

Abstract
Previous reports suggest that, in addition to its therapeutic effects, ionizing radiation (IR) increases the invasiveness of surviving cancer cells. Here, we demonstrate that this activity of IR in lung cancer cells is mediated by a signaling pathway involving p38 kinase, phosphoinositide 3‐kinase, Akt, and matrix metalloproteinase (MMP‐2). The invasion‐promoting doses of IR also increased and reduced the levels of vimentin and E‐cadherin, respectively, both of which are markers for the epithelial–mesenchymal transition (EMT). Interestingly, all of these malignant actions of IR were mimicked by the overexpression of Bcl‐XL, a pro‐survival member of the Bcl‐2 family, in lung cancer cells. Moreover, both RNA and protein levels of Bcl‐XL were elevated upon irradiation of the cells, and the prevention of this event using small‐interfering RNAs of Bcl‐XL reduced the ability of IR to promote invasion signals and EMT‐associated events. This suggests that Bcl‐XL functions as a signaling mediator of the malignant effects of IR. It was also demonstrated that IR enhances signal transducer and activator of transcription 3 (STAT3) phosphorylation, and the reduction of STAT3 levels via RNA interference prevented IR‐induced Bcl‐XL accumulation, and thus all the tested Bcl‐XL‐dependent events. Overall, the data suggest that IR induces Bcl‐XL accumulation via STAT3, which then promotes cancer cell invasion and EMT‐associated markers. Our findings demonstrate a novel function of Bcl‐XL in cancer, and also advance our understanding of the malignant actions of IR significantly. (Cancer Sci 2010)
Although ionizing radiation (IR) is widely utilized for cancer therapy,( 1 , 2 , 3 ) recent studies have shown that IR also exerts undesirable effects on cancer cells. For example, the irradiation of various types of cancer cells, including glioma,( 4 ) hepatocellular carcinoma,( 5 ) pancreatic cancer,( 6 ) and meningioma cells,( 7 ) with sublethal doses of IR resulted in an increase in their invasiveness in culture. Moreover, a large number of studies using animal models have demonstrated that local radiotherapy administered to primary tumors speeds their metastatic growth in vivo,( 8 , 9 , 10 ) thereby suggesting that besides its therapeutic effects, IR promotes the malignant behaviors of surviving cancer cells. This latter effect of IR is also consistent with the finding that local failure after radiotherapy increases the probability that distant metastasis will develop in cancer patients.( 11 ) Therefore, preventing such undesirable action of IR may be an essential component of efforts to improve the efficacy of radiotherapy.
In order to develop a strategy toward this objective, the mechanism by which IR promotes cancer cell invasion should be delineated. Previous studies using glioma cells have suggested that IR stimulates signaling components, such as p38 kinase, phosphoinositide 3‐kinase (PI3K), and Akt, leading to matrix metalloproteinase‐2 (MMP‐2) expression and thus enhanced cell invasion.( 4 ) The ability of IR to induce the PI3K/Akt‐dependent invasion pathway was also proposed using hepatocellular carcinoma cells.( 5 ) However, apart from these reports, limited information is currently available regarding the cellular components involved in the invasion‐promoting action of IR.
The Bcl‐2 family proteins are key regulators of cellular viability.( 12 , 13 , 14 , 15 ) While certain members of the family, such as Bax and Bak, promote apoptosis, other members, such as Bcl‐2, Bcl‐XL, and Bcl‐w bind directly to the pro‐apoptotic members to block their apoptotic actions, thus bolstering cell survival. Interestingly, evidence has been accumulating recently that suggests additional functions of the pro‐survival members in cancer. First, the overexpression of Bcl‐2,( 16 , 17 , 18 ) Bcl‐XL,( 19 , 20 , 21 ) or Bcl‐w( 22 , 23 ) in the specified cancer cells resulted in increases in the migratory and invasive potential of the cells. These effects of Bcl‐2( 16 , 17 ) and Bcl‐w( 22 , 23 ) appear to be the result of their ability to stimulate invasion‐promoting signaling events. It has also been demonstrated that the overexpression of Bcl‐2( 18 ) or Bcl‐XL ( 19 , 21 ) promotes the metastatic growth of cancer cells in an animal model. Therefore, it appears that the role of pro‐survival Bcl‐2 family members in cancer is not confined simply to the survival of cancer cells, but that they also support the invasion and metastasis of cancer cells. Importantly, this latter newly proposed function of pro‐survival members appears clinically relevant, considering that analyses of patient samples have revealed that Bcl‐w is expressed in gastric cancer( 24 ) and glioma multiforme,( 25 ) particularly in cancer cells evidencing infiltrative morphology. It was similarly reported that Bcl‐XL expression in cases of colon cancer was associated with lymph‐node metastasis of the cancer cells.( 26 )
Interestingly, certain, if not all, pro‐survival Bcl‐2 family members respond to IR. For example, cellular levels of Bcl‐2( 27 ) and Bcl‐XL ( 28 ) were shown to be elevated upon exposure of various cancer cell types to IR. Although the mechanism underlying this phenomenon remains largely unknown, the increase in Bcl‐2 and Bcl‐XL levels may contribute to the resistance of cancer cells to radiotherapy.( 29 , 30 ) In this regard, it might be speculated that IR‐induced pro‐survival members also mediate the ability of IR to promote cancer cell invasion. The current study was undertaken in order to investigate this possibility directly. In service of this purpose, we focused on Bcl‐XL as a model. Human lung cancer cells were used, given the frequent application of radiotherapy to lung cancer patients.( 31 )
Materials and Methods
Antibodies. Antibodies were purchased from the following institutions: anti‐Bcl‐XL, ‐Bcl‐2, ‐Bak, and ‐Akt from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti‐phospho‐Akt, ‐p38 kinase, ‐phospho‐p38 kinase, ‐signal transducer and activator of transcription 3 (STAT3), and ‐phospho‐STAT3 from Cell Signaling (Beverly, MA, USA), anti‐E‐cadherin and ‐vimentin from BD Pharmingen (San Diego, CA, USA), anti‐MMP‐2 from Calbiochem (La Jolla, CA, USA), Alexa Fluor‐488 (green) and Alexa Fluor‐555 (red) secondary antibodies from Molecular Probes (Carlsbad, CA, USA).
Cell culture, transfection, and treatment. Human A549 and H460 lung cancer cells were cultured in RPMI‐1640 medium supplemented with 10% heat‐inactivated fetal bovine serum and gentamicin (50 μg/mL). The expression constructs for Bcl‐XL were prepared using pcDNA3 vectors. These expression vectors, as well as small‐interfering RNAs (siRNAs) of Bcl‐XL and STAT3 (Ambion, Austin, TX, USA) were introduced into cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). To irradiate the cells/transfectants, they (3 × 105) were plated on 60 mm dishes, incubated until 70–80% confluence was reached, and then exposed to γ‐rays from a 137Cs γ‐ray source (Atomic Energy of Canada, Mississauga, ON, Canada) at a dose rate of 3 Gy/min. Where specified, cells were administered pharmacological inhibitors (Calbiochem) 30 min before irradiation. All of the treatments employed herein did not significantly influence cellular viability, as determined by flow cytometry( 24 ) or Trypan blue exclusion assays.
Invasion assay. Cells were seeded onto the upper surfaces of Matrigel‐coated polycarbonate filters (BD Biosciences, Bedford, MA, USA) in modified Boyden chambers (Corning, Corning, NY, USA), and analyzed for their invasiveness as described previously.( 22 )
Western blot analysis. The cell lysates were prepared via a previously reported method.( 32 ) To compare levels of secreted MMP‐2, conditioned media were prepared via 24 h of incubation of the cells in serum‐free medium. Proteins in the lysates or conditioned media were separated by SDS–PAGE, electrotransferred to Immobilon membranes (Millipore, Bedford, MA, USA), subsequently blotted using the specified antibodies, and visualized with the ECL detection system (Amersham, Uppsala, Sweden).
Immunofluorescence microscopy. Cells were grown on glass coverslips for 24 h. Where indicated, the cells were irradiated, and subsequently incubated for an additional 24 h. The cells were then fixed in 2% paraformaldehyde, permeabilized in PBS containing 0.2% Triton X‐100, blocked for 15 min with 1% normal goat serum in PBS at room temperature, and incubated for 16 h at 4°C with anti‐vimentin or ‐E‐cadherin. The cells were washed and incubated for 1 h at room temperature with Alexa Fluor‐488 (green) and Alexa Fluor‐568 (red) secondary antibodies, which were used for vimentin and E‐cadherin, respectively. The coverslips were again washed, mounted on slides with Fluorescence Mounting Medium (Dako Diagnostics, Mississauga, ON, Canada), and analyzed via confocal laser scanning microscopy (Zeiss LSM 710; Carl Zeiss, Oberkochen, Germany).( 33 )
Reverse transcription–polymerase chain reaction (RT‐PCR). Total RNA was extracted using Trizol reagent in accordance with the manufacturer’s instructions (Invitrogen). The RT reactions were performed using total RNA as a template and a StarScript RT kit (Stratagene, La Jolla, CA, USA). PCR amplification was conducted using the following primers: Bcl‐XL, 5′‐ATCAATGGCAACCCATCCTG‐3′ and 5′‐TTGTCTA‐CGCTTTCCACGCA‐3′; GAPDH, 5′‐CATCTCTGCCCCCTC‐TGCTGA‐3′ and 5′‐GGATGACCTTGCCCACAGCCT‐3′. PCR products were subjected to electrophoresis on 1.5% agarose gels, stained with ethidium bromide, and photographed.( 34 )
Statistical analysis. Results were analyzed for statistical significance via the Student’s t‐test. Differences were considered significant at P < 0.05.
Results
Ionizing radiation promotes malignant behaviors of A549 cells. It was previously reported that IR promoted glioma cell invasion by inducing MMP‐2 expression via p38 kinase, PI3K, and Akt.( 4 ) To confirm this action of IR in lung cancer cells, A549 cells were irradiated with sublethal doses of γ‐rays (10 Gy). This treatment induced an increase in the invasiveness of the cells, which was apparent at 16–24 h after irradiation, but not at 8 h or earlier (see below). Therefore, unless otherwise specified, 24 h was selected for further analyses of the irradiated cells. Western blot analysis showed that the irradiation also resulted in an increase in the phosphoryation levels of p38 kinase and Akt, as well as the protein levels of MMP‐2 (Fig. 1a). Moreover, the invasion‐promoting effect of IR was attenuated when the cells were irradiated in the presence of inhibitors for p38 kinase (SB202190), PI3K (LY294002), Akt (Akt inhibitor), or MMP‐2 (OA‐Hy). This confirms the involvement of these components in the IR‐induced invasion of A549 cells. Given that cancer cells’ invasiveness is related closely with their epithelial–mesenchymal transition (EMT),( 35 , 36 ) immunostaining (Fig. 1c) and western blotting (Fig. 1a) were conducted to analyze vimentin and E‐cadherin, markers for mesenchyma and epithelium, respectively. The irradiation indeed induced an increase and decrease in the levels of vimentin and E‐cadherin, respectively. This verifies the ability of IR to induce EMT‐associated features in A549 cells, as has also been proposed by other researchers.( 37 ) Overall, the data indicate that IR promotes malignant behaviors of lung cancer cells.
Figure 1.
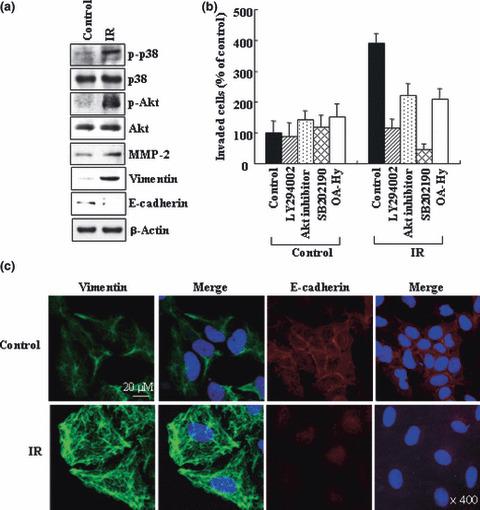
Ionizing radiation (IR) promotes malignant behaviors in A549 cells. (a) A549 cells were irradiated with 10 Gy of γ‐rays. After 24 h of incubation, cell lysates and conditioned media were prepared. Cell lysates were analyzed for p‐p38, p38, p‐Akt, Akt, vimentin, and E‐cadherin levels by western blotting using β‐actin as a loading control. The conditioned media were analyzed to compare the levels of secreted matrix metalloproteinase (MMP‐2). (b) A549 cells were incubated with or without inhibitors for phosphoinositide 3‐kinase (PI3K) (LY294002, 5 μM), Akt (Akt inhibitor I, 5 μM), p38 kinase (SB202190, 5 μM), or MMP‐2 (OA‐Hy, 5 μM) for 30 min, and then irradiated. After 24 h of incubation, cellular invasiveness was compared on Matrigel‐coated polycarbonate filters. The experiment was repeated three times, and the means and SDs were determined. (c) The control and irradiated cells were immunostained to analyze vimentin and E‐cadherin. DAPI staining to detect nuclei was also carried out. Stained cells were visualized with a confocal microscope.
Bcl‐XL is a common target of IR in multiple lung cancer cell types. In an effort to determine whether or not a pro‐survival Bcl‐2 family member is involved in this effect of IR, it was first necessary to determine which member of the family responds to IR in lung cancer cells. Figure 2 shows that levels of both Bcl‐XL and Bcl‐2 proteins were increased in the irradiated A549 cells, which became apparent at ∼8 h after irradiation and thus before the elevation of cellular invasiveness. Such effects appear to reflect a specific action of IR, as Bak was not detected regardless of irradiation. An increase in Bcl‐XL levels was also noted when H460 lung cancer cells were irradiated with sublethal doses of γ‐rays (1 Gy). By way of contrast, Bcl‐2 levels in H460 cells were not, or were only minimally, elevated by the same treatment. This suggests that Bcl‐XL is a more common responder to IR, at least in these experimental settings. Therefore, Bcl‐XL was selected as a model for further study.
Figure 2.
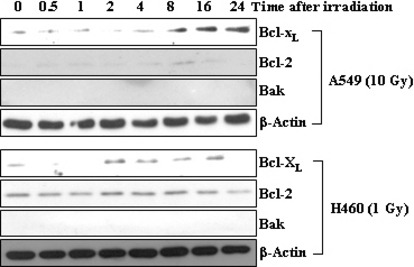
Bcl‐XL is a common target of ionizing radiation (IR) in both A549 and H460 cells. A549 and H460 cells were exposed to sublethal doses of IR, 10 and 1 Gy, respectively. At the indicated times after irradiation, the levels of Bcl‐XL, Bcl‐2, and Bak were compared via western blotting.
Bcl‐XL mimics IR to promote malignant features of A549 cells. To determine whether Bcl‐XL augments the malignant features of lung cancer cells, Bcl‐XL was overexpressed in A549 cells. This treatment indeed increased the phosphorylation levels of p38 kinase and Akt, protein levels of MMP‐2 (Fig. 3a), and the invasiveness of A549 cells (Fig. 3b). This latter event was again shown to be reduced by the use of inhibitors for p38 kinase, PI3K, Akt, and MMP‐2. Moreover, the overexpression of Bcl‐XL increased and decreased the levels of vimentin and E‐cadherin, respectively (Fig. 3a,c). These effects of Bcl‐XL are very reminiscent of those of IR, thereby indicating that Bcl‐XL mimics the actions of IR to stimulate malignant signals in A549 cells.
Figure 3.
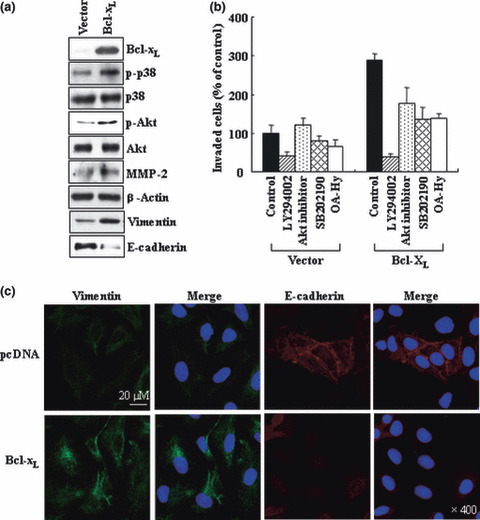
Bcl‐XL overexpression mimics the effects of ionizing radiation (IR). (a) A549 cells were transiently transfected with empty pcDNA3 vectors or vectors containing Bcl‐XL cDNA. The levels of the indicated components were compared via western blotting. (b) The control and Bcl‐XL transfectants were incubated with or without indicated inhibitors for 30 min, then analyzed for their invasiveness. (c) The transfectants were immuno‐stained and analyzed for vimentin and E‐cadherin.
Bcl‐XL mediates the malignant actions of IR. We next irradiated the control and Bcl‐XL transfectants with sublethal doses of IR. As compared to single treatment with either Bcl‐XL transfection or irradiation, combination treatment caused a further increase in Bcl‐XL levels, but induced no further increase in cellular invasiveness (Fig. 4a). Therefore, Bcl‐XL levels induced by either its transfection or irradiation might effectively promote cellular invasiveness. Our data support the hypothesis that Bcl‐XL operates in the IR‐induced signaling pathway. To more directly confirm this, IR‐induced Bcl‐XL accumulation was prevented via RNA interference. This treatment reduced the ability of IR to increase the phosphorylation levels of p38 kinase and Akt, MMP‐2 protein levels (Fig. 4b), and thus cellular invasiveness (Fig. 4c). Ionizing radiation also failed to increase the level of vimentin and reduce E‐cadherin levels when Bcl‐XL levels were reduced (Fig. 4b). Therefore, IR appears to induce all these malignant signals or events in a Bcl‐XL‐dependent manner. Overall, the data suggest that Bcl‐XL functions as a signaling component that mediates the actions of IR to promote cancer cell invasion and EMT.
Figure 4.
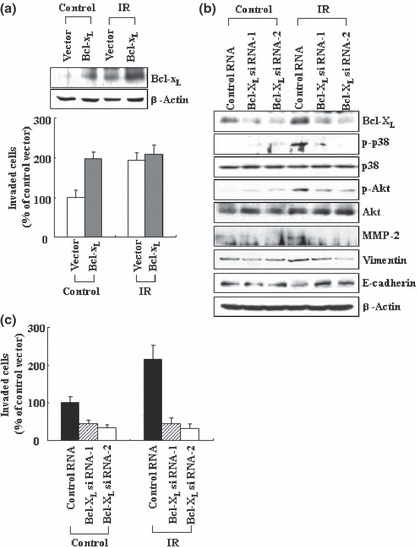
Bcl‐XL mediates the malignant actions of ionizing radiation (IR). (a) The transfectants were irradiated (10 Gy), and incubated for 24 h. Cellular levels of Bcl‐XL and cellular invasiveness were compared. (b) Control and Bcl‐XL siRNAs were introduced into A549 cells. After 24 h of incubation, the cells were irradiated, then incubated for an additional 24 h. Levels of the indicated components were compared via western blotting. (c) Cellular invasiveness was analyzed.
Signal transducer and activator of transcription 3 (STAT3) mediates IR‐induced Bcl‐XL accumulation. We subsequently assessed the cellular factors involved in IR‐induced Bcl‐XL accumulation. Reverse transcription–polymerase chain reaction (RT‐PCR) showed that the mRNA levels of Bcl‐XL increased in response to IR (Fig. 5a), suggesting that IR induces Bcl‐XL accumulation at the transcriptional level. This view led us to focus on STAT3, a transcription factor which can directly regulate the Bcl‐XL gene.( 38 , 39 ) Indeed, IR increased the phosphorylation levels of STAT3 without significantly affecting its protein levels (Fig. 5b). This verifies the ability of IR to activate STAT3, which was also proposed under other experimental conditions.( 40 , 41 ) Importantly, IR‐induced STAT3 phosphorylation was observed from 30 min after irradiation, and thus precedes IR‐induced Bcl‐XL accumulation. Consistent with this time sequence, the reduction of STAT3 levels by RNA interference attenuated the ability of IR to induce Bcl‐XL accumulation and thus Bcl‐XL‐dependent downstream events, including increases in phosphorylation or protein levels of p38 kinase, Akt, MMP‐2, and vimentin, as well as reductions in E‐cadherin levels (Fig. 5c). Moreover, IR failed to increase cells’ invasiveness when STAT3 levels were reduced (Fig. 5d). Overall, the data suggest that STAT3 mediates the ability of IR to induce invasion signals and EMT markers via the induction of Bcl‐XL accumulation.
Figure 5.
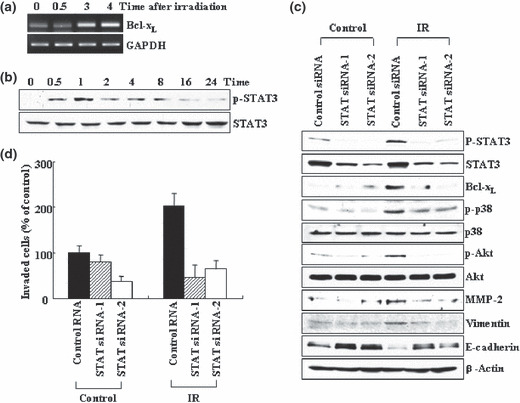
Signal transducer and activator of trans‐cription 3 (STAT3) mediates ionizing radiation (IR)‐induced Bcl‐XL accumulation. (a) A549 cells were irradiated. At the indicated times, the levels of Bcl‐XL mRNA were compared via RT‐PCR, using GAPDH as an internal control. (b) The irradiated cells were analyzed for levels of STAT3 protein and its phosphorylation via western blotting. (c) Control and STAT3 siRNAs were introduced into A549 cells. After 24 h of incubation, the cells were irradiated and incubated for an additional 24 h. Levels of the indicated components were compare via western blotting. (d) Cellular invasiveness was compared.
Transient action of IR. Figure 5(b) also shows that the levels of IR‐induced STAT3 phosphorylation were not stable, and decreased with continued incubation. This may reflect a downregulation of IR‐induced malignant signals. Indeed, Bcl‐XL, MMP‐2, vimentin, and E‐cadherin were restored to their original levels when incubation continued for up to 48–72 h (Fig. 6a). At this stage of post‐irradiation, the irradiated cells were not superior to untreated control cells in terms of their invasiveness (Fig. 6b). Therefore, the malignant effects of IR appear to be reversible.
Figure 6.
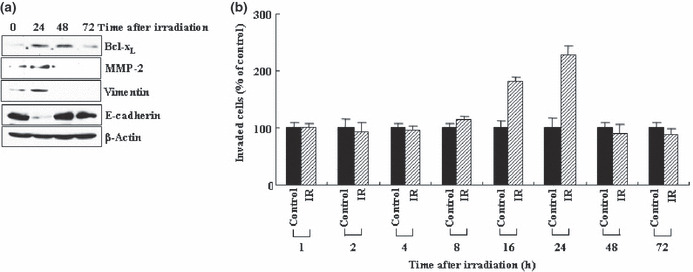
Ionizing radiation (IR) promotes invasion in a transient mode. (a) Levels of Bcl‐XL, matrix metalloproteinase‐2 (MMP‐2), vimentin, and E‐cadherin were analyzed by western blotting at the indicated times after irradiation. (b) Cellular invasiveness was compared at the times specified.
Cell‐type specificity of IR action. Given that IR induces Bcl‐XL accumulation not only in A549 cells but also in H460 cells (Fig. 2), we attempted to determine whether IR also functions in a Bcl‐XL‐dependent manner in the latter cell type. As in the case of A549 cells, the irradiation of H460 cells with doses of IR that elevated Bcl‐XL levels also resulted in an increase in the phosphorylation or protein levels of p38 kinase, Akt, MMP‐2, and vimentin, as well as a reduction in E‐cadherin levels (Fig. 7a). This confirms the ability of IR to stimulate invasion signals and EMT markers in H460 cells. Importantly, all of the effects of IR were mimicked by Bcl‐XL overexpression (Fig. 7b), and the reduction of Bcl‐XL accumulation by its RNA interference prevented the IR‐induced invasion of H460 cells (Fig. 7c), thereby suggesting that Bcl‐XL functions as a mediator of IR action in H460 cells. Moreover, irradiation also increased the levels of STAT3 phosphorylation, and the prevention of this event using STAT3 siRNAs abolished the actions of IR to stimulate or induce Bcl‐XL, p38 kinase, Akt, MMP‐2, and vimentin, to reduce E‐cadherin levels (Fig. 7d), and to promote the invasion of H460 cells (Fig. 7e). Therefore, STAT3 also appears to mediate IR‐induced Bcl‐XL accumulation in H460 cells. The data collectively suggest that Bcl‐XL and STAT3 mediate the ability of IR to induce invasion and EMT markers in multiple lung cancer cell types.
Figure 7.
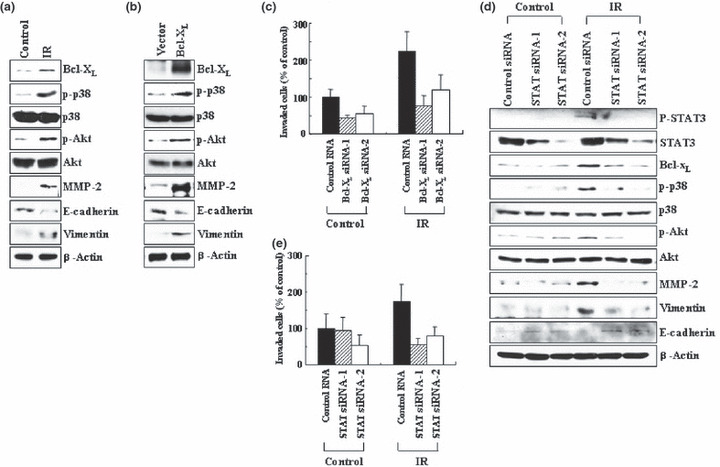
Role of Bcl‐XL and signal transducer and activator of transcription 3 (STAT3) in H460 cells. (a) H460 cells were irradiated (1 Gy), and incubated for 24 h. Levels of the indicated components were analyzed by western blotting. (b) H460 cells were transiently transfected with the control and Bcl‐XL expression vectors. Levels of the indicated components were compared. (c) Control and Bcl‐XL siRNAs were introduced into H460 cells. After 24 h of incubation, the cells were irradiated, incubated for an additional 24 h, and analyzed for invasiveness. (d) Control and STAT3 siRNAs were introduced into H460 cells. After 24 h of incubation, the cells were irradiated, incubated for an additional 24 h, and subsequently analyzed for the relevant components by western blotting. (e) Cellular invasiveness was compared.
Discussion
We have shown that IR enhances the invasiveness of lung cancer cells, A549 and H460, by stimulating a cellular pathway involving p38 kinase, Akt, and MMP‐2. These effects of IR were also accompanied by EMT‐associated events, including an increase in vimentin levels and a reduction in E‐cadherin levels. Therefore, we subsequently investigated the possible role of a pro‐survival Bcl‐2 family member in these malignant actions of IR in this system.
Our data do indeed suggest that Bcl‐XL functions as a signaling mediator of the malignant actions of IR. We focused on Bcl‐XL because levels of this molecule in both A549 and H460 cells were shown to be elevated upon irradiation with invasion‐promoting doses of IR. Similar results were obtained when the alternative lung cancer cells, H1299 and Calu‐1, were analyzed (data not shown), suggesting that Bcl‐XL is a common responder to IR in multiple lung cancer cell types. Moreover, the observation that the invasiveness of lung cancer cells is increased and decreased upon the overexpression and RNA interference (respectively) of Bcl‐XL confirms the ability of Bcl‐XL to promote cell invasion in this system. This function of Bcl‐XL was blocked by the use of inhibitors for p38 kinase, PI3K, Akt, and MMP‐2, suggesting that these components are also involved in Bcl‐XL‐induced invasion. Therefore, Bcl‐XL and IR appear to promote cell invasion via a commonly shared pathway. This view was supported further by the finding that Bcl‐XL overexpression mimicked the actions of IR in influencing phosphorylation or protein levels of p38 kinase, Akt, MMP‐2, vimentin, and E‐cadherin. Importantly, the ability of IR to influence all these components and thus to enhance cells’ invasiveness was attenuated in cases in which the levels of Bcl‐XL were reduced by RNA interference. Overall, our data indicate that IR promotes cell invasion and EMT‐associated events via the induction of Bcl‐XL accumulation. To the best of our knowledge, this is the first report to demonstrate directly that a pro‐survival Bcl‐2 family member can function as a signaling mediator of an extracellular stimulus. This role of Bcl‐XL can be contrasted with its anti‐apoptotic action, in which Bcl‐XL functions as a blocker of the apoptotic signal.( 42 , 43 ) It has been reported consistently that the ability of Bcl‐XL to suppress apoptosis and to enhance invasiveness can be dissociated in a manner dependent on the time after Bcl‐XL induction.( 20 )
We have also determined that STAT3 acts upstream of Bcl‐XL in the IR‐induced invasion pathway. The involvement of a transcription factor in IR‐induced Bcl‐XL accumulation was initially implied by the finding that IR increases not only Bcl‐XL protein levels, but also its mRNA levels. Given that STAT3 can induce Bcl‐XL expression under other experimental conditions,( 38 , 44 ) we focused on the role of STAT3 in this system. We confirmed that STAT3 responds to IR by showing that IR enhances STAT3 phosphorylation. This event preceded Bcl‐XL accumulation, suggesting that STAT3 is involved in IR‐induced Bcl‐XL accumulation. This latter event was indeed abolished when STAT3 levels were reduced as the result of RNA interference. Moreover, our finding that the reduction in STAT3 levels also attenuates the ability of IR to promote all of the Bcl‐XL‐dependent downstream events tested and the invasiveness of the cancer cells directly supports the notion that STAT3 mediates the ability of IR to induce invasion and EMT‐associated events via the induction of Bcl‐XL accumulation. In addition to these functions in IR‐induced invasion, STAT3 and Bcl‐XL may also contribute to the constitutive invasion activities of cancer cells. This latter view is based on the observation that phosphorylated STAT3 and Bcl‐XL exist in untreated control cells, and that the reduction of their levels via RNA interference can attenuate the invasiveness of the cells (4, 5). While STAT3 has been implicated in the regulation of the growth and survival of cancer cells,( 45 ) this study suggests its additional functions and illustrates a novel mechanism of its action.
In summary, we have demonstrated that IR induces Bcl‐XL accumulation via STAT3, which then promotes invasion and EMT‐associated events in lung cancer cells. This finding significantly advances our understanding not only of the role of Bcl‐XL in cancer, but also of the mechanism underlying the malignant actions of IR.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported by Nuclear Research & Development Program (M20702020002‐08N0202‐00210) of the National Research Foundation (NRF) of Korea, and in part by FG08‐21‐12 of the 21C Frontier Functional Human Genome Project and by Basic Science Research Program (R11‐2008‐014‐01001‐0) of the NRF.
References
- 1. Schmidt‐Ullrich RK. Molecular targets in radiation oncology. Oncogene 2003; 22: 5730–3. [DOI] [PubMed] [Google Scholar]
- 2. Barcellos‐Hoff MH, Park C, Wright EG. Radiation and the microenvironment‐tumorigenesis and therapy. Nat Rev Cancer 2005; 5: 867–75. [DOI] [PubMed] [Google Scholar]
- 3. Kumar P, Miller AI, Polverini PJ. Ap38 MAPK mediates γ‐irradiation‐induced endothelial cell apoptosis, and vascular endothelial growth factor protects endothelial cells through the phosphoinositide 3‐kinase‐Akt‐Bcl‐2 pathway. J Biol Chem 2004; 279: 43352–60. [DOI] [PubMed] [Google Scholar]
- 4. Park CM, Park MJ, Kwak HJ et al. Ionizing radiation enhances matrix metalloproteinase‐2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor‐mediated p38/Akt and phosphatidylinositol 3‐kinase/Akt signaling pathways. Cancer Res 2006; 66: 8511–9. [DOI] [PubMed] [Google Scholar]
- 5. Cheng JH, Chou CH, Kuo ML, Hsieh CY. Radiation‐enhanced hepatocellular carinoma cell invasion with MMP‐9 expression through PI3K/Akt/NF‐κB signal transduction pathway. Oncogene 2006; 25: 7009–18. [DOI] [PubMed] [Google Scholar]
- 6. Qian LW, Mizumoto K, Urashima T et al. Radiation‐induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin Cancer Res 2002; 8: 1223–7. [PubMed] [Google Scholar]
- 7. Kargiotis O, Chetty C, Gogineni V et al. uPA/uPAR downregulation inhibits radiation‐induced migration, invasion and angiogenesis in IOMM‐Lee meningioma cells and decreases tumor growth in vivo. Int J Oncol 2008; 33: 937–47. [PMC free article] [PubMed] [Google Scholar]
- 8. Canphausen K, Moses MA, Beecken WD, Khan MK, Folkman J, O’Reilly MS. Radiation therapy to a primary tumor accelerates metastatic growth in mice. Cancer Res 2001; 61: 2207–11. [PubMed] [Google Scholar]
- 9. Biswas S, Guix M, Rinehart C et al. Inhibition of TGF‐beta with neutralizing antibodies prevents radiation‐induced acceleration of metastatic cancer progression. J Clin Invest 2007; 117: 1305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Madani I, De Neve W, Mareel M. Does ionizing radiation stimulate cancer invasion and metastasis? Bull Cancer 2008; 95: 292–300. [DOI] [PubMed] [Google Scholar]
- 11. Suit HD. Local control and patient survival. Int J Radiat Oncol Biol Phys 1992; 23: 653–60. [DOI] [PubMed] [Google Scholar]
- 12. Cory S, Adams JM. The Bcl‐2 family: regulators of the cellular life‐or death switch. Nat Rev Cancer 2002; 2: 647–56. [DOI] [PubMed] [Google Scholar]
- 13. Belka C, Budach W. Anti‐apoptotic Bcl‐2 proteins: structure, function and relevance for radiation biology. Int J Radiat Biol 2002; 78: 643–58. [DOI] [PubMed] [Google Scholar]
- 14. Cory S, Huang DC, Adams JM. The Bcl‐2 family: roles in cell survival and oncogenesis. Oncogene 2003; 22: 8590–607. [DOI] [PubMed] [Google Scholar]
- 15. Yip KW, Reed JC. Bcl‐2 family proteins and cancer. Oncogene 2008; 27: 6398–406. [DOI] [PubMed] [Google Scholar]
- 16. Wick W, Wagner S, Kerkau S, Dichgans J, Tonn JC, Weller M. Bcl‐2 promotes migration and invasiveness of human glioma cells. FEBS Lett 1998; 440: 419–24. [DOI] [PubMed] [Google Scholar]
- 17. Choi J, Choi K, Benveniste EN et al. Bcl‐2 promotes invasion and lung metastasis by inducing matrix metalloproteinase‐2. Cancer Res 2005; 65: 5554–60. [DOI] [PubMed] [Google Scholar]
- 18. Trisciuoglio D, Desideri M, Ciuffreda L et al. Bcl‐2 overexpression in melanoma cells increases tumor progression‐associated properties and in vivo tumor growth. J Cell Physiol 2005; 205: 414–21. [DOI] [PubMed] [Google Scholar]
- 19. Espana L, Fernandez Y, Rubio N, Torregrosa A, Blanco J, Sierra A. Overexpression of Bcl‐xL in human breast cancer cells enhances organ‐selective lymph node metastasis. Breast Cancer Res Treat 2004; 87: 33–44. [DOI] [PubMed] [Google Scholar]
- 20. Weiler M, Bahr O, Hohlweg U et al. BCL‐xL: time‐dependent dissociation between modulation of apoptosis and invasiveness in human malignant glioma cells. Cell Death Differ 2006; 13: 1156–69. [DOI] [PubMed] [Google Scholar]
- 21. Du YC, Lewis BC, Hanahan D, Varmus H. Assessing tumor progression factors by somatic gene transfer into a mouse model: Bcl‐xL promotes islet tumor cell invasion. PLoS Biol 2007; 5: 2255–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bae IH, Park MJ, Yoon SH et al. Bcl‐w promotes gastric cancer cell invasion by inducing matrix metalloproteinase‐2 expression via phosphoinositide 3‐kinase, Akt, and Sp1. Cancer Res 2006; 66: 4991–5. [DOI] [PubMed] [Google Scholar]
- 23. Bae IH, Yoon SH, Lee SB, Park JK, Ho JN, Um HD. Signaling components involved in Bcl‐w‐induced migration of gastric cancer cells. Cancer Lett 2009; 277: 22–8. [DOI] [PubMed] [Google Scholar]
- 24. Lee HW, Lee SS, Lee SJ, Um HD. Bcl‐w is expressed in a majority of infiltrative gastric adenocarcinomas and suppresses the cancer cell death by blocking stress‐activated protein kinase/c‐Jun NH2‐terminal kinase activation. Cancer Res 2003; 63: 1093–1100. [PubMed] [Google Scholar]
- 25. Hoelzinger DB, Mariani L, Weis J et al. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia 2005; 7: 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang YL, Pang LQ, Wu Y, Wang XY, Wang CQ, Fan Y. Significance of Bcl‐xL in human colon carcinoma. World J Gastroenterol 2008; 14: 3069–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inayat MS, Chendil D, Mohiuddin M, Elford HL, Gallicchio VS, Ahmed MM. Didox (a novel ribonucleotide reductase inhibitor) overcomes bcl‐2 mediated radiation resistance in prostate cancer cell line PC‐3. Cancer Biol Ther 2002; 1: 539–45. [DOI] [PubMed] [Google Scholar]
- 28. Guo G, Wang T, Gao Q et al. Expression of ErbB2 enhances radiation‐induced NF‐κB activation. Oncogene 2004; 23: 535–45. [DOI] [PubMed] [Google Scholar]
- 29. Datta R, Manome Y, Taneja N et al. Overexpression of Bcl‐xL by cytotoxic drug exposure confers resistance to ionizing radiation‐induced inter‐nucleosomal DNA fragmentation. Cell Growth Differ 1995; 6: 363–70. [PubMed] [Google Scholar]
- 30. Aebersold DM, Kollar A, Beer KT, Laissue J, Greiner RH, Djonov V. Involvement of the hepatocyte growth factor/scatter factor receptor c‐met and of Bcl‐xL in the resistance of oropharyngeal cancer to ionizing radiation. Int J Cancer 2001; 96: 41–54. [DOI] [PubMed] [Google Scholar]
- 31. Das AK, Sato M, Story MD et al. Non‐small cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res 2006; 66: 9601–8. [DOI] [PubMed] [Google Scholar]
- 32. Kim DK, Cho ES, Seong JK, Um HD. Adaptive concentrations of hydrogen peroxide suppress cell death by blocking the activation of SAPK/JNK pathway. J Cell Sci 2001; 114: 4329–34. [DOI] [PubMed] [Google Scholar]
- 33. Naito A, Cook CC, Mizumachi T et al. Progressive tumor features accompany epithelial‐mesenchymal transition induced in mitochondrial DNA‐depleted cells. Cancer Sci 2008; 99: 1584–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petrella A, Ercolino SF, Festa M et al. Dexamethasone inhibits TRAIL‐induced apoptosis of thyroid cancer cells via Bcl‐xL induction. Eur J Cancer 2006; 42: 3287–93. [DOI] [PubMed] [Google Scholar]
- 35. Arias AM. Epithelial mesenchymal interactions in cancer and development. Cell 2001; 105: 425–31. [DOI] [PubMed] [Google Scholar]
- 36. Thiery JP. Epithelial‐mesenchymal transisions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 37. Jung JW, Hwang SY, Hwang JS, Oh ES, Park S, Han IO. Ionising radiation induces changes associated with epithelial‐mesenchymal transdifferentiation and increased cell motility of A549 lung epithelial cells. Eur J Cancer 2007; 43: 1214–24. [DOI] [PubMed] [Google Scholar]
- 38. Grad JM, Zeng XR, Boise LH. Regulation of Bcl‐xL: a little bit of this and a little bit of STAT. Curr Opin Oncol 2000; 12: 543–9. [DOI] [PubMed] [Google Scholar]
- 39. Lee TL, Yeh J, Friedman J et al. A signal network involving coactivated NF‐κB and STAT3 and altered p53 modulates BAX/BCL‐XL expression and promotes cell survival of head and neck squamous cell carcinomas. Int J Cancer 2008; 122: 1987–98. [DOI] [PubMed] [Google Scholar]
- 40. Singh‐Gupta V, Zhang H, Banerjee S et al. Radiation‐induced HIF‐1α cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int J Cancer 2009; 124: 1675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang HP, Long XH, Sun ZZ et al. Identification of differentially transcribed genes in human lymphoblastoid cells irradiated with 0.5 Gy of γ‐ray and the involvement of low dose radiation inducible CHD6 gene in cell proliferation and radiosensitivity. Int J Radiat Biol 2006; 82: 181–90. [DOI] [PubMed] [Google Scholar]
- 42. Labi V, Erlacher M, Kiessling S, Villunger A. BH3‐only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ 2006; 13: 1325–38. [DOI] [PubMed] [Google Scholar]
- 43. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 2009; 9: 501–6. [DOI] [PubMed] [Google Scholar]
- 44. Kunigal S, Lakka SS, Sodadasu PK, Estes N, Rao JS. Stat3‐siRNA induces Fas‐mediated apoptosis in vitro and in vivo in breast cancer. Int J Oncol 2009; 34: 1209–20. [PMC free article] [PubMed] [Google Scholar]
- 45. Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003; 101: 2940–54. [DOI] [PubMed] [Google Scholar]