analyzed by immunoblotting with an anti‐EGFR Ab. Cetuximab was found to precipitate the 140‐kDa EGFRvIII protein from the EGFRvIII‐expressing cells (Fig. 2a upper lane). There was no indication of an immunoreactive band migrating at the expected size of EGFRvIII in the mock control. The finding that the EGFRvIII protein was immunoprecipitated with cetuximab was confirmed by anti‐EGFR Ab recognizing this mutant receptor (Fig. 2a lower lane).
Figure 2.

(a) Immunoprecipitation showing cetuximab binding to epidermal growth factor receptor variant III (EGFRvIII). EGFRvIII‐expressing cells were washed and lyzed. Equal amounts of cell lysates were immunoprecipitated with cetuximab or anti‐EGFR antibody (Ab). The immunoprecipitates were probed by immunoblotting with anti‐EGFR Ab. A431 was used as a positive control for wild‐type (wt) EGFR expression, and EGFP as a negative control for EGFRvIII. (b) Immunofluorescence showing cetuximab reactivity with malignant glioma cells expressing EGFRvIII. EGFRvIII‐expressing cells were stained with cetuximab, then Texas Red–conjugated antihuman IgG secondary Ab. Staining of A431 or EGFP served as a reference for cetuximab reactivity with endogeneously expressed wt EGFR. Scale bar represents 100 µm.
Next, we performed immunofluorescence to investigate cetuximab reactivity with the EGFRvIII‐overexpressing glioma cells. In this assay, cetuximab was used as a primary Ab, and Texas Red–conjugated antihuman IgG as a secondary Ab. In the mock control expressing low amounts of wt EGFR, we observed very low levels of staining, most of which were close to the detection limit of the analysis (Fig. 2b). In contrast, strong staining was evident in the EGFRvIII‐overexpressing cells. Together these results demonstrate that cetuximab could recognize the deletion mutant EGFRvIII.
Cetuximab attenuates EGFRvIII expression and reduces phosphorylated EGFRvIII, but does not significantly inhibit Akt and MAPK signaling pathways. Based on the previous report that cetuximab‐binding leads to down‐regulation of EGFR expression, we investigated the effect of cetuximab on EGFRvIII expression by immunoblotting.( 13 ) On treatment with cetuximab at various concentrations, the expression levels of EGFRvIII protein decreased dramatically in a dose‐dependent manner (data not shown and Fig. 3a lower lane). In addition, we assessed the phosphorylation status of EGFRvIII by immunoprecipitation from the same lysates using a mAb recognizing EGFRvIII. The amount of immunoprecipitated EGFRvIII is shown in Fig. 3(a) (lower lane). A dose‐dependent decrease in the expression of phosphorylated EGFRvIII was also observed, and the levels of tyrosine‐phosphorylation corresponded to the expression of this receptor (Fig. 3a upper lane). Neither higher concentrations of cetuximab (= 100 µg/mL) nor prolonged exposure (72 h) augmented the effects on phosphorylated EGFRvIII expression (Fig. 3b and data not shown). Thus, cetuximab appeared to attenuate the EGFRvIII expression and reduce the phosphorylated EGFRvIII.
Figure 3.

Effect of cetuximab on epidermal growth factor receptor variant III (EGFRvIII) and its downstream signaling molecules. EGFRvIII‐expressing glioma cells were exposed to 0, 0.1, 1, 10, or 100 µg/mL cetuximab for 24 h. (a) 250 µg of total cell lysates were immunoprecipitated with an anti‐EGFR antibody (Ab). The immunoprecipitates were probed by immunoblotting with an antiphosphotyrosine Ab (upper lane) and the membranes were reblotted with an anti‐EGFR antibody (lower lane). (b) Equal amounts of cell lysates were immunoblotted with a specific antihuman antibody as the first antibody, and then with a horseradish peroxidase–conjugated secondary antibody.
Next, we examined the phosphorylation status of Akt and MAPK pathways. On treatment with cetuximab, phosphorylated Akt and MAPK mildly decreased in the cells at higher concentrations (Fig. 3b). The decreased levels were not so significant as that of phosphorylated EGFRvIII. In summary, these results would suggest that when treated with cetuximab, EGFRvIII expression was markedly attenuated, while its downstream pathways by Akt and MAPK remained activated in the EGFRvIII‐overexpressing glioma cells.
Cetuximab does not inhibit the growth of EGFRvIII‐overexpressing glioma cells. We examined the effects of cetuximab on the growth of EGFRvIII‐overexpressing glioma cells with an MTS assay. As can be observed in Fig. 4, cetuximab treatment did not produce a clear growth‐inhibitory effect in the EGFRvIII‐overexpressing cells even at the highest concentration tested. Also, treatment with cetuximab had a modest effect on the mock control. These data might support the findings determined by immunoblotting analyses.
Figure 4.
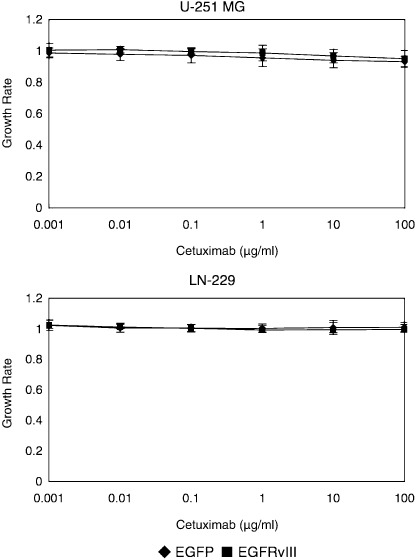
MTS assays showing the growth‐inhibitory effects of cetuximab on malignant glioma cells with and without epidermal growth factor receptor variant III (EGFRvIII) overexpression. 3000 glioma cells per well were placed onto a 96‐well tissue culture plate and incubated overnight. Cetuximab was added at concentrations of 0–100 µg/mL and incubated for 48 h before an MTS assay was performed. This figure is representative of three independent experiments. Bars, SD.
Cetuximab induces ADCC in the presence of human PBMCs against malignant glioma cells expressing EGFRvIII. In molecular targeting using mAb, ADCC or CDC activity should be considered as one of the potent antitumor mechanisms.( 16 ) First, we investigated the ADCC activity by morphological observation, PI stain, and an MTS assay. As our results in Fig. 5(a) show, cetuximab in the presence of PBMCs induced extensive lysis of the cells in culture. In contrast, treatment with cetuximab alone, human PBMCs alone, or PBMCs plus human IgG control (data not shown), did not apparently induce lysis of the cells. Correspondingly, PI nucleic acid stain revealed remarkable cell death in the cell population treated with cetuximab and PBMCs (Fig. 5b). To further evaluate cetuximab‐induced ADCC against EGFRvIII expressed in the cells, we performed the MTS assay in the presence of human PBMCs at an E/T ratio of 10. In the mock control with low expression levels of wt EGFR, mild ADCC activity was detected (Fig. 5c). On the other hand, cetuximab treatment significantly inhibited the proliferation of the EGFRvIII‐overexpressing glioma cells in a dose‐dependent manner. There was no significant percentage ADCC at the zero µg/mL of cetuximab in the EGFRvIII‐expressing cells as well as in mock controls (EGFP), suggesting that both cells could not be susceptible to PBMCs alone. Next, we examined CDC activity against these cells using the MTS assay in the presence of 25% human serum containing human complement. No CDC‐mediated cytolytic effect was observed even at the highest concentration of cetuximab tested (data not shown). These results suggest that this mAb also recognizes the EGFRvIII expressed on the cell surfaces and exerts potent ADCC activity against the glioma cells overexpressing this mutant receptor.
Figure 5.
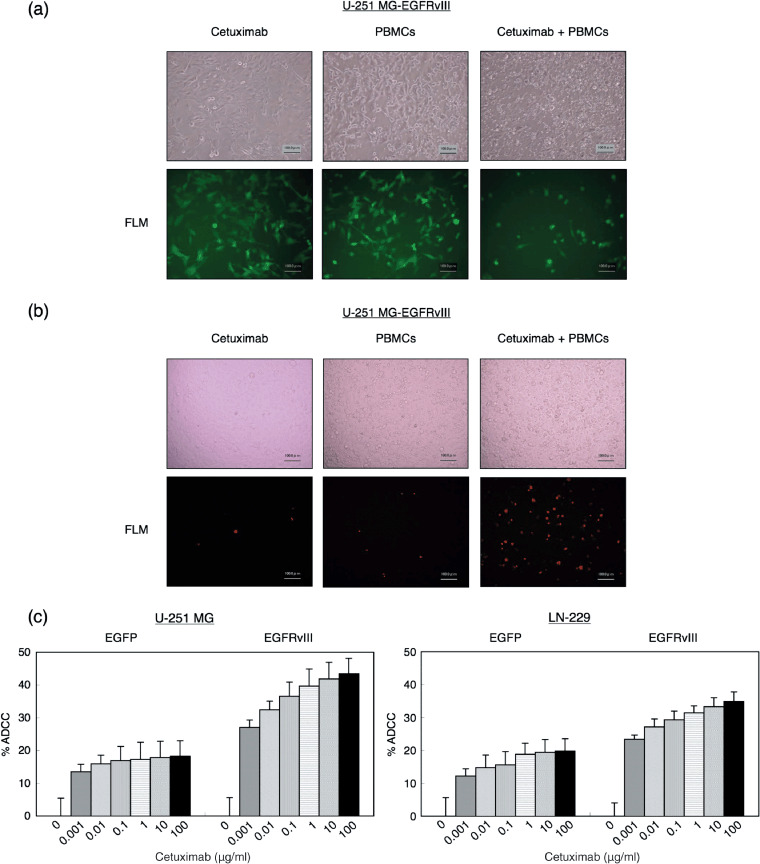
(a) Microscopic findings showing cetuximab‐mediated antibody‐dependent cell‐mediated cytotoxicity (ADCC) of glioma cells expressing epidermal growth factor receptor variant III (EGFRvIII). Monolayers of the glioma cells were treated with cetuximab (10 µg/mL) and human peripheral blood mononuclear cells (PBMCs) at an effector target ratio of 10:1 at 37°C for 24 h. Controls included treatment with cetuximab alone (left panels), PBMCs alone (middle panels), or PBMCs plus human IgG (data not shown). Note the extensive lysis of the cells in the presence of both cetuximab and PBMC (right panels) but not in any of the control cultures. (b) Propidium iodide (PI) stain showing cetuximab‐mediated ADCC of the EGFRvIII‐expressing glioma cells. Under the same condition as (a), PI solution was added and incubated for 15 min. Note the remarkable increase of the dead cells in the presence of both cetuximab and PBMC (right panels). (c) MTS assays showing the ADCC activity of cetuximab against malignant glioma cells with and without EGFRvIII expression. Under the same condition as in Fig. 4, human PBMCs were added at an effector target ratio of 10:1 and incubated for 48 h before an MTS assay was performed. This figure is representative of three independent experiments. Bars, SD.
Discussion
Overexpression of EGFR can be a promising characteristic as a molecular target for malignant glioma therapy.( 12 ) Malignant gliomas that are wt EGFR–positive may simultaneously overexpress EGFRvIII, which is reported to be associated with aggressive phenotypes and resistance to chemo‐ and radiotherapy.( 2 , 10 , 11 ) Although anti‐EGFR mAb cetuximab may play an important role and be hopefully evaluated in preclinical studies, limited data have been reported regarding the potential of cetuximab to target the EGFRvIII in malignant glioma cells.( 3 ) The present data provide evidence that cetuximab has a capability to recognize EGFRvIII as well as wt EGFR. Although cetuximab produces modest activities to block the EGFRvIII signaling or inhibit the growth of glioma cells with this receptor directly, this mAb has great potential to induce ADCC activity against the EGFRvIII‐expressing glioma cells. These results suggest that cetuximab therapy might be effective against malignant gliomas expressing EGFRvIII.
In any anti‐EGFR mAb strategy against EGFRvIII, it is of great importance to determine whether or not the mAb can recognize this deletion mutant receptor as the target. EGFR is composed of three major domains: extracellular domains including a ligand‐binding site, a hydrophobic transmembrane segment, and a tyrosine‐kinase containing cytoplasmic region.( 13 ) EGFRvIII is a mutant form of EGFR encoded by a mutated gene characterized by in‐frame deletion of 801 bp coding 6–273 amino acids from the extracellular domain.( 11 ) As the extracellular part is composed of domain I to IV, all of domain I and the amino‐terminal 2/3 of domain II are absent in this mutant receptor, while domain III and IV remain intact. Cetuximab has been produced as one of mAbs directed against the extracellular ligand‐binding domain of the EGFR, and recent structural studies have demonstrated that the interaction between cetuximab and the EGFR is with domain III of the receptor, not with other domains.( 18 ) Our experiments using immunoprecipitation and immunofluorescence revealed that cetuximab has the ability to bind to EGFRvIII. These findings suggest that EGFRvIII preserves the cetuximab‐binding structure regardless of its possible conformational change due to 268 amino acids deletion.( 18 , 23 ) There have been some recent reports on the possibility of cetuximab for detecting EGFRvIII.( 24 , 25 ) Aerts et al. developed cetuximab‐based imaging probe to target EGFR and demonstrated its potential as an imaging agent for not only wild type but also EGFRvIII.( 24 ) Yang's group used cetuximab as a delivery tool of radio‐isotope and evaluated boronated mAb for boron neutron capture therapy of a rat glioma expressing either wild‐type EGFR or EGFRvIII.( 25 ) This evidence reveals that cetuximab has a potential role in the advancement of anti‐EGFR strategy.
Cetuximab competes with ligands for binding to the EGFR.( 13 ) Binding of cetuximab to the EGFR prevents phosphorylation and activation of the receptor tyrosine kinase, resulting in the inhibition of its downstream signal transduction which controls cellular biology.( 13 , 14 ) This mode of action is considered as the primary mechanism for the antitumor activity of cetuximab. Therefore, EGFRvIII, constitutively activated regardless of ligand‐binding, may be insusceptible to such direct inhibition by cetuximab. Indeed, our experiments demonstrated that cetuximab, despite binding to the EGFRvIII, did not have clear inhibitory effects on the phosphorylation of EGFRvIII, Akt, and MAPK.
In mAb therapies, indirect growth‐inhibition by activating host immune effector cells is a hopeful mechanism for antitumor activity.( 16 ) Previously, we found that ADCC activity is a major mode of action of anti‐HER2/neu mAb trastuzumab for breast cancer cell lines.( 20 ) Therefore, even though cetuximab alone cannot inhibit the growth of the EGFRvIII‐expressing cells in culture, it may have cytolitic potential in the treatment of EGFRvIII‐expressing tumors in vivo by immunological mechanisms such as ADCC or CDC.( 26 ) ADCC has rarely been discussed as one of the antitumor mechanisms of cetuximab. Recently, it has been reported that this mAb causes EGFR‐expressing tumor cells to die through this mode of action and elicits effective ADCC activity against lung, head, and neck cancer.( 17 , 27 , 28 ) In the literature, the authors describe the potential of cetuximab to exhibit ADCC activity mediated by targeting EGFR expressed in tumor cells, but do not discuss in detail whether cetuximab‐mediated ADCC can be evoked against mutant EGFR, especially a deletion mutant form of extracellular domain that is vital for cetuximab binding, which is often discussed in malignant glioma. Our in vitro study showed that in the presence of human PBMCs, cetuximab induced strong ADCC, presumably due to additional activation of the immune effector functions by this antibody. A new insight drawn from our study is that even though the target is EGFRvIII, which has a partial deletion of EGF‐binding site, once cetuximab binds to the mutant receptor, ADCC could be substantially produced against malignant glioma cells. These findings suggest that in vivo treatment of cetuximab could generate antitumor activity through ADCC even against malignant gliomas expressing EGFRvIII, although it should be considered that the growth advantage conferred by EGFRvIII is mostly found in vivo, not in vitro.
Previous studies have shown that cetuximab binding results in internalization of the antibody‐receptor complex, which leads to down‐regulation of EGFR expression on the cell surface and the blockading of its downstream signaling.( 13 ) Patel and colleagues examined the ability of cetuximab as an effective drug for EGFRvIII‐expressing tumor cells and concluded that down‐regulation of EGFRvIII resulted in inhibition of cell proliferation.( 29 ) In our experimental conditions, cetuximab treatment attenuated EGFRvIII expression in a dose‐dependent manner. However, despite the decreased levels of this mutant receptor, cetuximab did not apparently inhibit the phosphorylation of Akt and MAPK and the growth of glioma cells with this receptor. Akt functions in one of the major signaling cascades, the phosphatidylinositol‐3‐kinase‐Akt pathway, and controls the balance between glioma cell survival and apoptosis.( 13 , 30 , 31 ) p44/42 MAPK functions in another major cascade, the ras–raf–MAPK pathway, which plays a critical role in cell growth and proliferation.( 13 ) There are several possible mechanisms to explain why cetuximab‐mediated attenuation of EGFRvIII did not produce antitumor activity in the EGFRvIII‐overexpressing glioma cells. The simplest explanation rests in the possibility that, because the EGFRvIII is not completely depleted even at the highest dose of cetuximab tested, EGFRvIII signaling still exists to maintain its downstream activation. Another potentially related explanation is that once constitutive activation of EGFRvIII is established in a cell, it may be difficult to disturb the constant downstream signaling. In any case, we expected cetuximab‐induced ADCC against EGFRvIII‐expressing cells and clearly demonstrated that cetuximab binding to EGFRvIII could exhibit ADCC activity, inducing glioma cell death. This evidence might hopefully have an impact in anti‐EGFR mAb therapy for malignant glioma.
When discussing the clinical relevance of cetuximab therapy against malignant glioma, there are two major points to be elucidated: (i) delivery of cetuximab to the central nervous system; and (ii) recruitment of immune‐effector cells into brain tumors. In chemotherapy for brain tumors including malignant glioma, it is necessary to consider whether the drug can effectively reach the tumor through the blood–brain barrier (BBB). Some encouraging findings regarding this problem have been reported. Eller et al. demonstrated that intraperitoneal injection of cetuximab significantly increased median survival in nude mice bearing intracranial xenografts of glioblastoma.( 3 ) Arwert and colleagues showed that intravenous injection of cetuximab resulted in a considerable reduction of intracranial glioma burden.( 32 ) This evidence suggests that systemic administration could achieve effective concentration in the brain. On the other hand, there is some information that intact antibodies do not reach significant levels in malignant gliomas after systemic administration. Therefore, in the clinical practice of mAb therapy for brain tumors, several methods to deliver the agent to tumors have been tried out, such as convection‐enhanced delivery with stereotactic infusion‐catheter placement and osmotic BBB disruption with selective intra‐arterial mannitol infusion, producing promising results. Recently, non‐invasive localized delivery of mAb to the mouse brain was reported by magnetic resonance imaging–guided focused ultrasound‐BBB disruption.( 33 ) These modalities would enable effective delivery of cetuximab to malignant gliomas in the brain. Next, recruitment of immune‐effector cells into the brain tumor is also vital for cetuximab to induce ADCC reaction against malignant glioma. In previous literature on mAb treatment for brain tumors, peritumoral infiltrates of macrophages were shown in mice treated with the mAb which was found to induce ADCC in vitro, whereas a paucity of T cells and natural killer cells was also described.( 34 ) Although it should be elucidated whether macrophages or microglia would be able to mediate the ADCC in the brain, the promising result in the paper suggested that the mAb‐mediated ADCC reaction could be evoked in in vivo models, which might be encouraging for cetuximab to produce an effective ADCC activity in vivo. In conclusion, the above information provides us with some hope that cetuximab may be used to treat patients with malignant glioma. As the next step, further investigation is needed in a clinical setting.
In summary, we have reported that cetuximab can target EGFRvIII and although this mAb appears to be less effective in direct inhibition of EGFRvIII activity, intervention of effector cells such as human PBMCs can produce antitumor efficacy of cetuximab even against EGFRvIII‐expressing glioma cells. In view of the concept that cetuximab has been previously shown to develop chemosensitizing and radiosensitizing effects, the use of this mAb may have great therapeutic potential against malignant gliomas.( 3 , 4 ) Moreover, conjugation of cytotoxic agents such as drugs or radioisotopes might produce enhanced antitumor activity.( 12 ) Thus, we emphasize that targeted therapy using the anti‐EGFR mAb cetuximab could play a significant role in the development of multidisciplinary treatment strategies for these tumors.
Acknowledgments
This work was partially supported by funds from the Third Term Comprehensive 10‐Year Strategy for Cancer Control (N.K. and K.F.) and Health and Labor Sciences Grants, Research on Advanced Medical Technology, H17‐Pharmaco‐006 (N.K. and K.F.).
We thank Brystol‐Myers Squibb (cetuximab) for providing the anti‐EGFR agents for experimental studies.
References
- 1. Von Deimling A, Louis DN, Wiestler OD. Molecular pathways in the formation of gliomas. Glia 1995; 15: 328–38. [DOI] [PubMed] [Google Scholar]
- 2. Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000; 60: 1383–7. [PubMed] [Google Scholar]
- 3. Eller JL, Longo SL, Kyle MM, Bassano D, Hicklin DJ, Canute GW. Anti‐epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery 2005; 56: 155–62. [DOI] [PubMed] [Google Scholar]
- 4. Eller JL, Longo SL, Hicklin DJ, Canute GW. Activity of anti‐epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery 2002; 51: 1005–14. [DOI] [PubMed] [Google Scholar]
- 5. Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA 1990; 87: 8602–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong AJ, Ruppert JM, Bigner SH et al . Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA 1992; 89: 2965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nishikawa R, Ji XD, Harmon RC et al . A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 1994; 91: 7727–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Batra SK, Castelino‐Prabhu S, Wikstrand CJ et al . Epidermal growth factor ligand‐independent, unregulated, cell‐transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ 1995; 6: 1251–9. [PubMed] [Google Scholar]
- 9. Huang HS, Nagane M, Klingbeil CK et al . The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 1997; 272: 2927–35. [DOI] [PubMed] [Google Scholar]
- 10. Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res 1996; 56: 5079–86. [PubMed] [Google Scholar]
- 11. Learn CA, Hartzell TL, Wikstrand CJ et al . Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res 2004; 10: 3216–24. [DOI] [PubMed] [Google Scholar]
- 12. Halatsch ME, Schmidt U, Behnke‐Mursch J, Unterberg A, Wirtz CR. Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat Rev 2006; 32: 74–89. [DOI] [PubMed] [Google Scholar]
- 13. Harding J, Burtness B. Cetuximab. An epidermal growth factor receptor chimeric human‐murine monoclonal antibody. Drugs Today 2005; 41: 107–27. [DOI] [PubMed] [Google Scholar]
- 14. Herbst RS, Shin DM. Monoclonal antibodies to a target epidermal growth factor receptor‐positive tumors. Cancer 2002; 94: 1593–611. [DOI] [PubMed] [Google Scholar]
- 15. Wakeling AE. Epidermal growth factor receptor tyrosine kinase inhibitors. Curr Opin Pharmacol 2002; 2: 382–7. [DOI] [PubMed] [Google Scholar]
- 16. Harris M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol 2004; 5: 292–302. [DOI] [PubMed] [Google Scholar]
- 17. Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody‐dependent cellular cytotoxicity of cetuximab against tumor cells with wild‐type or mutant epidermal growth factor receptor. Cancer Sci 2007; 98: 1275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7: 301–11. [DOI] [PubMed] [Google Scholar]
- 19. Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual‐agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti‐EGFR antibody with tyrosine kinase inhibitor. Cancer Res 2004; 64: 5355–62. [DOI] [PubMed] [Google Scholar]
- 20. Naruse I, Fukumoto H, Saijo N, Nishio K. Enhanced anti‐tumor effect of trastuzumab in combination with cisplatin. Jpn J Cancer Res 2002; 93: 574–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perera RM, Narita Y, Furnari FB et al . Treatment of human tumor xenografts with monoclonal antibody 806 in combination with a prototypical epidermal growth factor receptor‐specific antibody generates enhanced antitumor activity. Clin Cancer Res 2005; 11: 6390–9. [DOI] [PubMed] [Google Scholar]
- 22. Mishima K, Johns TG, Luwor RB et al . Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res 2001; 61: 5349–54. [PubMed] [Google Scholar]
- 23. Garrett TP, McKern NM, Lou M et al . Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002; 110: 763–73. [DOI] [PubMed] [Google Scholar]
- 24. Aerts HJ, Dubois L, Hackeng TM et al . Development and evaluation of a cetuximab‐based imaging probe to target EGFR and EGFRvIII. Radiother Oncol 2007; 83: 326–32. [DOI] [PubMed] [Google Scholar]
- 25. Yang W, Wu G, Barth RF et al . Molecular targeting and treatment of composite EGFR and EGFRvIII positive gliomas using boronated monoclonal antibodies. Clin Cancer Res 2008; 14: 883–91. [DOI] [PubMed] [Google Scholar]
- 26. Modjtahedi H, Moscatello DK, Box G et al . Targeting of cells expressing wild‐type EGFR and type‐III mutant EGFR (EGFRvIII) by anti‐EGFR MAb ICR62: a two‐pronged attack for tumour therapy. Int J Cancer 2003; 105: 273–80. [DOI] [PubMed] [Google Scholar]
- 27. Kurai J, Chikumi H, Hashimoto K et al . Antibody‐dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res 2007; 13: 1552–61. [DOI] [PubMed] [Google Scholar]
- 28. Astsaturov I, Cohen RB, Harari P et al . EGFR‐targeting monoclonal antibodies in head and neck cancer. Curr Cancer Drug Targets 2007; 7: 650–65. [DOI] [PubMed] [Google Scholar]
- 29. Patel D, Lahiji A, Patel S et al . Monoclonal antibody cetuximab binds to and down‐regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res 2007; 27: 3355–66. [PubMed] [Google Scholar]
- 30. Matar P, Rojo F, Cassia R et al . Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC‐C225): superiority over single‐agent receptor targeting. Clin Cancer Res 2004; 10: 6487–501. [DOI] [PubMed] [Google Scholar]
- 31. Mellinghoff IK, Wang MY, Vivanco I et al . Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005; 353: 2012–24. [DOI] [PubMed] [Google Scholar]
- 32. Arwert E, Hingtgen S, Figueiredo J‐L et al . Visualizing the dynamics of EGFR activity and antiglioma therapies in vivo . Cancer Res 2007; 67: 7335–42. [DOI] [PubMed] [Google Scholar]
- 33. Kinoshita M, McDannold N, Jolesz FA, Hynynen K. Noninvasive localized delivery of Herceptin to the mouse brain by MRI‐guided focused ultrasound‐induced blood–brain barrier disruption. Proc Nat Acad Sci USA 2006; 103: 11 719–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sampson JH, Crotty LE, Lee S et al . Unarmed, tumor‐specific monoclonal antibody effectively treats brain tumors. Proc Nat Acad Sci USA 2000; 103: 11 719–23. [DOI] [PMC free article] [PubMed] [Google Scholar]