

Abstract
Anaplastic large cell lymphoma (ALCL) and Hodgkin lymphoma (HL) express CD30 at high levels, but stimulation of this molecule has been reported to induce contradictory effects. To elucidate the molecular mechanism of CD30‐mediated apoptosis of ALCL, we compared the gene expression profiles of t(2;5)(p23;q35)‐positive ALCL with those of HL altered by CD30 agonistic stimulation. The results showed that BCL3, the high‐level expression of which in ALCL was previously reported, was further upregulated in response to CD30 stimulation, along with several pro‐apoptotic genes. Bcl‐3 protein was present as an intermediate phospho‐form in the resting‐state ALCL, becoming hyperphosphorylated (Bcl‐3P) upon stimulation. We next found that the stimulation promoted de novo synthesis of the nuclear factor (NF)‐κB2/p100 precursor as well as processing to p52, and a series of immunoprecipitation and western blotting analyses consistently showed association of Bcl‐3P with p52 in CD30‐stimulated ALCL. An electrophoretic mobility shift assay revealed the induction of κB binding activity of the p52 homodimer, and nuclear colocalization of Bcl‐3 and p52 was demonstrated in anaplastic lymphoma kinase‐positive ALCL tumor tissues by immunohistochemistry. As Bcl‐3 can act as an antirepressor or transactivator or both, we propose that the (p52)2/Bcl‐3P ternary complex, which is specifically induced in CD30‐stimulated ALCL, can modulate expression of apoptosis‐related genes regulated by NF‐κB, thereby accounting for CD30‐mediated apoptosis of ALCL. (Cancer Sci 2005; 96: 487–497)
Anaplastic large cell lymphoma (ALCL) was described initially as a subtype of aggressive lymphoma characterized by anaplastic cell morphology and the expression of CD30.( 1 ) In the late 1980s, cytogenetic studies identified a recurrent chromosomal translocation, t(2;5)(p23;q35), which was associated specifically with a fraction of ALCL.( 2 ) As a result of t(2;5)(p23;q35), the nucleophosmin (NPM) gene fuses upstream of the anaplastic lymphoma kinase (ALK) gene, encoding the NPM/ALK chimeric protein. NPM/ALK undergoes oligomerization, resulting in constitutive activation of the ALK tyrosine kinase function through tyrosine phosphorylation of NPM/ALK itself.( 3 ) Hodgkin lymphoma (HL), on the other hand, is characterized by the CD30‐positive Hodgkin/Reed–Sternberg (H/RS) cells, which are now considered to be derived from clonally expanded germinal center B cells.( 4 )
Anaplastic large cell lymphoma and HL share many clinicopathological and cellular features in addition to the expression of CD30.( 1 ) We showed previously that high‐level expression of the BCL3 gene, which encodes a nuclear protein belonging to the IκB family of inhibitors of nuclear factor (NF)‐κB transcriptional factors, differentiates t(2;5)(p23;q35)‐positive ALCL from HL.( 5 ) We found that NF‐κB components show a differential subcellular localization between ALCL and HL, and therefore proposed that high‐level expression of Bcl‐3 is responsible for the low level of NF‐κB binding activity in ALCL as compared with the constitutive activation of NF‐κB in HL.( 5 )
CD30 is a 120‐kDa transmembrane glycoprotein belonging to the tumor necrosis factor (TNF) receptor superfamily.( 6 , 7 ) The interaction of CD30 with its natural ligand, CD30L, is involved in the control of either cell survival or death by apoptosis.( 8 ) These contradictory effects induced by CD30 stimulation are highlighted by the effects on ALCL and HL; ALCL cells show apoptotic cell death in response to the stimulation of CD30, while the viability of HL cells is unaffected by the stimulation.( 8 , 9 , 10 ) The CD30‐mediated cell death of ALCL cells is suggested to be due to their inability to activate NF‐κB in response to the stimulation of CD30;( 10 ) however, the mechanism that links the stimulation with apoptosis is not yet fully understood.
We report here that the stimulation of CD30 in ALCL leads to the expression of Bcl‐3 at higher levels than in the resting state along with an upregulation of pro‐apoptotic genes. Furthermore, Bcl‐3 becomes hyperphosphorylated (Bcl‐3P) and synthesis of NF‐κB2/p100 precursors, as well as processing to p52, is promoted in ALCL upon stimulation of CD30. The significance of the interaction of Bcl‐3P with p52 in CD30‐stimulated ALCL cells will be discussed.
Materials and Methods
Cell lines
The ALCL cell lines Karpas 299, DEL and SR‐786 were provided by Drs Y. Matsuo (Fujisaki Cell Center, Okayama, Japan) and H. Drexler (DSMZ, Braunschweig, Germany).( 11 ) These ALCL cell lines carried t(2;5)(p23;q35) or its equivalent, and the presence of the NPM/ALK fusion genes was confirmed by long‐distance polymerase chain reaction.( 5 ) The HL cell line KM‐H2 and the CD30‐positive diffuse large B‐cell lymphoma (DLBCL) cell line KIS‐1 carrying t(9;14)(p13;q32) were established and characterized in our laboratory.( 12 , 13 ) All cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum under standard conditions.
Immobilization of CD30‐agonistic antibody, stimulation of cells and viability assays
The CD30‐agonistic antibody HeFi‐1 was provided by the Biological Resources Branch, National Cancer Institute (Rockville, MD, USA). Murine immunoglobulin G1 (IgG1) isotype control antibody was purchased from eBioscience (#16‐4714; San Diego, CA, USA). Immobilization of these antibodies was carried out according to the method described by Mir et al.( 10 ) Briefly, polystyrene six‐well plates were coated with either of the antibodies, which were suspended in phosphate‐buffered saline (PBS) at a concentration of 20 µg/mL, for 36 h at 4°C. Wells were washed and blocked with PBS containing 1% bovine serum albumin (PBS‐BSA) at 4°C overnight. Cells of interest were dispensed into wells coated with immobilized antibodies at a concentration of 1 × 106/mL, and incubated for 24 h at 37°C.
Cell viability was measured by flow cytometry using propidium iodide (PI) exclusion.( 10 ) The extent of cell death (%) due to the stimulation of CD30 was calculated as follows: ([1 − number of viable cells from wells coated with HeFi‐1 antibodies/number of viable cells from wells coated with isotype control antibodies] × 100). Experiments were carried out in triplicate.
cDNA array hybridization
Total cellular RNA was extracted from the cells using an RNeasy kit (Qiagen, Tokyo, Japan). After incubation with 5 U of RNase‐free DNase, the RNA was reverse transcribed in the presence of [32P]dATP using the Atlas Pure Total RNA Labeling System (Clontech, Palo Alto, CA, USA). Hybridization of the cDNA with an Atlas Human 1.2 Array (Clontech) was carried out according to the manufacturer's instructions. The membranes were exposed to a BAS2500 imaging analyzer (Fuji Photo Film, Tokyo, Japan) and the hybridization signals were quantified by ArrayGauge software version 1.2 (Fuji Photo Film). Data from each array were normalized using the median value to eliminate variability due to sample labeling.
Northern and western blot analyses and immunoprecipitation
Northern blotting was carried out as described previously.( 5 ) The cDNA probe for BCL3 was the clone cLK2 containing an 1.8‐kb cDNA fragment.( 14 ) Western blotting was carried out as described previously( 5 ) and the following antibodies were used: anti‐Bcl‐3 (sc‐185), anti‐β‐tubulin (sc‐5274), anti‐p50 (sc‐8414), anti‐p65 (sc‐8008, sc‐372), anti‐p52 (sc‐7386), anti‐RelB (sc‐226) and anti‐c‐Rel (sc‐71), which were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For immunoprecipitation, cells were lysed in Triton X lysis buffer (50 mmol/L Tris‐HCl [pH 7.4], 150 mmol/L NaCl, 1% Triton X, 5 mmol/L ethylenediaminetetracetic acid, 1 mmol/L phenylmethylsulfonyl fluoride, 3 mmol/L Na3VO4 and 50 mmol/L NaF), and rotated at 4°C with the antibody for 2 h and then overnight with protein G‐sepharose. The sepharose beads were pelleted, washed three times with lysis buffer, resuspended in sample buffer, heat‐denatured at 95°C for 5 min and subjected to western blot analysis.
Dephosphorylation of Bcl‐3 protein
Bcl‐3 was prepared in whole cell extracts. Cells were lysed in Nonidet P‐40 lysis buffer (20 mmol/L HEPES‐KOH [pH 7.8], 200 mmol/L NaCl, 0.2% Nonidet P‐40, 20% glycerol and 1 mmol/L dithiothreitol) containing a protease‐inhibitor cocktail. One unit of calf intestinal alkaline phosphatase (CIP; New England Biolabs, Beverly, MA, USA) was added to 30 µg of each cell lysate and incubated in CIP reaction buffer (60 mmol/L NaCl, 20 mmol/L HEPES‐KOH [pH 7.8], 2 mmol/L MgCl2 and 2 mmol/L dithiothreitol, with a protease‐inhibitor cocktail) with or without 3 mmol/L Na3VO4 and 50 mmol/L NaF for 1 h at room temperature. Then, 10 mmol/L ethylenediaminetetracetic acid was added to each sample at the end of the reaction and the samples were analyzed by western blotting.
Retroviral transduction of the BCL3 gene
A fragment of BCL3 cDNA was cloned into the BamHI site of a pFB‐IRES‐GFP vector.( 15 ) The 293T cells were then transfected with either pFB‐IRES‐GFP (mock) or pFB‐BCL3‐IRES‐GFP retroviral expression constructs using FuGene 6 Transfection Reagent (Roche, Mannheim, Germany) to obtain the recombinant virus. After 2 days of retroviral infection of HL cells, GFP‐positive cells were sorted by FACSVantage (Becton Dickinson, San Jose, CA, USA) and were maintained in bulk culture. After amplification in cell number, the cells were stimulated with HeFi‐1 for 24 h and were subjected to western blotting for Bcl‐3. Cell death was assessed by PI staining.
Electrophoretic mobility shift assay for nuclear factor‐κB activity
Electrophoretic mobility shift assay (EMSA) was carried out as described previously.( 5 ) The sequences of the oligonucleotide probes corresponding to a κB site of the human immunodeficiency virus promoter region (HIV κB) and MHC class I gene (H2 κB) were 5′‐AGTTGAGGGGACTTTCCCAGGC‐3′ and 5′‐CAGGGCTGGGGATTCCCATCTCCCACAGTTTCACTTC‐3′, respectively. For competition experiments, incubations were carried out with a 100‐fold excess of unlabeled probe. For antibody supershift assays, the lysates were preincubated for 10 min at 25°C with 4 µg of antibody before the addition of the radiolabeled gel shift probe. Antibodies used for the supershift assay were anti‐p50 (sc‐1190X), anti‐p65 (sc‐8008X), anti‐RelB (sc‐226) and anti‐c‐Rel (sc‐71) from Santa Cruz Biotechnology, and anti‐p52 (100‐4185) from Rockland (Gilbertsville, PA, USA).
Anaplastic large cell lymphoma tissues and immunohistochemical staining
Formalin‐fixed, paraffin‐embedded tissue sections were stained with hematoxylin and eosin, BerH2/CD30 antibody (Dako, Carpinteria, CA, USA), and ALK1 monoclonal antibody (Dako) according to the manufacturer's instructions. Monoclonal anti‐Bcl‐3 antibody (clone 1E8, Novocastra, Newcastle upon Tyne, UK) and monoclonal anti‐NF‐κB p52 antibody (#05‐361; Upstate Biotechnology, Charlottesville, VA, USA) were used as described previously.( 16 , 17 )
Results
CD30 stimulation by an agonistic antibody leads to apoptosis of anaplastic large cell lymphoma cells
We first compared the effect of CD30 stimulation on the five CD30‐positive lymphoma cell lines. The cells were incubated for 24 h with immobilized CD30‐agonistic antibody HeFi‐1 or isotype control antibody, and cell death was evaluated based on PI exclusion. As described by Mir et al.,( 10 ) significant fractions of ALCL cells stimulated with the agonistic antibody showed apoptotic cell death (Fig. 1a). The percentage of apoptotic cells after 24 h incubation ranged from 20% to 46% among the three ALCL cell lines (Fig. 1b). In contrast, KM‐H2 was resistant to the CD30‐mediated apoptosis, and viable cell numbers were not affected significantly by the stimulation as compared with the control. KIS‐1 was susceptible to the stimulation with HeFi‐1; the rate of cell death was comparable to or even higher than the rates for the ALCL cell lines.
Figure 1.
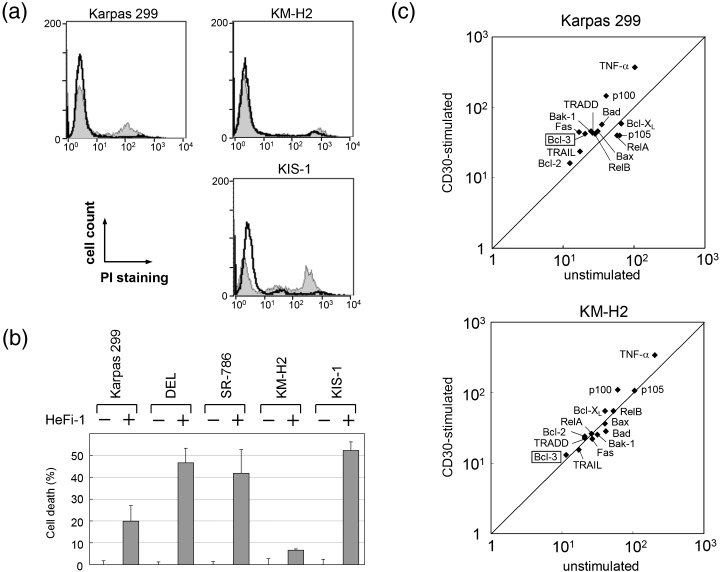
Apoptotic cell death of CD30‐positive lymphoma cell lines induced by agonistic stimulation of CD30. The cells were incubated for 24 h with either the immobilized CD30‐agonistic antibody HeFi‐1 or isotype‐matched control antibody, and (a) representative histograms of propidium iodide staining are shown. The cells stimulated by the control antibody are indicated by the bold line and CD30‐stimulated cells are indicated by the line forming the boundary of the grey space. (b) Cell death rate induced by CD30 stimulation. For each cell line, the viability of cells incubated with control antibody is defined as zero. Data represent the mean ± SD of three independent experiments. (c) Scatter plot analyses comparing the expression levels of representative genes between CD30‐stimulated Karpas 299 or KM‐H2 (y‐axis) and their unstimulated counterparts (x‐axis). After global normalization, log10 of the expression level was plotted for each gene.
Expression level of BCL3 mRNA in anaplastic large cell lymphoma cells is increased along with levels of pro‐apoptotic genes in response to stimulation of CD30
To elucidate the contradictory effects of stimulation of CD30 on ALCL and HL, we next compared changes in the gene expression profiles of Karpas 299 and KM‐H2 upon stimulation of CD30. 32P‐labeled cDNA prepared from the cells before and after stimulation with HeFi‐1 were hybridized with the Atlas Human 1.2 Array membranes, and the expression data were analyzed using the ArrayGauge software. After global normalization, we found that 162 genes in Karpas 299 and 115 genes in KM‐H2 were upregulated by more than 2.0‐fold upon stimulation of CD30, whereas 229 genes in Karpas 299 and 69 genes in KM‐H2 were downregulated by less than 0.7‐fold. A comparison of the affected genes between Karpas 299 and KM‐H2 revealed that as little as 19 and 26 genes, respectively, overlapped between the two cell lines. This differential alteration of expression profiles is most likely a reflection of the distinctive cellular response to stimulation of CD30 between ALCL and HL.
A scatter plot analysis comparing the levels of genes of interest before and after CD30 stimulation revealed that the genes for TNF‐α, Fas, TNF receptor 1‐associated death domain protein (TRADD) and TNF‐related apoptosis inducing ligand (TRAIL), all of which are involved in death signals transmitted by the TNF receptor family, were more upregulated in Karpas 299 than in KM‐H2 (Fig. 1c). Gene expression of Bax, Bak‐1 and Bad, which are members of the pro‐apoptotic Bcl‐2 family, was also increased in Karpas 299 cells. In contrast, Bcl‐XL, a member of the anti‐apoptotic Bcl‐2 family, was upregulated in KM‐H2 but not in Karpas 299. The expression levels of BCL2 mRNA, the basal level of which was higher in KM‐H2 than Karpas 299, was not significantly changed in either cell line. Thus, it is apparent that in Karpas 299, CD30 stimulation preferentially activates a series of genes involved in both intrinsic and extrinsic proapoptotic signaling pathways as compared with that in KM‐H2.
We showed previously that overexpression of the BCL3 gene differentiates ALCL from HL.( 5 ) Of particular interest, the level of BCL3 mRNA in Karpas 299 was upregulated 2.08‐fold upon stimulation of CD30, while the level in KM‐H2 remained low. With regard to the NF‐κB family, the expression level of NFKB2, which encodes the p100 precursor of the p52 protein, showed the most prominent change; being upregulated 3.61‐fold in Karpas 299 and 1.79‐fold in KM‐H2. The extent of the alteration of other NF‐κB members in Karpas 299 was as follows: RELA/p65, downregulated 0.69‐fold; RELB, upregulated 1.47‐fold; and NFKB1/p105, downregulated 0.63‐fold. The changes in the mRNA levels of these genes in KM‐H2 were not significant.
Stimulation of CD30 in anaplastic large cell lymphoma cells leads to enhanced BCL3 expression at both the mRNA and protein levels, and Bcl‐3 protein is hyperphosphorylated
To confirm the enhanced expression of BCL3 in ALCL cells induced by the stimulation of CD30, we carried out northern blotting using a BCL3 cDNA clone as the probe, and western blotting using an anti‐Bcl‐3 polyclonal antibody. The results showed clearly that the levels of BCL3 expression were enhanced at both the mRNA and protein levels in response to the stimulation of CD30 in the three ALCL cell lines (Fig. 2a,b). In contrast, BCL3 mRNA and protein levels were not significantly increased in KM‐H2 or KIS‐1.
Figure 2.
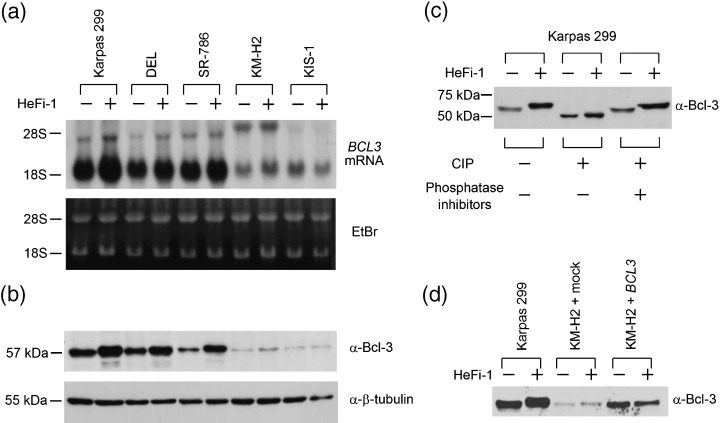
Alteration of the levels of BCL3 mRNA and Bcl‐3 protein, and phosphorylation status upon stimulation of CD30. (a) Northern blotting analysis of RNA extracted from CD30‐positive lymphoma cell lines with or without stimulation of CD30. Total RNA samples (10 µg) were electrophoresed on MOPS‐formaldehyde gel and hybridized with the BCL3 cDNA probe. The patterns of the 28S and 18S ribosomal RNA bands are indicated at the bottom to verify that comparable amounts of RNA were present in each lane. (b) CD30‐positive lymphoma cell lines with or without stimulation of CD30 were analyzed for Bcl‐3 protein expression by western blotting. Equal amounts (20 µg) of cell lysates were loaded onto sodium dodecylsulphate‐polyacrylamide gels. Western blotting for β‐tubulin is shown as a loading control. The samples are ordered as in (a). (c) Calf intestinal alkaline phosphatase (CIP) treatment of Bcl‐3 protein extracted from Karpas 299 with or without stimulation of CD30. Phosphatase inhibitors (NaF and Na3VO4) were added to the samples indicated to confirm that the degradation was a phosphatase‐specific reaction. (d) Comparison of Bcl‐3 protein expression among Karpas 299, mock‐transfected and BCL3‐transfected KM‐H2 with or without CD30 stimulation.
We noted that the Bcl‐3 proteins extracted from stimulated ALCL cells migrated more slowly than those extracted from the unstimulated cells. Bcl‐3 protein is composed of 15% serine, threonine or tyrosine residues (68 of 446 amino acids) and has been reported to be phosphorylated mainly at the carboxy‐terminus.( 14 , 18 ) We therefore speculated that the alteration in size reflects the extent to which the protein is phosphorylated. We prepared whole‐cell lysates of Karpas 299 with or without stimulation by HeFi‐1 and subjected them to treatment with CIP, and the change in the molecular size of Bcl‐3 was examined by western blotting. As shown in Fig. 2c, the Bcl‐3 protein extracted from unstimulated Karpas 299 had a molecular size of 57 kDa, whereas that from stimulated cells was larger in size. CIP treatment of both types of Bcl‐3 protein resulted in a reduction in size to 48 kDa, which matched the size of Bcl‐3 predicted from its cDNA sequence, and the dephosphorylation reaction was sufficiently repressed by phosphatase inhibitors. Thus, we concluded that Bcl‐3 protein in ALCL is present as intermediate phospho‐forms in the resting state, and the stimulation of CD30 leads to its hyperphosphorylation.
We next used a retroviral vector to introduce the BCL3 gene into KM‐H2 cells, in which the level of endogenous Bcl‐3 was much less than ALCL, and examined whether hyperphosphorylation of Bcl‐3 and CD30‐induced apoptosis are reproducible in the setting of HL. As shown in Fig. 2d, the size of the Bcl‐3 protein expressed in KM‐H2 was comparable to that in Karpas 299. Upon stimulation by CD30, however, the size of Bcl‐3 in transfected KM‐H2 was not altered. Accordingly, the cell death rate of BCL3‐transfected cells did not increase significantly upon stimulation as compared to that of mock‐transfected cells (increment: 1.5% and 1.0%, respectively). These observations were confirmed by additional experiments using another HL cell line, L428. It is therefore suggested that hyperphosphorylation of Bcl‐3 is a characteristic feature of ALCL and Bcl‐3P probably contributes to CD30‐mediated apoptosis of ALCL.
Stimulation of CD30 leads to de novo synthesis of the NF‐κB2/p100 precursor as well as processing to p52 in anaplastic large cell lymphoma
As described above, the cDNA array analysis showed that the expression level of NF‐κB2/p100 mRNA was elevated significantly upon stimulation of CD30 in ALCL cells, as well as in HL to a lesser extent. On the other hand, it has been reported that p100 undergoes inducible processing to p52 under certain conditions.( 17 , 19 , 20 , 21 ) We therefore examined the protein levels of the p100 precursor and p52 in CD30‐positive lymphoma cells with or without stimulation of CD30. Although there was no detectable level of p52 in the unstimulated state in any of the cell lines tested, the stimulation of CD30 led to the generation of p52 to varying degrees among the cell lines (Fig. 3a), and it was apparent that the ALCL cell lines consistently contained more p52 than KM‐H2 and KIS‐1. Furthermore, the levels of p100 were also elevated, indicating that the stimulation of CD30 in ALCL induces both de novo synthesis of the p100 precursor and processing to p52. On the other hand, KM‐H2 cells contained the largest amount of p100 in the resting state, probably reflecting the constitutive activation of NF‐κB;( 22 ) however, a minor fraction was processed to p52 in response to the stimulation of CD30.
Figure 3.
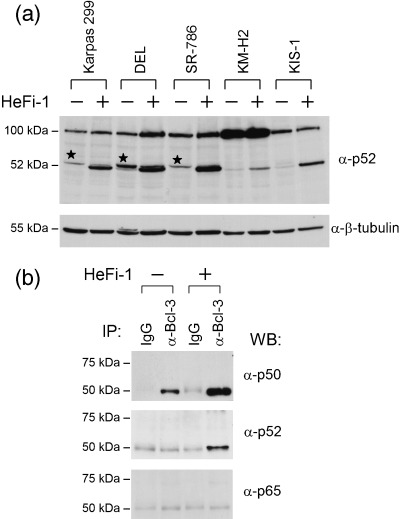
Induction of p52 upon stimulation of CD30, and interaction between Bcl‐3 and CD30‐induced p52 in anaplastic large cell lymphoma. (a) Western blot analysis of proteins from CD30‐positive lymphoma cell lines with or without stimulation of CD30 using an anti‐p52 monoclonal antibody. p100 and its processed product, p52, are indicated. The bands indicated by asterisks were detected reproducibly by another anti‐p52 antibody. Western blotting for β‐tubulin is shown as a loading control. (b) Cell lysates of Karpas 299 with or without stimulation of CD30 were immunoprecipitated with an anti‐Bcl‐3 antibody or a rabbit polyclonal IgG antibody, and western blotted with the antibodies indicated.
P52 is associated with Bcl‐3P as well as dimerized with p65 and RelB in stimulated anaplastic large cell lymphoma
Although Bcl‐3 belongs to the IκB family, Bcl‐3 is localized within the nucleus, and is reported to have high affinity for the (p50)2 and (p52)2 homodimers.( 23 , 24 ) Thus, the upregulation of Bcl‐3 as well as the induction of p52 upon stimulation of CD30 in ALCL cells prompted us to investigate the interaction between these two proteins. Cell lysates extracted from Karpas 299 with or without stimulation by HeFi‐1 were immunoprecipitated with an anti‐Bcl‐3 antibody and then western blotted with antibodies against NF‐κB components (Fig. 3b). As reported previously,( 5 ) Bcl‐3 was associated with p50 in the unstimulated cells, and the level of association was apparently increased in the CD30‐stimulated cells. Conversely, p52 was coprecipitated with Bcl‐3 after the stimulation. Other NF‐κB components with a Rel homology domain, that is, p65 (Fig. 3b), RelB and c‐Rel (data not shown), had no detectable interaction with Bcl‐3.
We next compared the association of p100/p52 with NF‐κB components among the three ALCL cell lines, KM‐H2 and KIS‐1. Cell lysates extracted from each cell line with or without stimulation of CD30 were subjected to immunoprecipitation with the anti‐p52 antibody, followed by western blotting with antibodies against NF‐κB components in addition to Bcl‐3 (Fig. 4a). The association was confirmed by a series of immunoprecipitation and western blotting analyses using the antibody combination in the reverse order (Fig. 4b). As shown in Fig. 4, the association between p52 and Bcl‐3 was induced exclusively in the CD30‐stimulated ALCL cell lines and the Bcl‐3 migrated to the hyperphosphorylated position. In addition, p52 was associated with p65 and RelB after the stimulation, while p100 was the partner of the two NF‐κB components in the unstimulated state. It was therefore concluded that the stimulation of CD30 in ALCL leads to the generation of a (p52)2/Bcl‐3P ternary complex as well as heterodimers of p65/p52 and RelB/p52.
Figure 4.
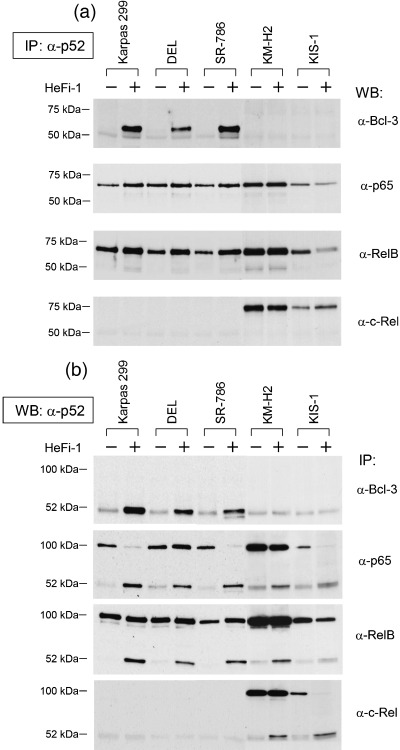
Patterns of association of p52 with Bcl‐3 and other nuclear factor (NF)‐κB components among CD30‐positive lymphoma cell lines. (a) Cell lysates of CD30‐positive lymphoma cell lines with or without stimulation of CD30 were immunoprecipitated with an anti‐p52 antibody and western blotted with antibodies against Bcl‐3 or other NF‐κB components. (b) Confirmation of the association of p100/p52 with Bcl‐3 or other NF‐κB components. Cell lysates were first immunoprecipitated with the antibodies indicated and then western blotted with an anti‐p52 antibody.
On the other hand, KM‐H2 cells contained heterodimers of p65/p100, RelB/p100 and c‐Rel/p100 in the resting state, and upon stimulation of CD30, a small fraction of p100 was processed to p52 to generate heterodimers with the three components. In KIS‐1 cells, a major fraction of p52 processed on stimulation of CD30 was bound to c‐Rel. These results indicated clearly that p52, whose production is induced by the stimulation of CD30, constitutes unique complexes in each cell line, potentially leading to differential NF‐κB activities among the three different types of CD30‐positive lymphomas.
CD30 stimulation promotes unique NF‐κB activities among CD30‐positive lymphomas
To better understand the CD30‐mediated NF‐κB activation among three different types of CD30‐positive lymphomas, we used an EMSA to compare the effects of CD30 stimulation on the NF‐κB‐binding activity. Whole‐cell lysates from lymphoma cells with or without stimulation by HeFi‐1 were incubated with the 32P‐labeled HIV κB probe. As indicated in Fig. 5a, the binding activities of the two ALCL cell lines, Karpas 299 and SR‐786, were increased in response to the stimulation, while those of DEL, KM‐H2 and KIS‐1 were not significantly affected. Supershift analyses with the anti‐p65, anti‐p50 and anti‐p52 antibodies demonstrated that the binding activities observed in CD30‐stimulated ALCL involved the p65/p50 heterodimer as well as (p50)2 homodimer (Fig. 5b); however, this study was not capable of detecting the DNA binding activity of p52.
Figure 5.
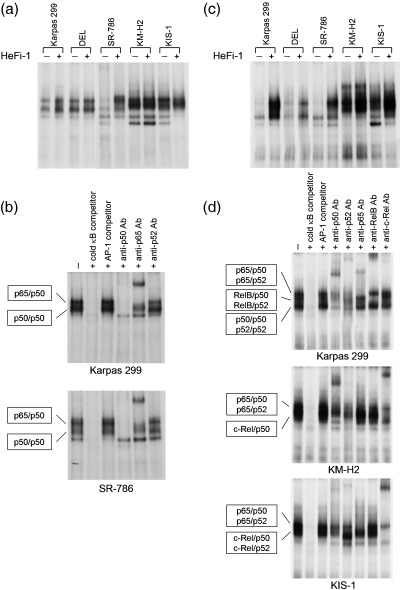
Comparison of nuclear factor (NF)‐κB binding activity among CD30‐positive lymphomas with or without stimulation of CD30. (a) Whole‐cell lysates of CD30‐positive lymphoma cell lines with or without stimulation of CD30 were incubated with the 32P‐labeled HIV κB probe, and κB binding was analyzed by electrophoretic mobility shift assay (EMSA). (b) Supershift assay of CD30‐stimulated Karpas 299 and SR‐786 using the antibodies indicated. Addition of anti‐Bcl‐3 antibodies produced no apparent supershifted bands (data not shown). The specificity of κB binding was determined by competition with an excess amount of unlabeled HIV κB probe or control unlabeled AP1 probe (5′‐CGCTTGATGAGTCAGCCGGAA‐3′). (c) NF‐κB DNA‐binding activities of p52‐containing components were compared among CD30‐positive lymphoma cell lines upon stimulation of CD30. The whole‐cell lysates were incubated with the 32P‐labeled H2 κB probe, and the binding was analyzed by EMSA. (d) Supershift assay of CD30‐stimulated Karpas 299, KM‐H2 and KIS‐1. The cell lysates were first incubated with antibodies against p50, p52, p65, RelB and c‐Rel, and the components of the NF‐κB‐binding complexes in each cell line were determined. The specificity of κB binding was determined by competition with an excess amount of unlabeled H2 κB probe or control unlabeled AP1 probe.
We next prepared the H2 κB probe that is known to bind to p52 with higher affinity than the HIV κB probe.( 25 ) The DNA binding activity in unstimulated ALCL cells was quite weak, while KM‐H2 and KIS‐1 showed high levels of activity (Fig. 5c). When stimulated with HeFi‐1, the cells showed increased levels of binding activity; however, the configuration of the binding by EMSA varied considerably among cell types. We carried out a series of supershift assays by adding antibodies against the five NF‐κB components (Fig. 5d). The results showed that the binding activities of CD30‐stimulated Karpas 299 cells involved three major complexes (i.e. p65/p50 and p65/p52 heterodimers, RelB/p50 and RelB/p52 heterodimers, and (p50)2 and (p52)2 homodimers). The remaining two ALCL cell lines seemed to lack measurable levels of DNA binding activity by the RelB‐containing components (data not shown).
In contrast, CD30‐stimulated KM‐H2 cells showed two identifiable bands on EMSA; the slower migrating band consisted of p65/p50 and p65/p52 heterodimers, while the c‐Rel/p50 heterodimer was the major component of the faster‐migrating band. KIS‐1 produced a smear‐like configuration, probably reflecting the apoptotic conditions after the stimulation. Nevertheless, the supershift assay revealed the DNA binding activities of the p65/p50 and p65/p52 heterodimers, and c‐Rel/p50 and c‐Rel/p52 heterodimers.
These results clearly indicated that the NF‐κB components identified by the immunoprecipitation and western blotting analyses are capable of binding to the κB sites of the DNA. Thus, stimulation of CD30 most likely leads to unique NF‐κB activities among CD30‐positive lymphomas, potentially accounting for the differential cellular responses induced by the stimulation.
Immunohistochemical detection of Bcl‐3 and p52 in lymphoma tissue of patients with ALK‐positive anaplastic large cell lymphoma
To confirm the nuclear colocalization of Bcl‐3 and p52 in lymphoma tissues, we used an immunohistochemical analysis of three patients with ALK‐positive ALCL (Fig. 6). All three cases were diagnosed as the common variant of ALCL; the lymphoma cells had typical kidney‐shaped nuclei and abundant cytoplasm. The lymphoma cells were strongly positive for CD30 on the cell membrane, and positive staining for ALK1 was observed in both the nucleus and cytoplasm (cases 1 and 2) or restricted to the cytoplasm (case 3). Thus, the last patient probably carried a variant translocation involving the ALK locus at 2p23 instead of t(2;5)(p23;q35).( 26 ) Case 1 was confirmed as positive for the NPM/ALK fusion gene.
Figure 6.
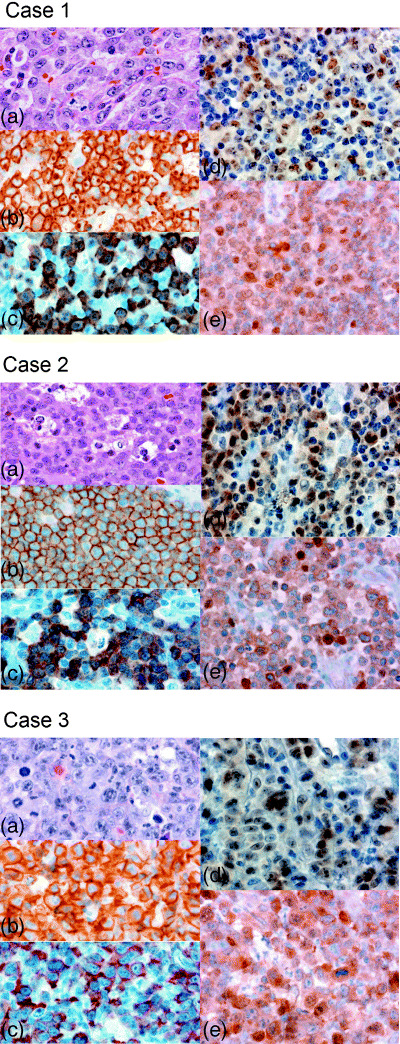
Immunohistochemistry of ALK‐positive anaplastic large cell lymphoma (ALCL) tumor tissues. Tumor biopsies of ALCL were analyzed sequentially with (a) hematoxylin and eosin, and with (b) anti‐CD30, (c) anti‐ALK, (d) anti‐Bcl‐3 and (e) anti‐p52 monoclonal antibodies. Original magnification, ×400.
As shown in Fig. 6, lymphoma cells of these three ALCL patients showed consistent strong nuclear staining by a monoclonal antibody raised against Bcl‐3. On the other hand, p52 was positive in both the cytoplasm and nucleus, and a fraction of lymphoma cells showed strong nuclear staining for anti‐p52. This study confirmed that the association of p52 and Bcl‐3, which was observed in CD30‐stimulated ALCL cell lines, does occur in a fraction of lymphoma cell nuclei in ALCL tumor tissues.
Discussion
The TNF receptor superfamily is a group of type I transmembrane proteins, comprising more than 20 members thus far identified in mammalian cells.( 27 , 28 ) Signal transduction events mediated by the members of this family have been shown to elicit a wide spectrum of cellular responses, ranging from stimulation of proliferation to initiation of the apoptotic cell death pathway.( 28 ) Several TNF receptor family members mediate apoptosis through their death domain (DD) composed of approximately 80 residues.( 29 ) Upon stimulation, the DD oligomerizes with DD‐containing adaptor molecules,( 30 , 31 ) and the complex then recruits a series of caspases, which are the central effector proteases of the cell death program. In contrast, the cytoplasmic tail of the TNF receptor molecules, including CD30, is associated with TNF receptor‐associated factors (TRAF), leading to the activation of NF‐κB transcription factors.( 32 ) As NF‐κB generally promotes the expression of anti‐apoptotic genes,( 33 ) the ultimate effect of TNF receptor‐mediated signals appears to be determined by the balance between the two contradictory responses.
CD30 was identified originally as being expressed at high levels on the surface of H/RS cells of HL,( 1 , 6 ) and the CD30‐TRAF‐NF‐κB pathway is activated constitutively through the ligand‐independent activation of CD30,( 34 ) probably contributing to the neoplastic cell growth of H/RS cells. However, CD30, which lacks DD, is capable of inducing apoptotic cell death,( 35 , 36 ) and the mechanism of apoptotic signaling initiated by CD30 has been elucidated. A proposed model of CD30‐mediated apoptosis of ALCL is a reduction of TRAF,( 10 ) which is in agreement with the very recent observation that the NPM/ALK chimeric protein can bind to TRAF to prevent its association with CD30.( 37 ) In the present study, we first confirmed that the stimulation of CD30 is capable of inducing apoptotic cell death in ALCL. In parallel, several pro‐apoptotic genes of both the TNF receptor and Bcl‐2 families were upregulated more in ALCL than HL. We next showed that the NF‐κB binding activity of ALCL cells is increased significantly in response to the stimulation of CD30. Thus, the CD30‐TRAF‐NF‐κB pathway may not be perturbed in CD30‐stimulated ALCL cells.
A peculiar finding of our study is that the stimulation of CD30 leads to an upregulation of BCL3 mRNA expression and enhancement of Bcl‐3 protein production in ALCL. These results imply that Bcl‐3 is likely to participate in the CD30 signaling pathway, and play a role in the cellular response induced by the stimulation in ALCL. It has been reported that the promoter sequence of the BCL3 gene carries two high‐affinity NF‐κB binding sites and the induction of BCL3 expression by TNF‐α is mediated by the downstream κB site.( 38 ) As ALCL cells express high levels of the BCL3 transcript in the resting state, the promoter area is presumably susceptible to transcriptional control by transcription factors. Thus, it is likely that the stimulation of CD30 activates NF‐κB, which in turn promotes the transcription of BCL3.
Another specific observation is that Bcl‐3 becomes hyperphosphorylated upon CD30 stimulation but the hyperphosphorylation was not reproduced in HL‐cells, suggesting that generation of Bcl‐3P occurs specifically in the setting of ALCL. This may be supported by a previous report in which the authors were not able to detect phosphorylation of Bcl‐3 following stimulation with TNF‐α from 293 and HeLa cells.( 39 ) Because t(2;5)(p23;q35)‐positive ALCL is characterized by increased tyrosine kinase activities induced by the NPM/ALK chimeric protein, we assumed initially that Bcl‐3 might be a target of this protein kinase. However, there was no physical interaction between Bcl‐3 and NPM/ALK observed in the coprecipitation studies, neither was immunoprecipitated Bcl‐3 protein detected by antiphosphotyrosine antibody. Instead, truncation of Bcl‐3 at a point within the C‐terminal domain abolished the modification of the protein (data not shown). It is therefore suggested that CD30‐induced protein kinase targets the serine residues of Bcl‐3, which are abundant in the C terminus. Very recently, Bcl‐3 of Karpas 299 was shown to be a substrate of glycogen‐synthase‐kinase (GSK) protein kinase, leading to degradation of Bcl‐3 through the proteasome pathway.( 40 ) At present, any possible link between the CD30‐induced hyperphosphorylation and GSK‐mediated phosphorylation of Bcl‐3 in ALCL remains unknown.
We demonstrated that stimulation of CD30 leads to both de novo synthesis of NF‐κB2/p100 precursor and processing to p52, and this response is most prominent in ALCL. The p100 precursor contains seven ankyrin repeats in its carboxy‐terminus and, like the IκB family of proteins, localizes to the cytoplasm where it functions as an inhibitor of other NF‐κB subunits.( 41 ) In contrast to p105, the processing of which is largely constitutive, p100 is expressed mostly in an unprocessed form, and recent studies have elucidated that its processing to p52 is promoted by NF‐κB‐inducing kinase( 42 ) through lymphotoxin β receptor( 19 ) or CD40.( 43 ) Thus, CD30‐induced p52 production may be mediated by similar mechanisms.
The most important observation of our study was that Bcl‐3, in response to CD30 stimulation, becomes associated with p52 and generates a (p52)2/Bcl‐3P ternary complex, while EMSA demonstrated the κB‐binding activity of the homodimer. The association was confirmed in ALCL tumor tissues, suggesting that the CD30‐mediated Bcl‐3–p52 association does occur in a fraction of neoplastic cell nuclei. Bcl‐3 can: (i) remove homodimers from κB sites so that transactivating NF‐κB heterodimers can bind and act as an antirepressor;( 44 ) (ii) form a complex with homodimers at κB sites and act as a transactivator;( 23 ) or (iii) enhance the binding of the homodimers to κB sites.( 45 ) These variable results may be related to the fact that the function of Bcl‐3 is modified by its phosphorylation status.( 18 , 46 ) The present study showed that in ALCL, Bcl‐3 is associated with the (p50)2 homodimer in the resting state, while Bcl‐3P can bind to the (p52)2 homodimer upon stimulation. It is therefore possible that the former complex acts favorably to promote cell survival as an oncogene, while generation of the latter complex is involved in apoptosis. As some members of the Bcl‐2 family, as well as other apoptosis‐related factors, are reported to be regulated by NF‐κB transcription factors,( 33 ) we propose that the (p52)2/Bcl‐3P complex, which is specifically induced in CD30‐stimulated ALCL, can modulate proapoptotic and antiapoptotic gene expression, thereby accounting for the CD30‐mediated apoptosis.
The physiological relevance of p52 and Bcl‐3 is supported by the finding that the phenotypes of p52‐deficient mice largely overlap with those of Bcl‐3 knockout mice.( 47 , 48 , 49 , 50 ) Both mice have reduced numbers of B cells, are unable to form germinal centers, are impaired in antibody responses to T‐cell‐dependent antigens, and lack follicular dendritic cell meshwork, although Bcl‐3‐deficient mice are reported to be less affected than p52‐deficient mice. Thus, Bcl‐3 seems to have the ability to boost the activity of p52, which cannot be replaced with p50. Of interest, NFKB2/LYT10 and BCL3 were both identified to be involved in recurrent chromosomal translocations of lymphoid neoplasms.( 14 , 51 )
t(2;5)(p23;q35)‐positive ALCL is characterized by the presence of the NPM/ALK fusion protein in both the nucleus and cytoplasm, expression of CD30 molecules on the cell surface and high‐level expression of Bcl‐3 in the nucleus. The present study revealed an association between CD30‐mediated signaling and Bcl‐3, which probably contributes to the apoptosis of ALCL cells. In contrast, the molecular and functional relationship of the NPM/ALK signaling cascade, which plays a pathogenetic role in ALCL, with the CD30–Bcl‐3 association remains to be determined. Finally, our study suggested that CD30‐mediated apoptosis may occur in a fraction of ALCL tumor tissues. Augmentation of this pathway by CD30‐agonistic antibodies may therefore be a promising therapeutic approach for this particular type of lymphoid neoplasm.
Acknowledgments
We are grateful to Ms Itsuko Koyanagi and Ms Yoko Sasaki for technical assistance in immunohistochemistry and cell sorting, respectively. We also thank Drs Kazunori Imada, Maki Ueda and Masakatsu Hishizawa for their considerate help. We are particularly indebted to Prof Tasuku Honjo for his support in preparing this manuscript.
References
- 1. Stein H, Foss HD, Durkop H et al. CD30+ anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood 2000; 96: 3681–95. [PubMed] [Google Scholar]
- 2. Le Beau MM, Bitter MA, Larson RA et al. The t(2;5)(p23;q35): a recurring chromosomal abnormality in Ki‐1‐positive anaplastic large cell lymphoma. Leukemia 1989; 3: 866–70. [PubMed] [Google Scholar]
- 3. Duyster J, Bai RY, Morris SW. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 2001; 20: 5623–37. [DOI] [PubMed] [Google Scholar]
- 4. Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed–Sternberg cells in Hodgkin's disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 1996; 184: 1495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishikori M, Maesako Y, Ueda C, Kurata M, Uchiyama T, Ohno H. High‐level expression of BCL3 differentiates t(2;5)(p23;q35)‐positive anaplastic large cell lymphoma from Hodgkin disease. Blood 2003; 101: 2789–96. [DOI] [PubMed] [Google Scholar]
- 6. Durkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin's disease. Cell 1992; 68: 421–7. [DOI] [PubMed] [Google Scholar]
- 7. Falini B, Pileri S, Pizzolo G et al. CD30 (Ki‐1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 1995; 85: 1–14. [PubMed] [Google Scholar]
- 8. Gruss HJ, Boiani N, Williams DE, Armitage RJ, Smith CA, Goodwin RG. Pleiotropic effects of the CD30 ligand on CD30‐expressing cells and lymphoma cell lines. Blood 1994; 83: 2045–56. [PubMed] [Google Scholar]
- 9. Masuda M, Ishida C, Arai Y et al. Dual action of CD30 antigen: anti‐CD30 antibody induced apoptosis and interleukin‐8 secretion in Ki‐1 lymphoma cells. Int J Hematol 1998; 67: 257–65. [DOI] [PubMed] [Google Scholar]
- 10. Mir SS, Richter BW, Duckett CS. Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood 2000; 96: 4307–12. [PubMed] [Google Scholar]
- 11. Drexler HG, Matsuo AY, MacLeod RA. Continuous hematopoietic cell lines as model systems for leukemia‐lymphoma research. Leuk Res 2000; 24: 881–911. [DOI] [PubMed] [Google Scholar]
- 12. Kamesaki H, Fukuhara S, Tatsumi E et al. Cytochemical, immunologic, chromosomal, and molecular genetic analysis of a novel cell line derived from Hodgkin's disease. Blood 1986; 68: 285–92. [PubMed] [Google Scholar]
- 13. Ohno H, Furukawa T, Fukuhara S et al. Molecular analysis of a chromosomal translocation, t(9;14)(p13;q32), in a diffuse large‐cell lymphoma cell line expressing the Ki‐1 antigen. Proc Natl Acad Sci USA 1990; 87: 628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ohno H, Takimoto G, McKeithan TW. The candidate proto‐oncogene bcl‐3 is related to genes implicated in cell lineage determination and cell cycle control. Cell 1990; 60: 991–7. [DOI] [PubMed] [Google Scholar]
- 15. Begum NA, Kinoshita K, Kakazu N et al. Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch. Science 2004; 305: 1160–3. [DOI] [PubMed] [Google Scholar]
- 16. Rassidakis GZ, Oyarzo MP, Medeiros LJ. BCL‐3 overexpression in anaplastic lymphoma kinase‐positive anaplastic large cell lymphoma. Blood 2003; 102: 1146–7. [DOI] [PubMed] [Google Scholar]
- 17. Eliopoulos AG, Caamano JH, Flavell J et al. Epstein–Barr virus‐encoded latent infection membrane protein 1 regulates the processing of p100 NF‐κB2 to p52 via an IKKγ/NEMO‐independent signalling pathway. Oncogene 2003; 22: 7557–69. [DOI] [PubMed] [Google Scholar]
- 18. Bundy DL, McKeithan TW. Diverse effects of BCL3 phosphorylation on its modulation of NF‐κB p52 homodimer binding to DNA. J Biol Chem 1997; 272: 33 132–9. [DOI] [PubMed] [Google Scholar]
- 19. Mordmuller B, Krappmann D, Esen M, Wegener E, Scheidereit C. Lymphotoxin and lipopolysaccharide induce NF‐κB‐p52 generation by a co‐translational mechanism. EMBO Rep 2003; 4: 82–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Atkinson PG, Coope HJ, Rowe M, Ley SC. Latent membrane protein 1 of Epstein–Barr virus stimulates processing of NF‐κB2 p100 to p52. J Biol Chem 2003; 278: 51 134–42. [DOI] [PubMed] [Google Scholar]
- 21. Xiao G, Cvijic ME, Fong A et al. Retroviral oncoprotein Tax induces processing of NF‐κB2/p100 in T cells: evidence for the involvement of IKKα. EMBO J 2001; 20: 6805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hinz M, Lemke P, Anagnostopoulos I et al. Nuclear factor κB‐dependent gene expression profiling of Hodgkin's disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J Exp Med 2002; 196: 605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bours V, Franzoso G, Azarenko V et al. The oncoprotein Bcl‐3 directly transactivates through κB motifs via association with DNA‐binding p50B homodimers. Cell 1993; 72: 729–39. [DOI] [PubMed] [Google Scholar]
- 24. Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D. The candidate proto‐oncogene Bcl‐3 encodes a transcriptional coactivator that activates through NF‐κB p50 homodimers. Genes Dev 1993; 7: 1354–63. [DOI] [PubMed] [Google Scholar]
- 25. Chang CC, Zhang J, Lombardi L, Neri A, Dalla‐Favera R. Mechanism of expression and role in transcriptional control of the proto‐oncogene NFKB‐2/LYT‐10. Oncogene 1994; 9: 923–33. [PubMed] [Google Scholar]
- 26. Falini B, Bigerna B, Fizzotti M et al. ALK expression defines a distinct group of T/null lymphomas (‘ALK lymphomas’) with a wide morphological spectrum. Am J Pathol 1998; 153: 875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, death. Cell 1994; 76: 959–62. [DOI] [PubMed] [Google Scholar]
- 28. Baker SJ, Reddy EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene 1996; 12: 1–9. [PubMed] [Google Scholar]
- 29. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281: 1305–8. [DOI] [PubMed] [Google Scholar]
- 30. Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain‐containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995; 81: 505–12. [DOI] [PubMed] [Google Scholar]
- 31. Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF‐dependent recruitment of the protein kinase RIP to the TNF receptor‐1 signaling complex. Immunity 1996; 4: 387–96. [DOI] [PubMed] [Google Scholar]
- 32. Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev 2003; 14: 193–209. [DOI] [PubMed] [Google Scholar]
- 33. Turco MC, Romano MF, Petrella A, Bisogni R, Tassone P, Venuta S. NF‐κB/Rel‐mediated regulation of apoptosis in hematologic malignancies and normal hematopoietic progenitors. Leukemia 2004; 18: 11–7. [DOI] [PubMed] [Google Scholar]
- 34. Horie R, Higashihara M, Watanabe T. Hodgkin's lymphoma and CD30 signal transduction. Int J Hematol 2003; 77: 37–47. [DOI] [PubMed] [Google Scholar]
- 35. Lee SY, Park CG, Choi Y. T cell receptor‐dependent cell death of T cell hybridomas mediated by the CD30 cytoplasmic domain in association with tumor necrosis factor receptor‐associated factors. J Exp Med 1996; 183: 669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amakawa R, Hakem A, Kundig TM et al. Impaired negative selection of T cells in Hodgkin's disease antigen CD30‐deficient mice. Cell 1996; 84: 551–62. [DOI] [PubMed] [Google Scholar]
- 37. Horie R, Watanabe M, Ishida T et al. The NPM‐ALK oncoprotein abrogates CD30 signaling and constitutive NF‐κB activation in anaplastic large cell lymphoma. Cancer Cell 2004; 5: 353–64. [DOI] [PubMed] [Google Scholar]
- 38. Brasier AR, Lu M, Hai T, Lu Y, Boldogh I. NF‐κB‐inducible BCL‐3 expression is an autoregulatory loop controlling nuclear p50/NF‐κB1 residence. J Biol Chem 2001; 276: 32 080–93. [DOI] [PubMed] [Google Scholar]
- 39. Heissmeyer V, Krappmann D, Wulczyn FG, Scheidereit C. NF‐κB p105 is a target of IκB kinases and controls signal induction of Bcl‐3‐p50 complexes. EMBO J 1999; 18: 4766–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Viatour P, Dejardin E, Warnier M et al. GSK3‐mediated BCL‐3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell 2004; 16: 35–45. [DOI] [PubMed] [Google Scholar]
- 41. Silverman N, Maniatis T. NF‐κB signaling pathways in mammalian and insect innate immunity. Genes Dev 2001; 15: 2321–42. [DOI] [PubMed] [Google Scholar]
- 42. Xiao G, Harhaj EW, Sun SC. NF‐κB‐inducing kinase regulates the processing of NF‐κB2 p100. Mol Cell 2001; 7: 401–9. [DOI] [PubMed] [Google Scholar]
- 43. Coope HJ, Atkinson PG, Huhse B et al. CD40 regulates the processing of NF‐κB2 p100 to p52. EMBO J 2002; 21: 5375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franzoso G, Bours V, Park S, Tomita‐Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl‐3 is an antagonist of p50/NF‐κB‐mediated inhibition. Nature 1992; 359: 339–42. [DOI] [PubMed] [Google Scholar]
- 45. Caamano JH, Perez P, Lira SA, Bravo R. Constitutive expression of Bc1‐3 in thymocytes increases the DNA binding of NF‐κB1 (p50) homodimers in vivo . Mol Cell Biol 1996; 16: 1342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dechend R, Hirano F, Lehmann K et al. The Bcl‐3 oncoprotein acts as a bridging factor between NF‐κB/Rel and nuclear co‐regulators. Oncogene 1999; 18: 3316–23. [DOI] [PubMed] [Google Scholar]
- 47. Franzoso G, Carlson L, Scharton‐Kersten T et al. Critical roles for the Bcl‐3 oncoprotein in T cell‐mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity 1997; 6: 479–90. [DOI] [PubMed] [Google Scholar]
- 48. Caamano JH, Rizzo CA, Durham SK et al. Nuclear factor (NF)‐κB2 (p100/p52) is required for normal splenic microarchitecture and B cell‐mediated immune responses. J Exp Med 1998; 187: 185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Franzoso G, Carlson L, Poljak L et al. Mice deficient in nuclear factor (NF)‐κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med 1998; 187: 147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Poljak L, Carlson L, Cunningham K, Kosco‐Vilbois MH, Siebenlist U. Distinct activities of p52/NF‐κB required for proper secondary lymphoid organ microarchitecture: functions enhanced by Bcl‐3. J Immunol 1999; 163: 6581–8. [PubMed] [Google Scholar]
- 51. Neri A, Chang CC, Lombardi L et al. B cell lymphoma‐associated chromosomal translocation involves candidate oncogene lyt‐10, homologous to NF‐κB p50. Cell 1991; 67: 1075–87. [DOI] [PubMed] [Google Scholar]