

Abstract
(Cancer Sci 2010; 101: 713–721)
Proteasome inhibition is a promising approach for cancer therapy. However, the mechanisms involved have not been fully elucidated. Gap junctions play important roles in the regulation of tumor cell phenotypes and mediation of the bystander effect in cancer therapy. Because the degradation of gap junction proteins involves the proteasome, we speculated that altered gap junctions might contribute to the antitumor activities of proteasome inhibition. Incubation of Hepa‐1c1c7 cells with the proteasome inhibitor MG132 elevated the levels of gap junction protein connexin 43 (Cx43) and promoted gap junctional intercellular communication. This was associated with a marked accumulation of ubiquitylated Cx43 and a significantly decreased rate of Cx43 degradation. The elevated Cx43 contributed to MG132‐induced cell apoptosis. This is shown by the observations that: (i) overexpression of Cx43 in the gap junction‐deficient LLC‐PK1 cells rendered them vulnerable to MG132‐elicited cell injury; (ii) fibroblasts derived from Cx43‐null mice were more resistant to MG‐132 compared with Cx43 wild‐type control; and (iii) the gap junction inhibitor flufenamic acid significantly attenuated cell damage caused by MG132 in Hepa‐1c1c7 cells. Further studies demonstrated that MG132 activates endoplasmic reticulum stress. Exposure of cells to the endoplasmic reticulum stress inducers thapsigargin and tunicamycin also led to cell apoptosis, which was modulated by Cx43 levels in a way similar to MG132. These results suggested that elevated Cx43 sensitizes cells to MG132‐induced cell apoptosis. Regulation of gap junctions could be an important mechanism behind the antitumor activities of proteasome inhibitors.
The proteasome plays a pivotal role in the maintenance of cell survival through controlling the degradation of important regulatory proteins. Disruption of proteasome function causes cell apoptosis, which has been explored as a novel therapeutic strategy for cancer.( 1 , 2 ) Preclinical trials have demonstrated that proteasome inhibition induces cell differentiation, suppresses cell proliferation, and promotes cell apoptosis.( 1 , 2 , 3 ) In addition, it sensitizes tumor cells to biotherapy, chemotherapy, and radiotherapy.( 4 , 5 , 6 , 7 ) The mechanisms underlying these effects are multiple, involving activation of pro‐death machineries and suppression of several cell survival signaling cascades.( 1 , 2 )
Activation of endoplasmic reticulum (ER) stress is one of the mechanisms mediating proteasome inhibition‐induced cell apoptosis.( 7 , 8 , 9 ) Interference with normal protein folding in the ER leads to accumulation of unfolded proteins. The clearance of these proteins requires a functional ER‐associated degradation pathway.( 10 , 11 ) Disruption of this pathway by proteasome inhibitors causes a build up of misfolded proteins, resulting in ER stress‐mediated cell death.( 7 , 8 , 9 )
Gap junctions, formed by specific proteins termed connexins (Cx), are intercellular channels that allow direct intercellular exchange of ions, nutrients, and small signaling molecules. Gap junctions play critical roles in the transmission of intercellular signals and in the control of cell growth, differentiation, migration, and survival.( 12 , 13 , 14 ) Dysfunction of gap junctions has been reported to be present in various tumors, and is well recognized as a promoting factor for carcinogenesis.( 15 , 16 , 17 , 18 , 19 ) Gap junctions have also been characterized as an important factor determining cell responses to conventional therapy and radiotherapy in cancer.( 16 , 17 ) The roles of gap junctions in the antitumor activities of proteasome inhibitors have not been investigated. However, several considerations have prompted us to investigate this possibility. First, the degradation of Cx involves the proteasome. As a short‐lived protein with the half‐life of only a few hours, the turnover of Cx is critically controlled by the proteasome.( 20 , 21 , 22 ) Second, gap junctions regulate cell phenotypes and govern cell fate against various stresses.( 23 , 24 , 25 ) The gap junction‐mediated bystander effect in suicide tumor gene therapy has been extensively documented.( 26 , 27 ) The antitumor activities of several chemotherapeutic agents such as retinoids and carotenoids have been described to be closely correlated with their ability to increase gap junction protein expression and function.( 17 , 28 , 29 ) Therefore, involvement of altered gap junctions in the therapeutic effects of proteasome inhibitors is highly possible.
Here, we present the first evidence that elevated Cx43 contributes to proteasome inhibitor MG132‐elicited cell death. Regulation of gap junctions could be a presently unrecognized mechanism behind the antitumor activities of proteasome inhibitors.
Materials and Methods
Reagents. Glucose‐regulated protein (GRP78), C/EBP homologous protein (CHOP), and ubiquitin antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA). Anti‐caspase‐3 antibody was from Cell Signaling (Beverly, MA, USA). FITC‐conjugated swine anti‐rabbit immunoglobulin was purchased from DAKO (Glostrup, Denmark). All other reagents, including MG132, anti‐connexin43 (Cx43), and anti‐β‐actin antibodies were obtained from Sigma‐Aldrich Japan (Tokyo, Japan).
Cells. The murine hepatoma cell line Hepa‐1c1c7 and porcine kidney epithelial cell line LLC‐PK1 were purchased from American Type Culture Collection (Manassas, VA, USA). Mouse embryonic fibroblasts were derived from the fetal offspring of mating pairs of heterozygous Cx43 knockout mice (B6,129‐Gja1 < tm1Kdr+/− mice; Jackson Laboratories, Bar Harbor, ME, USA), using a method described by Ehrlich et al. with minor modifications.( 30 ) Briefly, paired mouse forelimbs were taken from fetuses at day 18 of gestation, minced, and digested in DMEM/F12 containing 0.1% collagenase for 30 min. Freed cells were collected and cultured in DMEM/F12 medium containing 15% FBS. Cells at passages between 5 and 15 were used for this study. Genotypes of individual mice and established cell lines were analyzed by PCR.
Formazan assay. The number of viable cells was assessed by a formazan assay using Cell Counting Kit‐8 following the manufacturer`s instructions (Dojindo Laboratory, Kumamoto, Japan).
Scrape loading dye transfer assay. The scrape loading dye transfer (SLDT) assay was used to assess gap junctional intercellular communication (GJIC). Cells were exposed to culture medium containing 0.5% Lucifer Yellow. A scrape line on the monolayer was made with a surgical blade. After washing out background fluorescence, the cells were fixed and photographed with a digital camera attached to a fluorescence microscope (magnification ×200).
Immunocytochemistry. For immunochemistry, cells on glass slides were fixed in 4% formaldehyde for 10 min and permeabilized in 0.2% Triton‐X 100 for 5 min. Staining of Cx43 and ubiquitin was carried out using an anti‐Cx43 and anti‐ubiquitin antibody, respectively.( 31 , 32 )
Western blot analysis. Western blotting was carried out using the enhanced chemiluminescence system.( 31 , 32 ) Briefly, extracted cellular proteins were separated by 10% or 15% SDS‐polyacrylamide gels and electrotransferred onto polyvinylidine difluoride membranes. After blocking with 3% bovine serum albumin in PBS, the membranes were incubated with the antibodies. After washing, the filters were probed with horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse IgG, and the bands were visualized using the enhanced chemiluminescence system (Amersham Biosciences, Buckinghamshire, UK). To confirm equal loading of proteins, the filters were soaked in 62.5 mm Tris‐HCl (pH 6.8) containing 2% SDS and 100 mmβ‐mercaptoethanol for 30 min at 60°C and reprobed for β‐actin.
Immunoprecipitation. Hepa‐1c1c7 cells were treated with the indicated concentrations of MG132 for 12 h. The cells were lysed in RIPA buffer (50 mm Tris‐HCl, 150 mm NaCl, 5 mm EGTA containing 1% Triton, 0.5% deoxycholate, 0.1% SDS). The cellular lysates were homogenated, cleared, and immunoprecipitated using a rabbit polyclonal anti‐Cx43 antibody at 4°C overnight. Immune complexes were precipitated with protein‐A/G‐sephrose (Pharmacia, Piscataway, NJ, USA), and washed with RIPA buffer. The resulting pellets were resuspended in 2 × Laemmli buffer, and the proteins were resolved by electrophoresis on a 12% gradient SDS polyacrylamide gel, electrotransferred onto polyvinylidine difluoride membranes, and probed for ubiquitin using the enhanced chemiluminescence system, as described above.
Northern blot analysis. Total RNA was extracted by the single‐step method, and northern blot analysis was carried out as described previously.( 32 , 33 ) cDNA for Cx43, GRP78, and CHOP was used for the preparation of radiolabeled probes. Expression of GAPDH was used as a loading control.
Calcein AM–propidium iodide cell‐survival assay. Cell viability was evaluated by calcein AM–propidium iodide (PI) double staining following the manufacturer`s instructions (Dojindo Laboratory).
Transfection experiment. Hepa‐lclc7 cells in subconfluent culture were transfected with a ubiquitin–luciferase bioluminescence imaging reporter (Ub‐FL)( 34 ) by using Gene Juice according to the manufacturer`s instructions (Novagen, Madison, WI, US). Ub‐FL is a reporter construct that directs the production of a non‐hydrolyzable chain of ubiquitin peptides fused to firefly luciferase. Under normal conditions, Ub‐FL is rapidly degraded by the ubiquitin proteasome system. Proteasome inhibition stabilizes Ub‐FL and increases luciferase activity.( 34 ) In the present study, the transfected cells were exposed to the proteasome inhibitor MG132 and assayed for luciferase activity using the method previously described.( 31 , 33 ) LLC‐PK1 cells were transfected with pCx43‐EGFP1, control pEGFP‐N1, or mutated Cx43‐pEGFP vector.( 35 ) Clones with high levels of green fluorescent protein (GFP) were selected and used in this investigation.
Statistical analysis.
Values are expressed as means ± SE. Comparison of two populations was made by Student’s t‐test. For multiple comparisons, one‐way analysis of variance (ANOVA) followed by Dunnett’s test was employed. Both analyses were done using the SigmaStat statistical software (SPSS, Inc., Chicago, IL). P < 0.05 was considered to be a statistically significant difference.
Results
MG132 increases Cx43 protein levels and promotes GJIC. Incubation of Hepa‐1c1c7 cells with MG132 induced a time‐ and concentration‐dependent elevation in Cx43 protein levels (Fig. 1A,B). This effect of MG132 was rapid. A clear elevation of Cx43 was detectable within 3–6 h of cell exposure to 0.5 μg/mL MG132 and lasted for at least 24 h (Fig. 1A). MG132 at a concentration of 0.5 μg/mL caused a 2.5‐fold elevation in Cx43 protein level (Fig. 1C).
Figure 1.
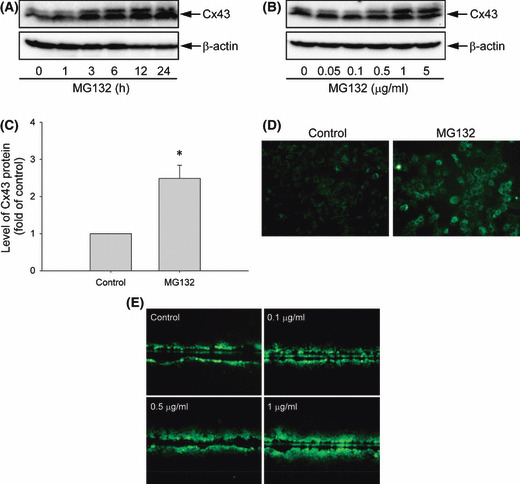
Effects of MG132 on connexin 43 (Cx43) protein expression, distribution, and function in Hepa‐1c1c7 cells. (A–C) Effects of MG132 on Cx43 protein levels. 1c1c‐7 cells were exposed to (A) 0.5 μg/mL MG132 for the indicated times or (B) various concentrations of MG132 for 12 h. The cellular protein was extracted and subjected to western blot analysis of Cx43. Expression of β‐actin is shown at the bottom as a loading control. (C) The intensities of Cx43 signal in cells treated with 0.5 μg/mL MG132 for 12 h were measured and expressed as fold induction relative to untreated control (mean ± SE, n = 5). *P < 0.01 versus untreated control. (D) Immunofluorescence staining of Cx43. Hepa‐1c1c7 cells were either left untreated or incubated with 0.5 μg/mL MG132 for 12 h, and then subjected to immunofluorescence staining of Cx43 (green). Note the obvious enhancement of Cx43 (green) at the perinuclear region and cell‐to‐cell contacts. Magnification, ×400. (E) Effects of MG132 on gap junctional intercellular communication measured by scrape loading dye transfer assay. Hepa‐1c1c7 cells were either left untreated or exposed to increasing concentrations of MG132 for 12 h. The micrographs of Lucifer Yellow diffusion into the cellular monolayer after scrape‐loading are shown. Magnification, ×200.
Elevated Cx43 was confirmed by immunofluorescence staining of cells with an anti‐Cx43 antibody. Normally, Cx43 molecules are localized at perinuclear and cell‐to‐cell contact regions (Fig. 1D). MG132 treatment pronouncedly augmented Cx43 staining at these areas.
The elevated Cx43 was associated with increased GJIC, when analyzed using SLDT assay. As shown in Figure 1E, more dye‐coupled cells were observed in MG132‐treated cells compared with the untreated control.
MG132 suppresses Cx43 degradation. To answer whether the elevated Cx43 was due to proteasome inhibition, the effects of MG132 on proteasome function and Cx43 degradation were examined. As shown in Figure 2A, MG132 induced a concentration‐dependent increase in luciferase activity in Hepa‐1c1c7 cells transiently transfected with Ub‐FL, indicating that MG132 suppresses 26S proteasome activity.( 34 ) Consistent with this result, MG132 caused a marked accumulation of ubiquitylated proteins in Hepa‐1c1c7 cells, as revealed by immunofluorescence staining (Fig. 2B) and western blot analysis using an anti‐ubiquitin antibody (Fig. 2C). The immunoprecipitation experiment demonstrated that MG132 caused a concentration‐dependent increase in Cx43 ubiquitylation (Fig. 2D).
Figure 2.
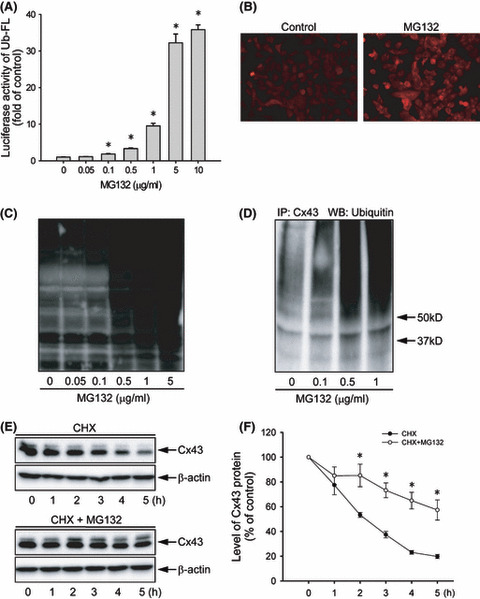
Effect of MG132 on proteasome function and connexin 43 (Cx43) degradation. (A) Effect of MG132 on ubiquitin–luciferase bioluminescence imaging reporter (Ub‐FL) activity. Hepa‐1c1c7 cells were transiently transfected with a Ub‐FL reporter and exposed to the indicated concentrations of MG132. The relative luciferase activity is expressed as fold induction over untreated control (mean ± SE, n = 4). *P < 0.01 versus untreated control. (B) Immunofluorescent staining for ubiquitin. Hepa‐1c1c7 cells were either left untreated or incubated with 0.5 μg/mL MG132 for 12 h, and then subjected to immunofluorescent staining of ubiqutin (red). Note the obvious enhanced intensity of ubiqutin (red) at the perinuclear region. Magnification, ×400. (C) Effect of MG132 on protein ubiqutiylation. Hepa‐1c1c7 cells were exposed to various concentrations of MG132 for 12 h. The cellular protein was extracted and subjected to western blotting analysis of ubiquitylated proteins. (D) Effect of MG132 on ubiquitylation of Cx43. Hepa‐1c1c7 cells were treated with the indicated concentrations of MG132 for 12 h. Cell lysates were subjected to immunoprecipitation (IP) with an anti‐Cx43 antibody and blotted with an anti‐ubiquitin antibody. (E) Effect of MG132 on Cx43 protein degradation. Hepa‐1c1c7 cells were exposed to 50 μg/mL cycloheximide (CHX) in the presence or absence of 0.5 μg/mL MG132 for the indicated times. Cellular proteins were analyzed by western blotting (WB) with an anti‐Cx43 antibody. A representative blot is shown in (E). (F) The intensity of each Cx43 signal in (E) was measured and the relative intensity of the band against its intensity at zero point are shown (mean ± SE, n = 4). *P < 0.01 versus untreated control.
To further assess the role of the proteasome in Cx43 turnover, the rate of Cx43 degradation was analyzed in the presence or absence of MG132. As shown in Figure 2E, blockade of protein synthesis using cycloheximide caused a gradual degradation of Cx43, which was largely blocked in the presence of MG132. Based on the rate of Cx43 degradation, the time required to achieve 50% degradation was estimated to be 2.2 and 5.0 h in control and MG132‐treated cells, respectively (Fig. 2F).
Involvement of elevated gap junction protein in MG132‐induced cell apoptosis in Hepa‐1c1c7 cells. During the immunochemistry experiment, we noticed that the elevated Cx43 in MG132‐treated 1c1c7 cells (Fig. 3A; upper panel) was accompanied by an increased number of apoptotic cells. Treatment of cells with 1 μg/mL MG132 for 12 h caused the appearance of rounded, shrunken, and loosely attached cells. These cells exhibited nuclear condensation and fragmentation, the typical features of apoptosis, when visualized by nuclear staining with DAPI. Quantitative analysis indicated that MG132 induced cell apoptosis in a concentration‐dependent manner (Fig. 3B).
Figure 3.
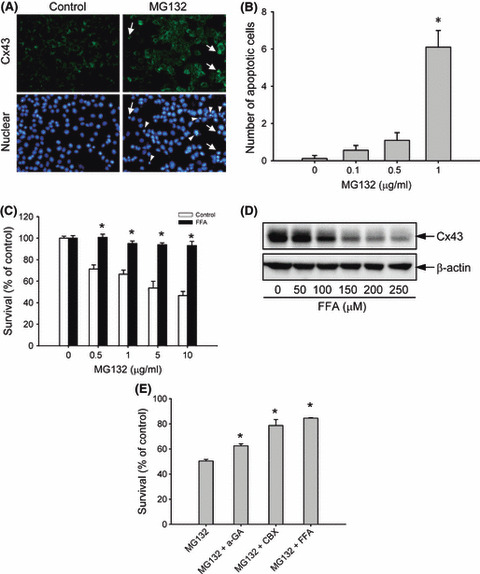
Involvement of gap junctions in MG132‐induced cell damage. (A) Concomitant induction of connexin 43 (Cx43) and cell apoptosis by MG132. Hepa‐1c1c7 cells were either left untreated or incubated with 1 μg/mL MG132 for 12 h, and then subjected to immunofluorescence staining of Cx43 (green; upper panel) and nuclei (DAPI stain, blue; lower panel). Arrow and arrow heads indicate apoptotic cells. Note the coexistence of elevated Cx43 and nuclear condensation in part of the indicated cells. Magnification, ×400. (B) The number of apoptotic cells after MG132 treatment. The cells with nuclear condensation and fragmentation were quantified and expressed as apoptotic cells per field (mean ± SE, n = 10). *P < 0.01 versus respective control. (C) Effect of the gap junction inhibitor flufenamic acid (FFA) on MG132‐elicited loss of cell viability. Hepa‐1c1c7 cells were exposed to the indicated concentrations of MG132 in the presence or absence of 150 μm FFA for 12 h. Cellular viability was determined by formazan assay. The data are expressed as a percentage of the control (mean ± SE, n = 4). *P < 0.01 versus respective control. (D) Effect of FFA on Cx43 protein levels. 1c1c‐7 cells were exposed to the indicated concentrations of FFA for 12 h. The cellular protein was extracted and subjected to western blot analysis of Cx43. Expression of β‐actin is shown at the bottom as a loading control. (E) Effects of several different gap junction inhibitors on MG132‐elicited loss of cell viability. Hepa‐1c1c7 cells were exposed to 5 μg/mL MG132 in the presence or absence of 10 μm 18‐α glycyrrhetinic (α‐GA), 10 μm carbenoxolone (CBX), or 150 μm FFA for 12 h. Cellular viability was determined by formazan assay. The data are expressed as a percentage of the control (mean ± SE, n = 4). *P < 0.01 versus MG132 alone.
To assess the role of elevated Cx43 in cell injury, MG132‐induced cytotoxicity in the presence or absence of the gap junction inhibitor was examined. As shown in Figure 3C, the gap junction inhibitor flufenamic acid (FFA) significantly attenuated the loss of cell viability induced by MG132. This effect of FFA was associated with a strong inhibition in Cx43 protein levels (Fig. 3D). Besides FFA, the gap junction inhibitors 18‐α glycyrrhetinic and carbenoxolone also greatly attenuated MG132‐elicited cell injury (Fig. 3E). These results indicated that gap junction contributes to MG132‐induced cell damage.
Expression of Cx43 in LLC‐PK1 cells renders them vulnerable to MG132‐elicited cell apoptosis. To further establish the role of elevated Cx43 in MG132‐elicited apoptosis, we transfected gap junction‐deficient LLC‐PK1 cells with a wild‐type Cx43 fused with enhanced GFP (Cx43‐EGFP)( 35 ) and examined the cell response to MG132‐induced cytotoxicity. As shown in Figure 4A, LLC‐PK1 cells expressing wild‐type Cx43‐EGFP displayed a liner localization of the fusion protein on the plasma membrane under fluorescence microscopy (Fig. 4A, lower panel). In the presence of MG132, the amount of fusion protein was markedly increased, as reflected by the enhanced EGFP fluorescence intensity and the widespread distribution. Western blot analysis confirmed the elevation of Cx43 (Fig. 4B). As a control, LLC‐PK1 cells were also transfected with a pEGFP construct (Fig. 4A, upper panel). The cellular expression and distribution of the control EGFP protein were not greatly affected by MG132.
Figure 4.
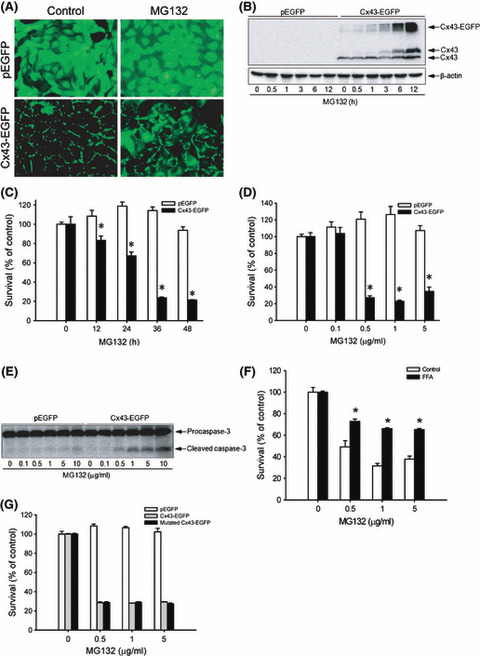
Overexpression of connexin 43 (Cx43) in LLC‐PK1 cells with MG132‐induced cell injury. (A) LLC‐PK1 cells were transfected with a vector encoding Cx43 (Cx43‐EGFP) or GFP protein (pEGFP) and clones expressing a high level of Cx43 and GFP were selected. The expression of Cx43 and GFP before and after treatment with 1 μg/mL MG132 for 12 h is shown. Note the linear distribution of Cx43‐GFP at the region of cell‐to‐cell contact in untreated cells and the increased intensity and widespread cellular distribution of Cx43‐GFP after MG132 treatment. Magnification, ×400. (B) LLC‐PK1 cells expressing pEGFP or Cx43‐EGFP were exposed to 3 μg/mL MG132 for 12 h. The cellular protein was extracted and subjected to western blot analysis of Cx43. Expression of β‐actin is shown at the bottom as a loading control. (C,D) Effects of MG132 on cellular viability. LLC‐PK1 cells were exposed to (C) 1 μg/mL MG132 for the indicated times or (D) different concentrations of MG132 for 36 h. Cellular viability was determined by formazan assay. The data are expressed as a percentage of the control (mean ± SE, n = 4). *P < 0.01 versus the respective control. (E) Cells were treated with the indicated concentrations of MG132 for 28 h. The cellular proteins were extracted and subjected to western blot analysis of caspase‐3. The top band represents procaspase‐3 (M r 35 000) and the bottom band indicates its cleaved, mature form (M r 17 000). (F) Effects of the gap junction inhibitor flufenamic acid (FFA) on MG132‐elicited loss of cell viability in Cx43‐expressing LLC‐PK1 cells. Cx43‐EGFP LLC‐PK1 cells were exposed to the indicated concentrations of MG132 in the presence or absence of 150 μm FFA for 36 h. Cellular viability was determined by formazan assay. The data are expressed as a percentage of the control (mean ± SE, n = 4). *P < 0.01 versus the respective control. (G) Expression of mutated Cx43‐EGFP in LLC‐PK1 cells on MG132‐initiated loss of cell viability. LLC‐PK1 cells expressing EGFP, Cx43‐EGFP, or communication‐free mutated Cx43‐EGFP were exposed to the indicated concentrations of MG132 for 36 h. Cellular viability was determined by formazan assay. The data are expressed as a percentage of the control (mean ± SE, n = 4).
Expression of Cx43 in LLC‐PK1 cells sensitized them to MG132‐triggered loss of cellular viability. As shown in Figure 4C,D, MG132 caused a time‐ and concentration‐dependent loss of cell viability in Cx43‐expressing LLC‐PK1 cells, but not in Cx43‐deficient control cells. In fact, MG132 at the lower concentrations tended to promote cell growth in Cx43‐deficient cells.
The increased susceptibility of Cx43‐expressing LLC‐PK1 cells to MG132 was associated with obviously increased levels of cleaved caspase‐3, suggesting that elevated Cx43 sensitizes cells to MG132‐induced apoptosis (Fig. 4E).
In further support of the role of gap junctions in MG132‐induced cell injury, the gap junction inhibitor FFA also significantly inhibited the loss of cell viability in Cx43‐EGFP LLC‐PK1 cells (Fig. 4F).
Gap junctions regulate cell phenotypes through either communication‐dependent or communication‐independent mechanisms.( 15 , 23 , 24 , 36 , 37 ) To distinguish which mechanism was responsible for sensitizing cells to MG132, LLC‐PK1 cells were transfected with a communication‐free mutated Cx43‐pEGFP( 35 ) and cell response to MG132 was evaluated. As shown in Figure 5G, expression of Δ130–137 (mutated) Cx43‐pEGFP in LLC‐PK1 cells elevated cell susceptibility to MG132 to an extent comparable to that caused by wild‐type Cx43 EGFP (Fig. 4G). This result indicated that the effect of elevated Cx43 on MG132‐elicited cell injury is GJIC‐independent.
Expression of Cx43 in LLC‐PK1 cells sensitizes cell to ER stress‐elicited cell damage. Given that ER stress mediates proteasome inhibition‐elicited cell apoptosis,( 7 , 8 , 9 ) we asked whether cell response to ER stress could also be influenced by gap junctions. To address this question, we first confirmed that MG132 was able to activate ER stress. As shown in Figure 5A, MG132 activated ER stress in LLC‐PK1 cells, as revealed by the elevated mRNA expression of GRP78 and CHOP. As expected, the ER stress inducers thapsigargin (TG) and tunicamycin (TM) also elevated GRP78 and CHOP (Fig. 5B). There was no obvious difference in the levels of GRP78 and CHOP between control and Cx43‐EGFP LLC‐PK1 cells in both basal and stimulated situations. We therefore proceeded to examine the influence of Cx43 on ER stress‐triggered cell death in LLC‐PK1 cells. Treatment of LLC‐PK1 cells with TG and TM resulted in a concentration‐dependent loss of cell viability, which was significantly more severe in Cx43‐expressing LLC‐PK1 cells compared with Cx43‐deficient control cells (Fig. 5C,D). Consistent with this, Cx43‐expressing LLC‐PK1 cells displayed a much stronger activation of caspase‐3 after TG treatment (Fig. 5E). These results indicated that elevated Cx43 sensitizes cells to ER stress‐elicited apoptosis.
Figure 5.
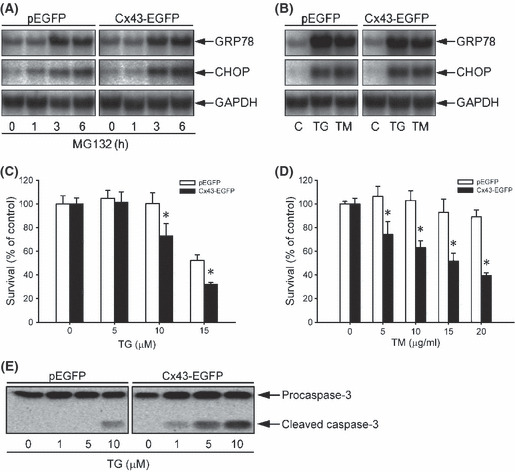
Induction of endoplasmic reticulum (ER) stress by MG132 and influence of connexin 43 (Cx43) expression on ER stress‐elicited cell injury in LLC‐PK1 cells. (A,B) Induction of ER stress by MG132, thapsigargin (TG), and tunicamycin (TM). LLC‐PK1 cells were treated with (A) 1 μg/mL MG132 for the indicated time or (B) 100 nm TG or 5 μg/mL TM for 6 h. Cellular RNA was extracted and subjected to northern blot analysis of GRP78 and CHOP. Expression of GAPDH is shown at the bottom as a loading control. (C,D) Induction of cytotoxicity in LLC‐PK1 cells expressing different amounts of Cx43. Cells were exposed to the indicated concentrations of (C) TG or (D) TM for 36 and 60 h respectively. Cellular viability was determined by formazan assay. The data are expressed as a percentage of cellular survival normalized against the untreated control (mean ± SE, n = 4). *P < 0.01 versus the respective control. (E) Different activation of caspase‐3 by TG in LLC‐PK1 cells expressing different amounts of Cx43. Cells were treated with the indicated concentrations of TG for 24 h. The cellular proteins were extracted and subjected to western blot analysis for caspase‐3.
Fibroblasts derived from wild‐type and Cx43 knockout littermates exhibit different response to MG132‐ and ER stress‐elicited cell damage. To further establish the role of elevated Cx43 in MG132‐ and ER stress‐elicited cell injury, we have examined the difference between fibroblasts derived from Cx43 wild‐type (+/+) and Cx43 knockout (−/−) littermates. As shown in Figure 6A, Cx43 was only detectable in Cx43+/+ and not in Cx43−/− fibroblasts. Consistent with this, only Cx43+/+ cells had functional GJIC, as analyzed by SLDT assay (data not shown). Incubation of wild‐type Cx43+/+ fibroblasts with MG132 elevated Cx43 protein levels. Longer incubation with a higher concentration of MG132 (5 μg/mL) led to the appearance of numerous shrunken and loosely adherent cells under a light microscope (data not shown). These cells were identified as either intense green (Fig. 6B, calcein AM) or red (Fig. 6B, PI) by calcein AM–PI staining, representing early apoptotic and dead cells, respectively.( 38 ) In contrast to the intensely stained and deformed apoptotic cells, the living cells were well spread and stained as light green. As shown in Figure 6B, far more intensely stained green and red cells were observed in Cx43+/+ fibroblasts after incubation with MG132, compared with Cx43−/− fibroblasts. The formazan assay demonstrated that MG132 caused a concentration‐dependent loss of cell viability in Cx43+/+ cells, but not in Cx43−/− cells. Interestingly, similar to the result obtained with Cx43‐deficient LLC‐PK1 cells, MG132 at the lower concentrations also promoted proliferation of Cx43−/− fibroblasts (Fig. 6C).
Figure 6.
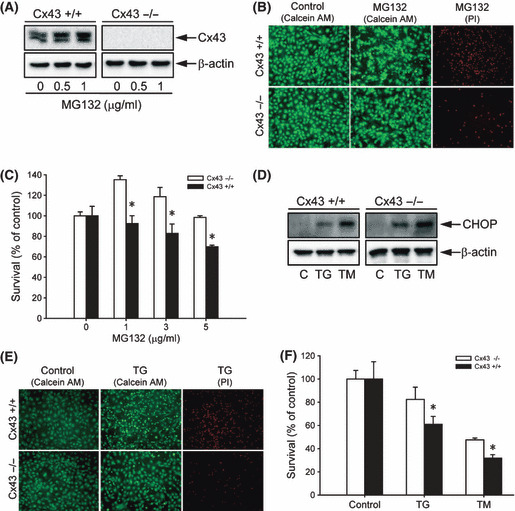
Influence of connexin 43 (Cx43) levels on cell response to MG132‐ and endoplasmic reticulum (ER) stress‐elicited cell injury. (A) Effects of MG132 on Cx43 protein levels in fibroblasts from Cx43+/+ and Cx43−/− littermates. Cx43+/+ and Cx43−/− fibroblasts were exposed to the indicated concentrations of MG132 for 12 h or left untreated. The expression of Cx43 was determined by western blot analysis. β‐Actin levels shown at the bottom of the blots indicate equal protein loading. (B,C) Cell viability of Cx43+/+ and Cx43−/− fibroblasts after MG132 treatment. (B) Cells were treated with 1 μg/mL MG132 for 36 h or left untreated (control). The living (green), early apoptotic (intense green), and dead cells (red) were identified by calcein AM/PI staining. (C) Fibroblasts were exposed to the indicated concentrations of MG132 for 36 h. Cell viability was evaluated by formazan assay. The data are expressed as percentage of the control (mean ± SE, n = 4). *P < 0.01 versus the respective control. (D) Induction of ER stress by MG132 in fibroblasts. Cx43+/+ and Cx43−/− fibroblasts were either exposed to 100 nm thapsigargin (TG) or 5 μg/mL tunicamycin (TM) for 12 h or left untreated. The expression of CHOP was determined by western blot analysis. (E) Cell viability in Cx43+/+ and Cx43−/− fibroblasts following ER stress as evaluated by calcein AM–propidium iodide (PI) double staining. Fibroblasts were treated with 5 μm TG for 36 h or left untreated. The living (green), early apoptotic (intense green), and dead cells (red; lower panel) were identified by calcein AM–PI staining. (F) Cell viability as evaluated by formazan assay. Cells were either exposed to 5 μm TG or 20 μg/mL TM for 48 h. The data are expressed as a percentage of the control (mean ± SE, n = 4). *P < 0.01 versus the respective control.
We also examined the fibroblast response to ER stress‐elicited cell injury. As shown in Figure 6D, the ER stress inducers TG and TM caused ER stress in fibroblasts, as evidenced by the elevated CHOP protein level. Exposure of fibroblasts to these chemicals resulted in cell injury, which was found to be far more severe in wild‐type Cx43+/+ fibroblasts, as revealed by calcein AM–PI staining (Fig. 6E) and formazan assay (Fig. 6F). These experiments further indicated that Cx43 levels regulate the cell response to MG132‐ and ER stress‐induced cell damage.
Discussion
In the present study, we demonstrated that upregulation of gap junctions is an important mechanism implicated in the antitumor activities of the proteasome inhibitor MG132. Exposure of Hepa‐1c1c7 cells to MG132 elevated Cx43 protein levels and promoted GJIC. This effect was caused by proteasome inhibition, because MG132 at the concentrations used effectively disrupted proteasome function, as evidenced by the significant elevation of Ub‐FL activity( 34 ) and marked accumulation of ubiquitylated Cx43. Furthermore, MG132 dramatically decreased the rate of Cx43 degradation and prolonged the half‐life of Cx43. This evidence thus support a predominant role of the proteasome in Cx43 degradation. This conclusion is consistent with previous studies in several other cell types.( 20 , 21 , 22 ) Of note, increased levels of Cx43 protein could also result from the enhanced Cx43 synthesis. However, the rapid elevation of Cx43 following MG132 addition (within 3–6 h) made this alternative less likely. Indeed, we did not find an increase in Cx43 mRNA levels after treatment of Hepa‐1c1c7 cells with MG132 (data not shown).
The elevated Cx43 contributed to MG132‐elecited cell apoptosis. Several observations supported this idea. First, MG132‐induced cytotoxicity in Hepa‐1c1c7 cells was associated with elevated levels of Cx43 protein and increased GJIC. Inhibition of gap junctions with FFA, 18‐α glycyrrhetinic, or carbenoxolone significantly attenuated the cytotoxic effects of MG132. Second, overexpression of Cx43 in gap junction‐deficient LLC‐PK1 cells sensitized them to MG132‐induced cell apoptosis. In contrast, fibroblasts from Cx43 knockout mice were resistant. Several investigators have reported that ER stress mediates proteasome inhibitor‐induced cell injury.( 7 , 8 , 9 ) Consistent with these reports, MG132 activated ER stress in all of the cell types tested in the current investigation (data not shown). In addition, ER stress‐elicited cell apoptosis was enhanced by Cx43 in a manner similar to MG132.
The mechanisms involved in the effects of gap junction are presently unclear. The possibilities include: (1) GJIC‐dependent and/or independent regulation of the cell survival signaling pathway; and (2) induction or exaggeration of ER stress by Cx43 overexpression. Under several pathological situations, gap junctions are known to be able to transfer molecules like superoxide and calcium ions to propagate a toxic response.( 15 , 23 , 24 ) A similar scenario could occur in the current investigation, because production of superoxide and induction of ER Ca2+ release by proteasome inhibitors and their roles in induction of cell apoptosis have been reported.( 9 , 39 ) However, contradictory to this speculation, transfection of LLC‐PK1 cells with a communication‐free mutated Cx43 enhanced cell susceptibility to MG132 to an extent comparable to wild‐type Cx43, suggesting that the effect of Cx43 was communication‐independent. At present, the mechanisms underlying the communication‐independent effect of Cx43 are unclear. Given that Cx43 molecules are transported to the cell surface via the conventional secretory pathway,( 40 , 41 ) one would expect that Cx43 overexpression could increase the load of protein in the ER, inducing or exaggerating ER stress. However, our data also did not support this speculation. No difference was found in the levels of the ER stress markers GRP78 and CHOP between Cx43‐expressing and control LLC‐PK1 cells. It appears that Cx43 regulated the downstream response of cells to ER stress, rather than the level of ER stress itself. In line with this conclusion, caspase 3, a protease critically involved in the initiation of ER‐induced apoptosis, was activated by MG132 and TG in Cx43‐expressing LLC‐PK1 cells. In addition, a previous study by Huang et al. demonstrated that overexpression of Cx43 increases cell susceptibility to chemotherapeutic agents in a communication‐independent manner via inhibition of the apopotosis inhibitor bcl‐2.( 42 )
Recently, we have reported that ER stress downregulates gap junction protein expression and function.( 43 ) Interestingly, although MG132 also induced ER stress in Hepa‐1c1c7 cells, it elevated rather than suppressed gap junctions in the current investigation. This obvious discrepancy may be explained by the fact that ER stress‐elicited reduction of gap junction proteins was largely due to the accelerated degradation of Cx molecules following activation of the ER‐associated degradation pathway, that is, the proteasome degradation pathway. Indeed, in our previous studies, we have observed that inhibition of proteasomes with MG132 could largely prevent TG‐ and TM‐induced reduction of Cx43 levels.( 43 )
Multiple mechanisms have been shown to be involved in the antitumor activities of proteasome inhibitors. Apart from induction of ER stress‐reactive oxygen species,( 44 ) proteasome inhibition also results in release of cytochrome c,( 45 ) suppression of nuclear factor‐κB activity,( 5 , 6 ) activation of the death receptor pathway,( 46 ) and sensitization of cells to killing by tumor necrosis factor.( 4 ) Interestingly, gap junctions are also able to transmit and propagate cellular apoptosis triggered by tumor necrosis factor‐α and intracellular injection of cytochrome c.( 47 , 48 ) Gap junctions may sensitize cells to proteasome inhibitor‐induced apoptosis via propagation and amplification of the effects of multiple pro‐death machineries. It is worth mentioning that gap junctions have also been documented to alleviate cellular injury in several pathological situations.( 25 ) The reasons for the conflicting effects are presently unclear. It is possible that the effects of gap junctions on cell damage may differ, depending on the property of stimuli, the magnitude of injury, as well as the cell and tissue type involved.
Proteasome inhibition sensitizes cancer cells to biotherapy, chemotherapy, and radiotherapy.( 4 , 5 , 6 , 7 ) However, the mechanisms involved have not been fully elucidated. Gap junctions may mediate the synergistic effects of proteasome inhibitors in combinational cancer therapy. In support of this speculation, dysfunction of gap junctions in tumor cells has been documented to be closely related to drug resistance. Upregulation of gap junctions sensitizes cells to a variety of cancer chemotherapeutic agents.( 24 , 28 , 48 , 49 , 50 )
Our findings may have important clinical implications for therapeutic utilization of proteasome inhibitors in tumors. First, we characterized gap junctions as a novel mechanism underlying the antitumor activities of proteasome inhibitors. Given that gap junctions exert multiple tumor‐suppressing effects on various tumors,( 15 , 16 , 17 , 18 , 19 , 51 ) upregulation of the gap junction itself could have significant implications. The concomitant induction of gap junction protein expression and several pro‐death machineries such as ER stress by proteasome inhibition may explain why proteasome inhibitors have potent antitumor activities, even when they are used alone. Second, our findings indicate that gap junctions are a critical factor governing the cell response to proteasome inhibitors. Several human tumors are less susceptible to proteasome inhibitor‐induced apoptosis.( 1 , 2 ) However, the molecular mechanisms regulating cell responses to proteasome inhibitors are largely unknown. The different cellular expression levels of gap junction protein could underlie the varied cell response to proteasome inhibitors. From a therapeutic standpoint, enhancement of gap junction protein expression and function might represent a novel approach to sensitize cancer cells toward proteasome inhibitors. Third, our findings also indicate that gap junctions might mediate the synergistic effects of proteasome inhibitors in combinational cancer therapy. In fact, modulation of gap junctions has been described to increase the efficacy of chemotherapy, radiotherapy, and gene therapy in cancer.( 15 , 16 , 17 , 24 , 28 , 51 ) The rapid, potent, and cell type‐non‐specific induction of gap junctions by proteasome inhibitors indicates that proteasome inhibition could be an ideal approach to modulate gap junctions and to increase the efficacy of tumor therapies.( 22 )
In conclusion, our study revealed that upregulation of gap junction protein expression and function is a presently unrecognized mechanism underlying the antitumor activities of proteasome inhibitors. Gap junctions influence cell susceptibility to proteasome inhibitors, and modulation of gap junctions could be a promising way to increase the therapeutic efficacy of proteasome inhibitors.
Acknowledgments
This work was supported in part by grants from the Takeda Science Foundation, Japan–China Medical Association, and Grants‐in‐Aid for Scientific Research no. 17659255 and 20590953 to J. Yao from the Ministry of Education, Science, Sports, and Culture, Japan.
References
- 1. Daviet L, Colland F. Targeting ubiquitin specific proteases for drug discovery. Biochimie 2008; 90: 270–83. [DOI] [PubMed] [Google Scholar]
- 2. Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004; 5: 417–21. [DOI] [PubMed] [Google Scholar]
- 3. Hideshima T, Richardson P, Chauhan D et al. The proteasome inhibitor PS‐341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61: 3071–6. [PubMed] [Google Scholar]
- 4. Conticello C, Adamo L, Giuffrida R et al. Proteasome inhibitors synergize with tumor necrosis factor‐related apoptosis‐induced ligand to induce anaplastic thyroid carcinoma cell death. J Clin Endocrinol Metab 2007; 92: 1938–42. [DOI] [PubMed] [Google Scholar]
- 5. Cusack JC Jr, Liu R, Houston M et al. Enhanced chemosensitivity to CPT‐11 with proteasome inhibitor PS‐341: implications for systemic nuclear factor‐kappaB inhibition. Cancer Res 2001; 61: 3535–40. [PubMed] [Google Scholar]
- 6. Russo SM, Tepper JE, Baldwin AS Jr et al. Enhancement of radiosensitivity by proteasome inhibition: implications for a role of NF‐kappaB. Int J Radiat Oncol Biol Phys 2001; 50: 183–93. [DOI] [PubMed] [Google Scholar]
- 7. Nawrocki ST, Carew JS, Pino MS et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress‐mediated apoptosis. Cancer Res 2005; 65: 658–66. [DOI] [PubMed] [Google Scholar]
- 8. Nawrocki ST, Carew JS, Dunner K Jr et al. Bortezomib inhibits PKR‐like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res 2005; 65: 510–9. [DOI] [PubMed] [Google Scholar]
- 9. Fribley A, Zeng Q, Wang C‐Y. Proteasome inhibitor PS‐341 induces apoptosis through induction of endoplasmic reticulum stress‐reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol 2004; 24: 9695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res 2005; 569: 29–63. [DOI] [PubMed] [Google Scholar]
- 11. Kitamura M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am J Physiol Renal Physiol 2008; 295: F323–34. [DOI] [PubMed] [Google Scholar]
- 12. Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev 2003; 83: 1359–400. [DOI] [PubMed] [Google Scholar]
- 13. Yao J, Oite T, Kitamura M. Gap junctional intercellular communication in the juxtaglomerular apparatus. Am J Physiol Renal Physiol 2009; 296: F939–46. [DOI] [PubMed] [Google Scholar]
- 14. Yao J, Zhu Y, Morioka T, Oite T, Kitamura M. Pathophysiological roles of gap junction in glomerular mesangial cells. J Membr Biol 2007; 217: 123–30. [DOI] [PubMed] [Google Scholar]
- 15. Chipman JK, Mally A, Edwards GO. Disruption of gap junctions in toxicity and carcinogenicity. Toxicol Sci 2003; 71: 146–53. [DOI] [PubMed] [Google Scholar]
- 16. Mesnil M, Crespin S, Avanzo JL, Zaidan‐Dagli M‐L. Defective gap junctional intercellular communication in the carcinogenic process. Biochim Biophys Acta 2005; 1719: 125–45. [DOI] [PubMed] [Google Scholar]
- 17. King TJ, Bertram JS. Connexins as targets for cancer chemoprevention and chemotherapy. Biochim Biophys Acta 2005; 1719: 146–60. [DOI] [PubMed] [Google Scholar]
- 18. McLachlan E, Shao Q, Wang H‐l, Langlois S, Laird DW. Connexins act as tumor suppressors in three‐dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res 2006; 66: 9886–94. [DOI] [PubMed] [Google Scholar]
- 19. Shao Q, Wang H, McLachlan E, Veitch GIL, Laird DW. Down‐regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res 2005; 65: 2705–11. [DOI] [PubMed] [Google Scholar]
- 20. Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc Res 2004; 62: 256–67. [DOI] [PubMed] [Google Scholar]
- 21. Laing JG, Beyer EC. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J Biol Chem 1995; 270: 399–403. [DOI] [PubMed] [Google Scholar]
- 22. Musil LS, Le A‐CN, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem 2000; 275 (25): 207–15. [DOI] [PubMed] [Google Scholar]
- 23. Lin JH, Weigel H, Cotrina ML et al. Gap‐junction‐mediated propagation and amplification of cell injury. Nat Neurosci 1998; 1: 494–500. [DOI] [PubMed] [Google Scholar]
- 24. Azzam EI, De Toledo SM, Little JB. Direct evidence for the participation of gap junction‐mediated intercellular communication in the transmission of damage signals from alpha ‐particle irradiated to nonirradiated cells. Proc Natl Acad Sci U S A 2001; 98: 473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andrade‐Rozental AF, Rozental R, Hopperstad MG, Wu JK, Vrionis FD, Spray DC. Gap junctions: the “kiss of death” and the “kiss of life”. Brain Res Rev 2000; 32: 308–15. [DOI] [PubMed] [Google Scholar]
- 26. Hamel W, Magnelli L, Chiarugi VP, Israel MA. Herpes simplex virus thymidine kinase/ganciclovir‐mediated apoptotic death of bystander cells. Cancer Res 1996; 56: 2697–702. [PubMed] [Google Scholar]
- 27. Mesnil M, Yamasaki H. Bystander effect in herpes simplex virus‐thymidine kinase/ganciclovir cancer gene therapy: role of gap‐junctional intercellular communication. Cancer Res 2000; 60: 3989–99. [PubMed] [Google Scholar]
- 28. Hossain MZ, Wilkens LR, Mehta PP, Loewenstein W, Bertram JS. Enhancement of gap junctional communication by retinoids correlates with their ability to inhibit neoplastic transformation. Carcinogenesis 1989; 10: 1743–8. [DOI] [PubMed] [Google Scholar]
- 29. Claudia G, Sáez LV, Montoya M, Eugenín E, Alvarez MG. Increased gap junctional intercellular communication is directly related to the anti‐tumor effect of all‐trans‐retinoic acid plus tamoxifen in a human mammary cancer cell line. J Cell Biochem 2003; 89: 450–61. [DOI] [PubMed] [Google Scholar]
- 30. Ehrlich HP, Gabbiani G, Meda P. Cell coupling modulates the contraction of fibroblast‐populated collagen lattices. J Cell Physiol 2000; 184: 86–92. [DOI] [PubMed] [Google Scholar]
- 31. Yao J, Kitamura M, Zhu Y et al. Synergistic effects of PDGF‐BB and cAMP‐elevating agents on expression of connexin43 in mesangial cells. Am J Physiol Renal Physiol 2006; 290: F1083–93. [DOI] [PubMed] [Google Scholar]
- 32. Yao J, Hiramatsu N, Zhu Y et al. Nitric oxide‐mediated regulation of connexin43 expression and gap junctional intercellular communication in mesangial cells. J Am Soc Nephrol 2005; 16: 58–67. [DOI] [PubMed] [Google Scholar]
- 33. Yokouchi M, Hiramatsu N, Hayakawa K et al. Atypical, bidirectional regulation of cadmium‐induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ 2007; 14: 1467–74. [DOI] [PubMed] [Google Scholar]
- 34. Luker GD, Pica CM, Song J, Luker KE, Piwnica‐Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat Med 2003; 9: 969–73. [DOI] [PubMed] [Google Scholar]
- 35. Oyamada Y, Zhou W, Oyamada H, Takamatsu T, Oyamada M. Dominant‐negative connexin43‐EGFP inhibits calcium‐transient synchronization of primary neonatal rat cardiomyocytes. Exp Cell Res 2002; 273: 85–94. [DOI] [PubMed] [Google Scholar]
- 36. Qin H, Shao Q, Curtis H et al. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J Biol Chem 2002; 277 (29): 132–8. [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y‐W, Kaneda M, Morita I. The gap junction‐independent tumor‐suppressing effect of connexin 43. J Biol Chem 2003; 278: 852–6. [DOI] [PubMed] [Google Scholar]
- 38. Gatti R, Belletti S, Orlandini G, Bussolati O, Dall’Asta V, Gazzola GC. Comparison of Annexin V and Calcein‐AM as early vital markers of apoptosis in adherent cells by confocal laser microscopy. J Histochem Cytochem 1998; 46: 895–900. [DOI] [PubMed] [Google Scholar]
- 39. Nawrocki ST, Carew JS, Pino MS et al. Aggresome disruption: a novel strategy to enhance bortezomib‐induced apoptosis in pancreatic cancer cells. Cancer Res 2006; 66: 3773–81. [DOI] [PubMed] [Google Scholar]
- 40. VanSlyke JK, Musil LS. Dislocation and degradation from the ER are regulated by cytosolic stress. J Cell Biol 2002; 157: 381–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thomas T, Jordan K, Simek J et al. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J Cell Sci 2005; 118: 4451–62. [DOI] [PubMed] [Google Scholar]
- 42. Huang RP, Hossain MZ, Huang R, Gano J, Fan Y, Boynton AL. Connexin 43 (cx43) enhances chemotherapy‐induced apoptosis in human glioblastoma cells. Int J Cancer 2001; 92: 130–8. [PubMed] [Google Scholar]
- 43. Huang T, Wan Y, Zhu Y et al. Downregulation of gap junction expression and function by endoplasmic reticulum stress. J Cell Biochem 2009; 107: 973–83. [DOI] [PubMed] [Google Scholar]
- 44. Emanuele S, Calvaruso G, Lauricella M et al. Apoptosis induced in hepatoblastoma HepG2 cells by the proteasome inhibitor MG132 is associated with hydrogen peroxide production, expression of Bcl‐XS and activation of caspase‐3. Int J Oncol 2002; 21: 857–65. [DOI] [PubMed] [Google Scholar]
- 45. Wagenknecht B, Hermisson M, Groscurth P, Liston P, Krammer PH, Weller M. Proteasome inhibitor‐induced apoptosis of glioma cells involves the processing of multiple caspases and cytochrome c release. J Neurochem 2000; 75: 2288–97. [DOI] [PubMed] [Google Scholar]
- 46. Liu X, Yue P, Chen S et al. The proteasome inhibitor PS‐341 (bortezomib) up‐regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL‐induced apoptosis despite up‐regulation of c‐FLIP and survivin expression in human NSCLC cells. Cancer Res 2007; 67: 4981–8. [DOI] [PubMed] [Google Scholar]
- 47. Frank DK, Szymkowiak B, Josifovska‐Chopra O, Nakashima T, Kinnally KW. Single‐cell microinjection of cytochrome c can result in gap junction‐mediated apoptotic cell death of bystander cells in head and neck cancer. Head Neck 2005; 27: 794–800. [DOI] [PubMed] [Google Scholar]
- 48. Wang M, Berthoud VM, Beyer EC. Connexin43 increases the sensitivity of prostate cancer cells to TNFα‐induced apoptosis. J Cell Sci 2007; 120: 320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saez CG, Velasquez L, Montoya M, Eugenin E, Alvarez MG. Increased gap junctional intercellular communication is directly related to the anti‐tumor effect of all‐trans‐retinoic acid plus tamoxifen in a human mammary cancer cell line. J Cell Biochem 2003; 89: 450–61. [DOI] [PubMed] [Google Scholar]
- 50. Sato H, Senba H, Virgona N et al. Connexin 32 potentiates vinblastine‐induced cytotoxicity in renal cell carcinoma cells. Mol Carcinog 2007; 46: 215–24. [DOI] [PubMed] [Google Scholar]
- 51. Trosko JE, Chang CC. Mechanism of up‐regulated gap junctional intercellular communication during chemoprevention and chemotherapy of cancer. Mutat Res 2001; 480‐481: 219–29. [DOI] [PubMed] [Google Scholar]